
Abstract
Sepsis-associated encephalopathy is a common neurological complication of sepsis and is responsible for higher mortality and poorer long-term outcomes in septic patients. Sepsis-associated encephalopathy symptoms can range from mild delirium to deep coma, which occurs in up to 70% of patients in intensive care units. The pathological changes in the brain associated with sepsis include cerebral ischaemia, cerebral haemorrhage, abscess and progressive multifocal necrotic leukoencephalopathy. Several mechanisms are involved in the pathogenesis of sepsis-associated encephalopathy, such as blood–brain barrier dysfunction, cerebral blood flow impairment, glial cell activation, leukocyte transmigration, and neurotransmitter disturbances. These events are interrelated and influence each other, therefore they do not act as independent factors. This review is focused on new evidence showing the pathological process of sepsis-associated encephalopathy.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Sepsis and severe sepsis (sepsis accompanied by acute multiple organ dysfunction syndrome) are defined as life-threatening organ dysfunction caused by a dysregulated host response to infection [1]. This condition is the most common and leading cause of intensive care unit mortality worldwide. According to a recent international study, approximately 31.5 million sepsis deaths, 19.4 million severe sepsis deaths, and 5.3 million deaths have been reported annually [2]. Therefore, sepsis, especially severe sepsis, is an important public health problem and frequently a fatal condition of patients in intensive care units. Multiple organ system dysfunction, especially in the nervous system, occurs with sepsis. This indirectly increases the risk of sepsis-associated encephalopathy (SAE). A retrospective analysis showed that the incidence of neurological dysfunction in patients with sepsis is as high as 48.9%, second only to cardiac dysfunction [3].
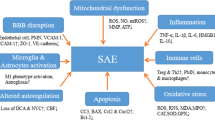
Sepsis-associated encephalopathy, considered multifocal brain dysfunction because of a dysregulated host response without primary central nervous system (CNS) infection, is the most common cause of encephalopathy in intensive care units [4]. Survivors of SAE exhibit long periods of neurological sequelae, particularly neurocognitive deterioration [5], and the principal clinical manifestations of SAE may range from mild symptoms, such as aprosexia and disorientation to delirium or coma [6]. Due to the lack of early diagnostic criteria, SAE patients are more serious, have a higher risk of death, and are prone to long-term cognitive dysfunction than non-SAE patients [7]. However, the pathogenesis underlying SAE remains unclear. Emerging evidence shows that some mechanisms, such as blood–brain barrier (BBB) dysfunction [5], cerebral blood flow (CBF) impairment [8], glial cell activation [9], leukocyte transmigration [10], and neurotransmitter disturbances [11], have been considered potential causative factors. The combination and synergism of all these contributors could be the underlying mechanism of SAE, but the exact interplay and connection between them is unclear.
BBB dysfunction
The BBB is a regulated interface separating the CNS from the peripheral circulation, which prevents the entry of toxic substances into the CNS and maintains CNS homeostasis [12]. The BBB is composed of vascular endothelial cells surrounded by the basal membrane, tight junction proteins, pericytes, end-feet of astrocytes and microglia [13]. During the early stages of sepsis, BBB alterations were observed among septic animal models [14]. Systemic inflammation and oxidative stress are crucial in mediating BBB integrity loss during sepsis.
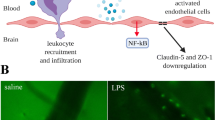
Under septic conditions, inflammatory cytokines such as IL-1β and TNF-α are systemically elevated [15]. Moreover, BBB damage and tight junction protein downregulation are closely related to the severity of sepsis and systemic inflammation [16]. The interaction of lipopolysaccharide (LPS) with TLR4 in endothelial cells can activate NF-κB through the MyD88 signalling pathway and GEF-H1-RhoA signalling pathway [17], and NF-κB is responsible for the activation of genes that encode proinflammatory cytokines and chemokines, such as TNF-α, IL-1β, and IL-6 [18]. LPS and proinflammatory cytokines can result in a significant decrease in occludin expression via the p38MAPK/JNK pathways and induce alterations in cell morphology and permeability [19]. Moreover, polymerase δ-interacting protein 2 has been reported to mediate LPS-induced BBB disruption by regulating NF-κΒ subunit p65 activation and Cox-2 as well as prostaglandin E2 induction [5]. In response to systemic inflammation, inflammatory mediators can disrupt the BBB and enter the brain to promote the activation of microglia [20]. Persistent microglial activation contributes to the generation of inflammatory cytokines and reactive oxygen species, which perpetuates a vicious cycle and aggravates BBB dysfunction in patients with sepsis [21] (Fig. 1).

Proposed pathological process of BBB dysfunction during SAE. The BBB plays an integral role in separating the CNS from the peripheral circulation under healthy conditions. In sepsis, systemic inflammation can destroy barrier functional integrity and promote the activation of microglia. Activation of microglial cells and astrocytes induces the generation of inflammatory cytokines and reactive oxygen species to aggravate BBB dysfunction. In addition, proteolytic enzymes derived from leukocyte transmigration into the brain parenchyma can induce BBB impairment
In addition to the abovementioned factors, there are many other contributors to BBB failure under sepsis conditions, such as Drp1-Fis1-mediated mitochondrial dysfunction [22], alteration of sphingolipid metabolism in endothelial cells [23], tight junction downregulation mediated by matrix metalloproteinases [24] and detachment of pericytes from the basal lamina [14]. There is other evidence to indicate that the Omi/HtrA2 pathway manipulates LPS-induced endothelial cell apoptosis by translocating from mitochondria to the cytosol and inducing X-linked inhibitor of apoptosis protein degradation. In addition, Omi/HtrA2 also participated in the decline of occludin, claudin-5 and ZO-1 expression [25].
