
Abstract
Atrial fibrillation (AF) is a common cardiac arrhythmia and a major risk factor for stroke, heart failure, and premature death. AF has a strong genetic predisposition. This review highlights the recent findings on the genetics of AF from genome-wide association studies (GWAS) and high-throughput sequencing studies. The consensus from GWAS implies that AF is both polygenic and pleiotropic in nature. With the advent of whole-genome sequencing and whole-exome sequencing, rare variants associated with AF pathogenesis have been identified. The recent studies have contributed towards better understanding of AF pathogenesis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Atrial fibrillation (AF) is the most common cardiac arrhythmia that affects nearly 34 million people across the globe. AF can lead to stroke, heart failure, dementia, and death [1]. In addition to clinical risk factors such as advanced age, hypertension, diabetes, obesity, and the male gender [2, 3], many studies have underlined the substantial impact of genetic risk factors in AF.
AF is heritable, with affected individuals having a strong genetic predisposition [4]. It has been shown that one in four AF affected individuals has a first degree relative with AF [5]. Genome-wide association studies (GWAS) have identified more than 100 AF-associated common loci [6, 7] and more than 35 genes have been identified as susceptibility loci by candidate gene studies [8]. More recently, genome- and exome-sequencing studies have found rare genetic variants associated with AF [9, 10].
The current lack of highly effective therapies is due to the lacunae in understanding the mechanisms leading to AF. Uncovering the genetic predisposition to AF can offer great improvement of risk prediction models and algorithms that are currently backed by clinical variables, enable a better understanding of the discrete subtypes of AF and open novel therapeutic options [11, 12].
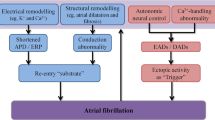
The different subsets of AF could be associated with a specific set of genes. For example, lone and early-onset AF is mostly found to be due to variations in genes involved in electric signaling that cause electrical disturbances that manifest as AF [13]. Genes involved structurally and functionally with ion channels, intercellular signaling, and homeostatic control are widely implicated with AF pathogenesis. Figure 1 illustrates different mechanisms implicated in AF pathogenesis.

Atrial fibrillation pathogenesis. The illustration depicts the four functional mechanisms involved in the pathogenesis of atrial fibrillation. Mutations in transcription factors lead to abnormalities in cardiac development. Structural gene mutations affect sarcomere architecture. Mutations in cardiac ion channels alter electrical properties resulting in refractoriness and action potential duration (APD) shortening. Mutations in genes encoding calcium handling proteins affect contractility
Though AF can be monogenic as studied in large families, the majority of AF presents as a common complex trait with a significant interplay between the predisposing genes and environmental factors [13, 14]. It is interesting to note that AF susceptibility is influenced by both rare variants and common single nucleotide polymorphisms in the common sets of genes. Table 1 summarizes the biological functions of well-established genes associated with AF across many studies. Genome-wide association studies (GWAS) require no prior hypothesis and are designed to identify common disease-associated variants selectively fabricated on SNP microarrays. GWAS along with the high throughput whole-genome sequencing (WGS) and whole-exome sequencing (WES) studies that discover potential rare variants have led to a paradigm shift in studies of human genetics with their comprehensive approach and their ability to delve deeper into the genetic content. This review focuses on GWAS and high throughput sequencing studies on AF published in the last decade to give an insight into the latest findings.
Genes identified by GWAS
The earliest GWAS of this decade in 2010 by Ellinor et al. that sought to identify common variants associated with lone AF included 1355 patients and 12,844 referents of European ancestry. The study defined lone AF as AF with an onset before 66 years of age and without a preceding history of myocardial infarction, heart failure, or known left ventricular systolic dysfunction. A significant association was identified with intronic variant rs13376333 in the KCNN3 gene that encodes a potassium intermediate/small conductance calcium-activated channel (subfamily N, member 3) and is involved in atrial repolarization [26].