Taken together, various factors can result in BBB dysfunction, whereas BBB abnormalities in turn change amino acid transportation [26] and promote neuronal damage, apoptosis and brain oedema [27]. In addition, SAE can occur in the absence of BBB breakdown and is accompanied by increased water diffusion anisotropy and altered glial cell morphology in the white matter of the brain [28].
CBF impairment
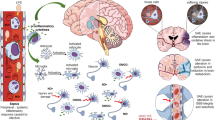
CBF is strictly regulated to ensure energy and oxygen supply in the brain, which is determined by cerebral perfusion pressure, cardiac output and small cerebral vascular tone [29]. In the early phase of sepsis, cerebral microcirculatory impairment occurs even after restoration of adequate global haemodynamics [30]. Hypotensive episodes and dysregulated autoregulation also contribute to cerebral hypoperfusion [31], which occurs first in the cerebral cortex [28]. Decreased cerebral oxygenation leads to neuronal anoxia and apoptosis [32]. Furthermore, hypoperfusion of the brain can cause elevated PaCO2. In hypercapnia, extracellular pH is reduced, and acid-sensing ion channel-1 A is activated by extracellular acidosis. The activation of acid-sensing ion channel-1 A is associated with CO2-induced NO production as well as vasodilation and subsequent increases in CBF [33, 34]. Increased vascular bed perfusion leads to increased vascular hydrostatic pressure, which leads to cerebral oedema and aggravates brain injury [35]. In addition, studies have shown that severe hypercapnia (PaCO2 100-120 mmHg) can result in higher AQP4 levels and brain oedema, ultimately aggravating brain damage [36]. Cerebrospinal fluid (CSF) decreases to maintain stable intracranial pressure when CBF increases during hypercapnia [37]. CSF plays a vital role in transporting nutrients and protein clearance in the CNS [38]. Therefore, we hypothesized that decreased CSF in hypercapnia may lead to the accumulation of metabolites and ultimately aggravate the formation of encephalopathy (Fig. 2).

Schematic view of pathological changes observed in SAE. They include BBB dysfunction, CBF impairment, glial cell activation, leukocyte transmigration and neurotransmitter disturbances
Glial cell activation
The neurovascular unit is composed of neurons, capillaries, microglia, oligodendrocytes and extracellular matrix [39]. In particular, microglia play an important defensive role in response to various pathogens and neuronal injury and are resident and immunocompetent cells of the CNS [40]. Increasing evidence indicates that endothelium–microglia interactions are associated with a variety of inflammation-associated brain diseases [41]. When the BBB is destroyed, resting microglia are activated swiftly after cellular damage appears, and subsequently some inflammatory cytokines such as TNF-α, IL-6 and IL-1β are released to eliminate toxins from the extracellular space [42]. This part mainly focuses on the mechanism of glial cell activation and its effect on brain dysfunction in sepsis.
Microglial cells, acting as antigen-presenting cells, express a variety of receptors, such as TLRs, major histocompatibility complex, CX3CR1 chemokine receptor and CD11b/CD45 [43]. Therefore, LPS, other pathogen components and inflammation in the peripheral blood can activate microglia when they pass through the increased permeability of the BBB [44] and then increase the levels of inflammatory factors in sepsis [45]. The combination of LPS and TLR4 on microglia can trigger a proinflammatory program, which includes the production of TNF-α and the increased secretion of glutamate through connexin channels and the cystine/glutamate antiporter system, finally promoting the deregulation of calcium influx and inducing neuronal dysfunction [46]. Moreover, TNF-α and glutamate can enhance the production of each other [47]. Microglial cell activation also induces the synthesis and upregulation of IL-6 and IL-1β via the expression of CD40 and ligand [48] as well as the activation of transcription factors such as NF-κB, contributing to the perpetuation of the inflammatory challenge [49]. IL-1β secreted by activated microglia might suppress axon development and synapse formation through activation of the p38-MAPK signalling pathway associated with memory impairments in septic patients [50]. Additionally, Rachid et al. showed that LPS could induce endothelial cell death and increase BBB permeability by activating microglia. The mechanism of this effect appears to be mediated by NF-kB, JAK-STAT and JNK. These factors could then lead to the upregulation of iNOS and NADPH oxidase, which then generate NO and superoxide, respectively. These factors alone or together with peroxynitrite are cytotoxic to endothelial cells [51]. Therefore, inhibiting microglial activation could improve long-term cognitive performance in sepsis survivors [52].
In addition to microglia, astrocytes also play an important role in the onset of SAE. Under experimental sepsis conditions, mitochondrial biogenesis and ATP levels of astrocytes were significantly elevated to be suitable for the high-energy requirement and recover the ultrastructure of the mitochondria [53], which was mediated via the IL-6/AMPK signalling pathway [54]. Previous studies have clearly demonstrated that sepsis impaired astrocytic clearance of dehydroascorbic acid from the extracellular fluid and decreased the intracellular ascorbate concentration, which could upregulate iNOS and decrease glutamate uptake by astrocytes [55]. Hasegawa-Ishii et al. uncovered cytoskeletal and morphological alterations in hippocampal astrocytes after LPS injection, so astrocytes prepare for receiving cytokine signals via receptors expressed on the end-feet and then produce their own cytokines, including CXCL10, CCL11, and G-CSF, to change the hippocampal microenvironment [56]. For instance, astrocyte-derived CCL11 and G-CSF may stimulate microglia [57] and enhance the proliferation of microglia [56] in the hippocampus, resulting in learning and memory impairment [58]. Furthermore, the increased release of TNF-α and IL-1β by astrocytes aggravates inflammatory injury after the injection of LPS [59]. In the context of systemic inflammation, astrocytes can also regulate the phenotype of microglia. TLR4 stimulation and the costimulation of dopamine receptor D3 in astrocytes can promote the acquisition of proinflammatory features, ultimately promoting microglial activation (M1 microglia increase) and neuroinflammation [60].
Leukocyte transmigration
The transmigration of inflammatory leukocytes is a significant element of the innate immune response. Neutrophils are the predominant immune cells mediating much of the tissue injury during the progression of inflammation [61]. In the normal brain, there are no neutrophils in the brain parenchyma [62]. Under septic conditions, blood-borne proinflammatory mediators are produced, along with the activation of endothelial cells and the expression of ICAM-1 and VCAM-1; these outcomes contribute to neutrophil adhesion and recruitment into brain microvessels [63, 64]. The CXCR2 is a G-protein-coupled receptor for the well-known studied CXC chemokines including CXCL1, CXCL2, and CXCL5. It is widely expressed on hematopoietic cells and non-hematopoietic cells such as endothelial cells. Interactions between CXCR2 and its ligands play essential roles in leukocyte recruitment cascade in cerebral microvessels [65]. During CNS inflammation, astrocytes secrete significantly higher levels of CXCL1. Both endothelial CXCR2 and astrocyte-derived CXCL1 are crucial effectors mediating adhesion molecules expression on endothelial cells, resulting in robust neutrophil infiltration and leukocyte–endothelial cell interactions in the brain [66]. In addition, KC or MIP-2 produced by microglia also guide neutrophil transmigration into the brain parenchyma [67]. Subsequently, neutrophils accumulating in CNS jeopardize brain cells by increasing in cytokine levels, MPO activity and promoting oxidative damage [68]. Furthermore, neutrophils also generate many pro-inflammatory cytokines, ROS and proteolytic enzymes, including NOX, MPO, MMPs, elastase and cathepsins, which have hazardous effects on BBB integrity and allow passage of neutrophils into the brain [69]. It has been shown that ROS lead to junction proteins downregulation and the endothelial cytoskeleton reorganization through MLCK, PKC, MAPK, and Rho GTPases signaling pathways [70]. MMPs can dissolve the extracellular matrix, basal lamina and potentiate BBB disruption [71]. Persistent accumulation of neutrophils is related to cell death, brain oedema and tissue loss [72]. Therefore, the vicious cycle between neutrophil recruitment and BBB impairment aggravates neuronal dysfunction in sepsis.
In addition to neutrophils, other immune cells, such as inflammatory monocytes, are also recruited through the chemokine receptor CCR2, which plays an important role in SAE-induced long-term cognitive impairment [73].
Neurotransmitter disturbances
Most physiological and pathological processes in the brain involve multiple neurotransmitters. Inflammatory and metabolic alterations have been perceived as contributing to changes in cerebral neurotransmitters [74]. Thus, elucidating the role of different neurotransmitters could be beneficial to better therapeutic approaches to treat related diseases. Disorders of multiple neurotransmitters underlie the pathobiology of SAE. In the current research, a causal relationship has been demonstrated between the development of SAE and changes in neurotransmitter release or concentrations, such as acetylcholine [75], dopamine [76], serotonin [77], norepinephrine [78], gamma-aminobutyric acid (GABA) and its derivatives [79].
Acetylcholine dysfunction
Cholinergic signals mainly regulate cognitive function, movement, learning and memory by both nicotinic and muscarinic receptors [80]. The dysfunction of cholinergic signals could contribute to the occurrence of delirium, including inattention, confusion and perceptual disturbances [81]. Cholinergic signals also play a critical anti-inflammatory role, mainly suppressing endotoxin-inducible proinflammatory cytokines and TNF through interaction with peripheral α7 subunit-containing nicotinic acetylcholine receptors expressed on macrophages [82]. In 2010, Willem and colleagues demonstrated that peripheral inflammation could degenerate cholinergic neurons in the basal forebrain by activating microglia, while cholinergic inhibition of microglia could reduce neuronal dysfunction [74]. To study the effect of neuroinflammation on cholinergic transmission in the basal forebrain of sepsis patients, researchers examined cholinergic neuronal bodies in the basal forebrain and molecular cholinergic components in the cortex and hippocampus. They found that microglia were activated, and Iba1, IL-1β, and IL-6 gene expression in the cortex and hippocampus was upregulated. Choline acetyltransferase-positive neurons were significantly decreased in the basal forebrain of sepsis survivors. In the hippocampus, acetylcholinesterase (AChE) activity was enhanced, and the expression of the gene encoding the M1 muscarinic acetylcholine receptor, Chrm1, was decreased [75, 83]. As expected, microglial activation was associated with choline acetyltransferase protein expression and AChE activity. Consistent with a sepsis-induced cholinergic deficiency in the CNS, increasing acetylcholine receptor activity or using AChE inhibitors can prolong the lifetime of acetylcholine, thus attenuating proinflammatory cytokine release by microglia and improving the survival of sepsis patients [84]. The underlying mechanism is that increasing cholinergic activity can restore endotoxaemia-induced deficits in synaptic plasticity by decreasing small conductance calcium-activated potassium channels or decreasing calcium influx [85]. Therefore, cholinergic hypofunction and microglial activation may be significant underlying events leading to cognitive dysfunction among sepsis survivors. Furthermore, cholinergic signalling can protect neurons in the striatum, hippocampus, and cortex from neurotoxicity triggered by excitotoxic amino acids and other toxic substances [86]. Notably, plasma AChE activity can reflect acetylcholine levels in the brain; however, some studies have previously shown that serum AChE activity is not related to delirium in septic patients, and there are no clinical trials demonstrating the beneficial role of cholinergic agonists in the treatment of delirium [87].