In 2017, Lee et al. conducted a two-stage GWAS in Korean patients with early-onset AF. Stage 1 consisted of 672 patients who underwent radiofrequency catheter ablation (RFCA) and 3700 controls. The stage 2 replication cohort consisted of 200 independent cases and 1812 controls. The study identified genes for critical transcription factors (PRRX1, PITX2, TBX5, ZFHX3, HAND2) in addition to NEURL, and PPFIA1 [39]. PRRX1 encodes a homeodomain transcription factor highly expressed in the developing heart. A PRRX1 knockout study found impaired fetal pulmonary vasculature development [34]. Paired-like homeodomain 2 (PITX2) encodes a cardiac transcription factor located at 4q25, the locus identified by the first GWAS to be significantly associated with AF in Europeans and Chinese [30]. The region 4q25 has been identified to harbor strong AF susceptibility signals by a study that interrogated nine established AF associated loci in more than 60,000 individuals of European ancestry [40]. The role of PITX2, in specifying pulmonary venous myocardium is considered important, given the fact that pulmonary veins are the sources of AF trigger in most cases [41, 42]. Adult and larval zebrafish pitx2c (cardiac-specific isoform of pitx2) loss-of-function models displayed atrial conduction defects, altered cardiac metabolism and structural remodelling phenotypes similar to that observed in human AF patients. The study demonstrated the role of PITX2 in regulating expression of ion channels involved in cardiac electrical conduction properties by RNA-seq analysis of loss- and gain-of-function embryonic hearts in zebrafish. Pitx2c−/− adult atria exhibit loss of cardiac tissue integrity, electrophysiological defects leading to arrhythmia, fibrosis and other features of AF. The findings of this study support the hypothesis that underlying cardiomyopathy predisposes to AF [43]. TBX5 plays a critical role in cardiac development. It reduces fibrosis and improves cardiac function by reprogramming non-myocytes. Previously gain-of-function mutations in TBX5 have been associated with early-onset AF [44].
The co-regulation activity of the transcriptional factors TBX5 and PITX2 is required for cardiac calcium regulations and the transcriptional regulatory network that they govern is considered essential for atrial development [45]. ZFHX3 is a transcription factor associated with JAK/STAT signaling pathway could play a role in electrical and structural atrial remodeling mediated via the inflammatory process [38]. The transcription factor HAND2 is involved in the reprogramming of cardiac fibroblasts into functional cardiac-like myocytes and is associated with heart repair [22].
The NEURL gene encodes an E3 ubiquitin ligase. A functional study using embryonic zebrafish showed that knockdown of the NEURL ortholog specifically altered atrial action potential duration (APD) without affecting cardiac contractile function or heart rate [15]. The study also supports the role of NEURL in atrial repolarization and hence, AF. NEURL has been found to interact with PITX2 [39]. PPFIA4 encodes a member of the evolutionarily conserved liprin protein family that regulates the disassembly of focal cell adhesion to aid cell–matrix interactions [33].
In 2017, a Japanese study by Low et al. carried out a well-powered GWAS with 8180 AF cases and 28,612 controls and an additional follow up of 3120 cases and 125,064 controls, identified many of the previously reported AF risk loci (KCNN3, PRRX1, CAND2, PITX2, GJA1, CAV1, C9orf3, SYNPO2L, NEURL, CUX2, TBX5, SYNE2, HCN4, ZFHX3) and new loci near KCND3, PPFIA4, SLC1A4, HAND2, NEBL, SH3PXD2A [46]. The majority of the genetic variants identified by GWAS are found in the non-coding regions of the genome [6, 47]. These variants could most likely be involved in gene regulation. An expression quantitative trait locus (eQTL) can be defined as “a locus that explains a fraction of the genetic variance of a gene expression phenotype”. eQTL analysis involves testing the association between such variants and gene expression levels concerning the phenotype [48]. By expression quantitative trait loci (eQTL) analysis Low et al. found that the locus 1q31.1 that includes PPF1A4 contains an SNP rs17461925 that was found to be significantly associated with increased expression of PPF1A4 in the aorta. The number of different loci identified in the study showcases the polygenic nature of AF implicating genes with diverse functions as is described below. Most of the genes encode ion channel proteins involved in conduction and cell coupling. GJA1 encodes a cardiac gap junction protein connexin43 involved in cell–cell electrical coupling. This connexin protein is found altered in cardiac anomalies [21]. CAV1 selectively expressed in the atria encodes caveolin-1, a cellular membrane protein involved in signal transduction. It negatively regulates a potassium channel protein KCNH2, involved in cardiac repolarization [18, 25]. Hyperpolarization-activated cyclic nucleotide-gated channel HCN4, is a cardiac ion channel protein highly expressed in the sinoatrial node. HCN4 is the predominant gene that encodes pacemaker channel in the heart [49]. Mutations in HCN4 are associated with various forms of sinus nodal dysfunction [23, 50].