Amine abnormalities
Dopamine is a neurotransmitter with multiple functions, and it is considered a major regulator of inflammation via D1-like DA receptors (D1 and D5) and D2-like receptors (D2, D3 and D4) [88, 89]. In the brain, dopamine is critical for the maintenance of working memory and the regulation of emotion [90], and substantial evidence demonstrates that an overdose of dopamine has been associated with the development of SAE [91]. In 1985, Freund et al. observed high levels of dopamine in the hippocampus, striatum and pons-medulla, along with low levels of breakdown products (HVA and 3MT), suggesting decreased turnover of dopamine during sepsis [77]. Compared with non-SAEs, severely septic animals and encephalopathy exhibited obviously lower levels of dopamine [92]. In contrast, the findings of the Oytun Erbass study exhibited an evident increase in brain HVA levels in septic animals compared with the sham group, confirming an increase in brain dopamine turnover during sepsis. It was also indicated that there was a significant positive correlation between brain dopaminergic activity and stereotypic behavioural scores [76]. Shimizu found that the concentrations of dopamine in the hypothalamus and striatum did not differ significantly between the septic group and the control group. Striatal dopamine metabolites tended to decrease 48 h after the induction of sepsis [93]. In addition, dopamine receptor D3 expressed in astrocytes but not microglia can regulate the dynamics of the acquisition of proinflammatory and anti-inflammatory phenotypes by astrocytes and microglia. Upon systemic LPS challenge, TLR4 stimulation and the costimulation of dopamine receptor D3 induced astrocyte activation and decreased the production of the anti-inflammatory protein Fizz1 by M2 microglia, thus favouring the function of M1 microglia and promoting neuroinflammation [60]. The data in Nolan’s study showed that dopamine could activate the NF-κB pathway in macrophages and prime the NLRP3 inflammasome, eventually inducing the production of inflammatory cytokines. These effects may be a vital mechanism for neuroplasticity in dopaminergic brain regions [94]. Additionally, recent research suggested that administration of a small dose of l-dopamine at an early stage in sepsis can limit neuroinflammation, improve neuroplasticity and reverse sepsis-induced decreases in hippocampal dopamine levels [95]. Clearly, there are inconsistent opinions on changes in dopamine activity in sepsis pathology. It is possible that dopamine activity may be related to the severity of sepsis. Combined with the above findings, these findings indicate that dopaminergic alterations are at least partly responsible for the progression of SAE.
Serotonin, or 5-hydroxytryptamine, a modulator of various functions in the CNS, is associated with prosocial behaviour and affective disorders [96]. The role of serotonin in the pathogenesis of encephalopathy has been extensively studied. According to reports, concentrations of brain and plasma tryptophan were increased in septic rats, which is the basis of increased brain serotonin metabolism in sepsis [92]. In addition, Herbert R. Freund et al. discovered that concentrations of the serotonin precursor and its metabolite 5-hydroxyindoleacetic acid (5-HIIA) were initially increased in most tissues in mild septic animals. The 5-HIIA/serotonin ratio was also increased significantly, indicating an increased turnover of serotonin [77], but eventually declined with the progression of severe sepsis [92]. In 1999, Shimizu also proposed that the level of 5-HIIA and the 5-HIIA/serotonin ratio were increased in sepsis compared with the sham group in the cortex, striatum, and hippocampus both 24 and 48 h after the operation, whereas the hypothalamic 5-HIIA did not change significantly [93]. In addition, in contrast to the increase in the serotonin synthesis rate observed in the above reports, Finn Bengtsson’s experiment demonstrated that there were no major changes in the CNS serotonin synthesis rate following 12 or 24 h of sepsis [97], and it has been demonstrated that serum serotonin levels can be used as a peripheral indicator for central serotonin levels, but there are no correlations of serum serotonin levels with delirium [87]. It is difficult to explain why there are regional differences in changes in serotonergic activity.
Norepinephrine, acting as a critical neuromodulator, is known to play important roles in regulating multiple brain functions, including attention, learning, and memory. The hippocampus receives noradrenergic innervation and expresses β-adrenergic receptors, which are involved in modulating the induction of long-lasting forms of synaptic potentiation [98]. Dysfunctional noradrenergic signalling is correlated with a number of cognitive impairments, such as Alzheimer’s disease [99] and depression [100]. In 1984, Freund found that brain norepinephrine decreases with sepsis and SAE [92]. Instead, in 1985, he discovered that most of the dopamine in the striatum is converted to norepinephrine, which leads to high levels of norepinephrine [77]. As with serotonin, there were also regional differences in norepinephrine expression. Hypothalamic norepinephrine was decreased at 24 h after septic operation. Cortical norepinephrine was increased 48 h after the septic operation; however, there was no significant change in hippocampal norepinephrine [93]. Research shows that both increases and decreases in norepinephrine might cause dysregulation of norepinephrine-dependent functions and lead to a pathological state under sepsis conditions [78]. On the other hand, researchers observed that the levels of hippocampal β2-adrenoceptor were significantly decreased after sepsis, accompanied by increased proinflammatory cytokines and downregulated CREB/BDNF and synaptic protein expression; eventually, septic mice exhibited cognitive deficits. Intriguingly, β2-adrenoceptor activation alleviates sepsis-induced cognitive impairments by modulating microglial phenotypes and reversing neuroinflammation and synaptic plasticity [101]. In summary, abnormalities in the noradrenergic transmission system play a vital role in SAE.
GABA alteration
GABA, a nonprotein amino acid formed by decarboxylation of glutamic acid [102], is the principal brain inhibitory neurotransmitter that regulates inflammatory and neurodegenerative diseases [103]. In a previous study, it was reported that GABA receptor density in the brain was altered in animal studies of metabolic encephalopathy [104]. In septic patients, elevated IL-1β together with a reduced release of IL-1α increases Akt-mediated GABAergic activity, which contributes to the alteration of synaptic strength and cognitive dysfunction [105]. In an acute inflammation model, increased tonic α5 GABAAR current and surface levels in hippocampal neurons by IL-1β through a p38-MAPK signalling pathway are critical for inflammation-induced memory deficits [106].
In summary, these alterations in neurotransmitter metabolism may be an important reason for the development of SAE. Notably, it would probably be incorrect to interpret that any single neurotransmitter dysfunction should be responsible for SAE, which involves multifactorial mechanisms and multiple neurotransmitter interactions [77].
Conclusions
Sepsis-associated encephalopathy is a common and detrimental neurological complication of sepsis but lacks diagnostic criteria and target-directed treatments, additionally, it is prone to be overlooked in clinical practice. The pathogenesis of SAE is complex and multifactorial. The increased understanding of pathogenesis is beneficial for developing new preventive and therapeutic strategies to reduce sequelae of sepsis. This paper summarizes the pathogenesis of SAE as mentioned above. The pathomechanisms of SAE are parallel, influence each other, and contribute to neuronal dysfunction. A thorough understanding of the pathogenesis of CNS dysfunction is helpful to reduce the incidence of SAE. Future efforts should be directed towards understanding the crosstalk of these mechanisms, not each of them alone.
Data availability
All authors declares that all data support their published claims and comply with field standards.
Abbreviations
- SAE:
-
Sepsis-associated encephalopathy
- CNS:
-
Cerebral blood flow
- CSF:
-
Cerebrospinal fluid
- BBB:
-
Blood–brain barrier
- LPS:
-
Lipopolysaccharide
- GABA:
-
Gamma-aminobutyric acid
- AChE:
-
Acetylcholinesterase
- 5-HIIA:
-
5-Hydroxyindoleacetic acid
References
Singer M, Deutschman CS, Seymour CW et al (2016) The third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA 315(8):801–810
Fleischmann C, Scherag A, Adhikari NK et al (2016) Assessment of global incidence and mortality of hospital-treated sepsis. current estimates and limitations. Am J Respir Crit Care Med 193(3):259–272
Schuler A, Wulf DA, Lu Y et al (2018) The impact of acute organ dysfunction on long-term survival in sepsis. Crit Care Med 46(6):843–849
Chung HY, Wickel J, Brunkhorst FM et al (2020) Sepsis-associated encephalopathy: from delirium to dementia? J Clin Med 9(3):703
Kikuchi DS, Campos ACP, Qu H et al (2019) Poldip2 mediates blood–brain barrier disruption in a model of sepsis-associated encephalopathy. J Neuroinflamm 16(1):241
Kuperberg SJ, Wadgaonkar R (2017) Sepsis-associated encephalopathy: the blood–brain barrier and the sphingolipid rheostat. Front Immunol 8:597
Zhao L, Wang Y, Ge Z et al (2021) Mechanical learning for prediction of sepsis-associated encephalopathy. Front Comput Neurosci 15:739265
Ai ML, Huang L, Feng Q et al (2019) The clinical significance of transcranial Doppler in early diagnosis of sepsis-associated encephalopathy. Zhonghua nei ke za zhi 58(11):814–818
Shulyatnikova T, Verkhratsky A (2020) Astroglia in sepsis associated encephalopathy. Neurochem Res 45(1):83–99
Ren C, Yao RQ, Zhang H et al (2020) Sepsis-associated encephalopathy: a vicious cycle of immunosuppression. J Neuroinflamm 17(1):14
Czempik PF, Pluta MP, Krzych LJ (2020) Sepsis-associated brain dysfunction: a review of current literature. Int J Environ Res Public Health 17(16):5852
Keaney J, Campbell M (2015) The dynamic blood–brain barrier. FEBS J 282(21):4067–4079
Jin X, Liu J, Liu W (2014) Early ischemic blood brain barrier damage: a potential indicator for hemorrhagic transformation following tissue plasminogen activator (tPA) thrombolysis? Curr Neurovasc Res 11(3):254–262
Nishioku T, Dohgu S, Takata F et al (2009) Detachment of brain pericytes from the basal lamina is involved in disruption of the blood–brain barrier caused by lipopolysaccharide-induced sepsis in mice. Cell Mol Neurobiol 29(3):309–316
Andreasen AS, Krabbe KS, Krogh-Madsen R et al (2008) Human endotoxemia as a model of systemic inflammation. Curr Med Chem 15(17):1697–1705
Erikson K, Tuominen H, Vakkala M et al (2020) Brain tight junction protein expression in sepsis in an autopsy series. Crit Care 24(1):385
Guo F, Tang J, Zhou Z et al (2012) GEF-H1-RhoA signaling pathway mediates LPS-induced NF-kappaB transactivation and IL-8 synthesis in endothelial cells. Mol Immunol 50(1–2):98–107
Weighardt H, Holzmann B (2007) Role of Toll-like receptor responses for sepsis pathogenesis. Immunobiology 212(9–10):715–722
Ni Y, Teng T, Li R et al (2017) TNFalpha alters occludin and cerebral endothelial permeability: role of p38MAPK. PLoS ONE 12(2):e0170346
Sankowski R, Mader S, Valdes-Ferrer SI (2015) Systemic inflammation and the brain: novel roles of genetic, molecular, and environmental cues as drivers of neurodegeneration. Front Cell Neurosci 9:28
Nwafor DC, Brichacek AL, Mohammad AS et al (2019) Targeting the blood–brain barrier to prevent sepsis-associated cognitive impairment. J Cent Nerv Syst Dis 11:1179573519840652
Haileselassie B, Joshi AU, Minhas PS et al (2020) Mitochondrial dysfunction mediated through dynamin-related protein 1 (Drp1) propagates impairment in blood–brain barrier in septic encephalopathy. J Neuroinflamm 17(1):36
Vutukuri R, Brunkhorst R, Kestner RI et al (2018) Alteration of sphingolipid metabolism as a putative mechanism underlying LPS-induced BBB disruption. J Neurochem 144(2):172–185
Qin LH, Huang W, Mo XA et al (2015) LPS induces occludin dysregulation in cerebral microvascular endothelial cells via MAPK signaling and augmenting MMP-2 levels. Oxid Med Cell Longev. 2015:120641
Hu Y, Bi Y, Yao D et al (2019) Omi/HtrA2 protease associated cell apoptosis participates in blood–brain barrier dysfunction. Front Mol Neurosci 12:48
Dahl RH, Berg RMG, Taudorf S et al (2018) A reassessment of the blood–brain barrier transport of large neutral amino acids during acute systemic inflammation in humans. Clin Physiol Funct Imaging 38(4):656–662
Hughes CG, Patel MB, Pandharipande PP (2012) Pathophysiology of acute brain dysfunction: what’s the cause of all this confusion? Curr Opin Crit Care 18(5):518–526
Griton M, Dhaya I, Nicolas R et al (2020) Experimental sepsis-associated encephalopathy is accompanied by altered cerebral blood perfusion and water diffusion and related to changes in cyclooxygenase-2 expression and glial cell morphology but not to blood-brain barrier breakdown. Brain Behav Immun 83:200–213
Willie CK, Tzeng YC, Fisher JA et al (2014) Integrative regulation of human brain blood flow. J Physiol 592(5):841–859
Taccone FS, Su F, Pierrakos C et al (2010) Cerebral microcirculation is impaired during sepsis: an experimental study. Crit Care 14(4):R140
Ferlini L, Su F, Creteur J et al (2020) Cerebral autoregulation and neurovascular coupling are progressively impaired during septic shock: an experimental study. Intensive Care Med Exp 8(1):44
Taccone FS, Su F, De Deyne C et al (2014) Sepsis is associated with altered cerebral microcirculation and tissue hypoxia in experimental peritonitis. Crit Care Med 42(2):e114–e122
Faraci FM, Taugher RJ, Lynch C et al (2019) Acid-sensing ion channels: novel mediators of cerebral vascular responses. Circ Res 125(10):907–920
Hoiland RL, Fisher JA, Ainslie PN (2019) Regulation of the cerebral circulation by arterial carbon dioxide. Compr Physiol 9(3):1101–1154
Taccone FS, Castanares-Zapatero D, Peres-Bota D et al (2010) Cerebral autoregulation is influenced by carbon dioxide levels in patients with septic shock. Neurocrit Care 12(1):35–42
Zhou Q, Cao B, Niu L et al (2010) Effects of permissive hypercapnia on transient global cerebral ischemia-reperfusion injury in rats. Anesthesiology 112(2):288–297
Van Der Kleij LA, De Vis JB, De Bresser J et al (2020) Arterial CO2 pressure changes during hypercapnia are associated with changes in brain parenchymal volume. Eur Radiol Exp 4(1):17
Bothwell SW, Janigro D, Patabendige A (2019) Cerebrospinal fluid dynamics and intracranial pressure elevation in neurological diseases. Fluids Barriers CNS 16(1):9
Zhao Z, Nelson AR, Betsholtz C et al (2015) Establishment and dysfunction of the blood–brain barrier. Cell 163(5):1064–1078
Aguzzi A, Barres BA, Bennett ML (2013) Microglia: scapegoat, saboteur, or something else? Science 339(6116):156–161
Da Fonseca AC, Matias D, Garcia C et al (2014) The impact of microglial activation on blood–brain barrier in brain diseases. Front Cell Neurosci 8:362
Moraes CA, Zaverucha-Do-Valle C, Fleurance R et al (2021) Neuroinflammation in sepsis: molecular pathways of microglia activation. Pharmaceuticals (Basel) 14(5):416
Hickman S, Izzy S, Sen P et al (2018) Microglia in neurodegeneration. Nat Neurosci 21(10):1359–1369
Melief J, Koning N, Schuurman KG et al (2012) Phenotyping primary human microglia: tight regulation of LPS responsiveness. Glia 60(10):1506–1517
Heming N, Mazeraud A, Verdonk F et al (2017) Neuroanatomy of sepsis-associated encephalopathy. Crit Care 21(1):65
Michels M, Steckert AV, Quevedo J et al (2015) Mechanisms of long-term cognitive dysfunction of sepsis: from blood-borne leukocytes to glial cells. Intensive Care Med Exp 3(1):30
Kaushal V, Schlichter LC (2008) Mechanisms of microglia-mediated neurotoxicity in a new model of the stroke penumbra. J Neurosci 28(9):2221–2230
Michels M, Danieslki LG, Vieira A et al (2015) CD40–CD40 ligand pathway is a major component of acute neuroinflammation and contributes to long-term cognitive dysfunction after sepsis. Mol Med 21:219–226
Danielski LG, Giustina AD, Badawy M et al (2018) Brain barrier breakdown as a cause and consequence of neuroinflammation in sepsis. Mol Neurobiol 55(2):1045–1053
Han Q, Lin Q, Huang P et al (2017) Microglia-derived IL-1beta contributes to axon development disorders and synaptic deficit through p38-MAPK signal pathway in septic neonatal rats. J Neuroinflamm 14(1):52
Kacimi R, Giffard RG, Yenari MA (2011) Endotoxin-activated microglia injure brain derived endothelial cells via NF-kappaB, JAK-STAT and JNK stress kinase pathways. J Inflamm (Lond) 8:7
Michels M, Vieira AS, Vuolo F et al (2015) The role of microglia activation in the development of sepsis-induced long-term cognitive impairment. Brain Behav Immun 43:54–59
Zhao YZ, Gao ZY, Ma LQ et al (2017) Research on biogenesis of mitochondria in astrocytes in sepsis-associated encephalopathy models. Eur Rev Med Pharmacol Sci 21(17):3924–3934
Chen XL, Wang Y, Peng WW et al (2018) Effects of interleukin-6 and IL-6/AMPK signaling pathway on mitochondrial biogenesis and astrocytes viability under experimental septic condition. Int Immunopharmacol 59:287–294
Korcok J, Wu F, Tyml K et al (2002) Sepsis inhibits reduction of dehydroascorbic acid and accumulation of ascorbate in astroglial cultures: intracellular ascorbate depletion increases nitric oxide synthase induction and glutamate uptake inhibition. J Neurochem 81(1):185–193
Hasegawa-Ishii S, Inaba M, Umegaki H et al (2016) Endotoxemia-induced cytokine-mediated responses of hippocampal astrocytes transmitted by cells of the brain-immune interface. Sci Rep 6:25457
Parajuli B, Horiuchi H, Mizuno T et al (2015) CCL11 enhances excitotoxic neuronal death by producing reactive oxygen species in microglia. Glia 63(12):2274–2284
Villeda SA, Luo J, Mosher KI et al (2011) The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature 477(7362):90–94
Fernandes A, Silva RF, Falcao AS et al (2004) Cytokine production, glutamate release and cell death in rat cultured astrocytes treated with unconjugated bilirubin and LPS. J Neuroimmunol 153(1–2):64–75
Montoya A, Elgueta D, Campos J et al (2019) Dopamine receptor D3 signalling in astrocytes promotes neuroinflammation. J Neuroinflamm 16(1):258
Rossaint J, Margraf A, Zarbock A (2018) Role of platelets in leukocyte recruitment and resolution of inflammation. Front Immunol 9:2712
Giles JA, Greenhalgh AD, Denes A et al (2018) Neutrophil infiltration to the brain is platelet-dependent, and is reversed by blockade of platelet GPIbalpha. Immunology 154(2):322–328
Margraf A, Ley K, Zarbock A (2019) Neutrophil recruitment: from model systems to tissue-specific patterns. Trends Immunol 40(7):613–634
Peng X, Luo Z, He S et al (2021) Blood–brain barrier disruption by lipopolysaccharide and sepsis-associated encephalopathy. Front Cell Infect Microbiol 11:768108
Wu F, Chen X, Zhai L et al (2020) CXCR2 antagonist attenuates neutrophil transmigration into brain in a murine model of LPS induced neuroinflammation. Biochem Biophys Res Commun 529(3):839–845
Wu F, Zhao Y, Jiao T et al (2015) CXCR2 is essential for cerebral endothelial activation and leukocyte recruitment during neuroinflammation. J Neuroinflamm 12:98
Zhou H, Andonegui G, Wong CH et al (2009) Role of endothelial TLR4 for neutrophil recruitment into central nervous system microvessels in systemic inflammation. J Immunol 183(8):5244–5250
Zarbato GF, De Souza Goldim MP, Giustina AD et al (2018) Dimethyl fumarate limits neuroinflammation and oxidative stress and improves cognitive impairment after polymicrobial sepsis. Neurotox Res 34(3):418–430
Jin R, Yang G, Li G (2010) Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol 87(5):779–789
Krizbai IA, Bauer H, Bresgen N et al (2005) Effect of oxidative stress on the junctional proteins of cultured cerebral endothelial cells. Cell Mol Neurobiol 25(1):129–139
Turner RJ, Sharp FR (2016) Implications of MMP9 for blood brain barrier disruption and hemorrhagic transformation following ischemic stroke. Front Cell Neurosci 10:56
Kenne E, Erlandsson A, Lindbom L et al (2012) Neutrophil depletion reduces edema formation and tissue loss following traumatic brain injury in mice. J Neuroinflamm 9:17
Andonegui G, Zelinski EL, Schubert CL et al (2018) Targeting inflammatory monocytes in sepsis-associated encephalopathy and long-term cognitive impairment. JCI Insight 3(9):1–8
Van Gool WA, Van De Beek D, Eikelenboom P (2010) Systemic infection and delirium: when cytokines and acetylcholine collide. Lancet 375(9716):773–775
Zaghloul N, Addorisio ME, Silverman HA et al (2017) Forebrain cholinergic dysfunction and systemic and brain inflammation in murine sepsis survivors. Front Immunol 8:1673
Erbas O, Taskiran D (2014) Sepsis-induced changes in behavioral stereotypy in rats; involvement of tumor necrosis factor-alpha, oxidative stress, and dopamine turnover. J Surg Res 186(1):262–268
Freund HR, Muggia-Sullam M, Lafrance R et al (1986) Regional brain amino acid and neurotransmitter derangements during abdominal sepsis and septic encephalopathy in the rat. The effect of amino acid infusions. Arch Surg 121(2):209–216
Gyoneva S, Traynelis SF (2013) Norepinephrine modulates the motility of resting and activated microglia via different adrenergic receptors. J Biol Chem 288(21):15291–15302
Leon A, Lepouse C, Floch T et al (2006) Brain injury during severe sepsis. Ann Fr Anesth Reanim 25(8):863–867
Acharya S, Kim KM (2021) Roles of the functional interaction between brain cholinergic and dopaminergic systems in the pathogenesis and treatment of schizophrenia and Parkinson’s disease. Int J Mol Sci 22(9):1–8
Bugiani O (2021) Why is delirium more frequent in the elderly? Neurol Sci 42(8):3491–3503
Pavlov VA, Wang H, Czura CJ et al (2003) The cholinergic anti-inflammatory pathway: a missing link in neuroimmunomodulation. Mol Med 9(5–8):125–134
Santos-Junior NN, Catalao CHR, Costa LHA et al (2018) Experimental sepsis induces sustained inflammation and acetylcholinesterase activity impairment in the hypothalamus. J Neuroimmunol 324:143–148
Hofer S, Eisenbach C, Lukic IK et al (2008) Pharmacologic cholinesterase inhibition improves survival in experimental sepsis. Crit Care Med 36(2):404–408
Zivkovic AR, Sedlaczek O, Von Haken R et al (2015) Muscarinic M1 receptors modulate endotoxemia-induced loss of synaptic plasticity. Acta Neuropathol Commun 3:67
Van Eijk MM, Roes KC, Honing ML et al (2010) Effect of rivastigmine as an adjunct to usual care with haloperidol on duration of delirium and mortality in critically ill patients: a multicentre, double-blind, placebo-controlled randomised trial. Lancet 376(9755):1829–1837
Tomasi CD, Salluh J, Soares M et al (2015) Baseline acetylcholinesterase activity and serotonin plasma levels are not associated with delirium in critically ill patients. Rev Bras Ter Intensiva 27(2):170–177
Liu A, Ding S (2019) Anti-inflammatory effects of dopamine in lipopolysaccharide (LPS)-stimulated RAW264.7 cells via inhibiting NLRP3 inflammasome activation. Ann Clin Lab Sci 49(3):353–360
Rangel-Barajas C, Coronel I, Floran B (2015) Dopamine receptors and neurodegeneration. Aging Dis 6(5):349–368
Bissonette GB, Roesch MR (2016) Development and function of the midbrain dopamine system: what we know and what we need to. Genes Brain Behav 15(1):62–73
Sommer BR, Wise LC, Kraemer HC (2002) Is dopamine administration possibly a risk factor for delirium? Crit Care Med 30(7):1508–1511
Freund HR, Muggia-Sullam M, Peiser J et al (1985) Brain neurotransmitter profile is deranged during sepsis and septic encephalopathy in the rat. J Surg Res 38(3):267–271
Shimizu I, Adachi N, Liu K et al (1999) Sepsis facilitates brain serotonin activity and impairs learning ability in rats. Brain Res 830(1):94–100
Nolan RA, Reeb KL, Rong Y et al (2020) Dopamine activates NF-kappaB and primes the NLRP3 inflammasome in primary human macrophages. Brain Behav Immun Health 2:17
Li F, Zhang B, Duan S et al (2020) Small dose of L-dopa/benserazide hydrochloride improved sepsis-induced neuroinflammation and long-term cognitive dysfunction in sepsis mice. Brain Res 1737:146780
Beis D, Holzwarth K, Flinders M et al (2015) Brain serotonin deficiency leads to social communication deficits in mice. Biol Lett 11(3):1–8
Bengtsson F, Bugge M, Hansson L et al (1987) Serotonin metabolism in the central nervous system following sepsis or portacaval shunt in the rat. J Surg Res 43(5):420–429
O’dell TJ, Connor SA, Guglietta R et al (2015) beta-Adrenergic receptor signaling and modulation of long-term potentiation in the mammalian hippocampus. Learn Mem 22(9):461–471
Xu H, Rajsombath MM, Weikop P et al (2018) Enriched environment enhances beta-adrenergic signaling to prevent microglia inflammation by amyloid-beta. EMBO Mol Med 10(9):1–7
Manczak EM, Dougherty B, Chen E (2019) Parental depressive symptoms potentiate the effect of youth negative mood symptoms on gene expression in children with asthma. J Abnorm Child Psychol 47(1):99–108
Zong MM, Zhou ZQ, Ji MH et al (2019) Activation of beta2-adrenoceptor attenuates sepsis-induced hippocampus-dependent cognitive impairments by reversing neuroinflammation and synaptic abnormalities. Front Cell Neurosci 13:293
Obata K (2013) Synaptic inhibition and gamma-aminobutyric acid in the mammalian central nervous system. Proc Jpn Acad Ser B 89(4):139–156
Sallam MY, El-Gowilly SM, Abdel-Galil AG et al (2016) Central GABAA receptors are involved in inflammatory and cardiovascular consequences of endotoxemia in conscious rats. Naunyn Schmiedebergs Arch Pharmacol 389(3):279–288
Dadsetan S, Balzano T, Forteza J et al (2016) Infliximab reduces peripheral inflammation, neuroinflammation, and extracellular GABA in the cerebellum and improves learning and motor coordination in rats with hepatic encephalopathy. J Neuroinflamm 13(1):245
Serantes R, Arnalich F, Figueroa M et al (2006) Interleukin-1beta enhances GABAA receptor cell-surface expression by a phosphatidylinositol 3-kinase/Akt pathway: relevance to sepsis-associated encephalopathy. J Biol Chem 281(21):14632–14643
Wang DS, Zurek AA, Lecker I et al (2012) Memory deficits induced by inflammation are regulated by alpha5-subunit-containing GABAA receptors. Cell Rep 2(3):488–496
Acknowledgements
First and foremost, I would like to show my deepest gratitude to my supervisor, Professor Wenlan Liu, a kind, responsible and resourceful scholar, who has provided me with valuable guidance in every stage of scientific research. In addition, I would like to thank the anonymous reviewers for their helpful remarks.
Funding
The work was supported by Guangdong Innovation Platform of Translational Research for Cerebrovascular Diseases and grants from National Natural Science Foundation of China (Grant No. 81760227, 81873747), Guangdong Basic and Applied Basic Research Foundation (Grant No. 2019A1515010311) and Science and Technology Innovation Commission of Shenzhen Municipality (Grant Nos. GJHZ20190820115001765, JCYJ20180507184656626).
Author information
Authors and Affiliations
Contributions
KY had the idea for the article and drafted the work. JQC and TW performed the literature search, and YZ critically wrote and revised the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no relevant financial or nonfinancial interests to disclose.
Ethical approval
This article does not contain any studies with human participants performed by any of the authors.
Consent for publication
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Yang, K., Chen, J., Wang, T. et al. Pathogenesis of sepsis-associated encephalopathy: more than blood–brain barrier dysfunction. Mol Biol Rep 49, 10091–10099 (2022). https://doi.org/10.1007/s11033-022-07592-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-022-07592-x