KCND3 also encodes an ion channel protein with diverse functions relevant to atrial fibrillation pathogenesis such as regulating neurotransmitter release, neural excitability, heart rate, and smooth muscle contraction [46]. A previous study found a gain-of-function mutation in KCND3 to be associated with the early onset of persistent lone AF [24]. CAND2 on the other hand has been shown to prolong the atrial action potential duration similar to NEURL [15]. C9orf3 encodes aminopeptidase that cleaves angiotensin III to generate angiotensin IV which is involved in the renin-angiotensin pathway (NCBI Gene ID: 84909). SYNPO2L encodes Synaptopodin 2-Like Protein, an actin-associated protein with a role implicated in heart morphogenesis and sarcomere organization by gene ontology (NCBI Gene ID: 79933). A zebrafish knockdown study has shown the important role of SYNPO2L in the structural development and function of the sarcomere [51]. Similarly, SYNE2 encodes the isoforms of nesprin-2, expressed in the heart and skeletal muscle. Nesprin-2 is found localized throughout the sarcomere where it anchors the nucleus to the cytoskeleton and maintains nuclear structural integrity [36]. Low et al. in this study identified a common missense variant in the NEBL gene that encodes a nebulin-like protein expressed in cardiac muscle that binds to actin, which interacts with thin filaments and Z-line-associated proteins in striated muscle [46, 52].
A recent study found that inhibiting the function of SH3PXD2A, a scaffolding protein led to the altered regulation of extracellular matrix degradation and axon guidance by growth cone invadosomes [53]. The study by Low et al. on the Japanese has thus added a new dimension to the understanding of the molecular pathogenesis of AF pointing to genes involved in axon guidance and neural crest development [46].
In 2018, a GWAS in the Norwegian population by Nielsen et al. that included 6337 AF cases and 61,407 controls and an independent replication cohort consisting of 30,679 AF cases and 2,78,895 controls identified two AF loci of which the region 2q31 was linked to an important gene TTN gene [54], that encodes sarcomeric protein titin, the largest human protein highly expressed in cardiac tissues and makes up the striated heart muscle [37]. The other locus 1p32 was previously found to be associated on a genome-wide scale with ECG-derived measurements (QRS amplification and duration) that depict cardiac structure and function, most likely associated with the development of AF. Pathway and functional enrichment analyses of most of the risk variants at AF associated loci in the study point to regions of open chromatin state during fetal heart development implying that impaired cardiac development in the fetus can lead to critical AF risk factors in adult life such as altered elastic properties of atrium resulting in increased left atrial pressure that can facilitate premature depolarizations from pulmonary vein, the potential AF trigger [54].
A large GWAS study that included 60,620 AF cases and 9,70,216 controls from biobanks of samples of European ancestry by Nielsen et al. in 2018 identified risk variants at 111 genomic loci and prioritized 151 candidate genes that included those that are involved in cardiac and skeletal muscle function and integrity (AKAP6, CFL2, MYH6, MYH7, MYO18B, MYO1C, MYOCD, MYOT, MYOZ1, MYPN, PKP2, RBM20, SGCA, SSPN, SYNPO2L, TTN, TTN-AS, WIPF1); mediation of developmental events (ARNT2, EPHA3, FGF5, GATA4, GTF2I, HAND2, LRRC10, NAV2, NKX2–5, PITX2, SLIT3, SOX15, TBX5) along with genes likely to be involved in intracellular calcium handling in the heart (CALU, CAMK2D, CASQ2, PLN), angiogenesis (TNFSF12, TNFSF12-TNFSF13), hormone signaling (CGA, ESR2, IGF1R, NR3C1, THRB), and function of cardiac ion channels (HCN4, KCND3, KCNH2, KCNJ5, KCNN2, KCNN3, SCN10A, SCN5A, SLC9B1). Most of the AF-associated risk variants were present in the non-coding regions suggesting their importance in transcriptional regulation in the adult heart and the development of fetal heart or probably both [55].
A more recent GWAS identified 18 loci associated with left atrium (LA) volume and function demonstrating an association between LA traits derived from cardiac magnetic resonance (CMR) and the development of AF [17]. The study provides evidence for an intrinsic relationship between genetic underpinnings of AF and compromised LA structure and function. The study identified genes linked with cardiomyopathy (DSP, SIX5, CILP) and arrhythmia (MYO18B, TTN, CASQ2, C9OFR3). Mutations in DSP which encodes desmoplakin are associated with cardiomyopathies [56, 57]. SIX5 is localized to a chromosomal region that harbors genes linked with myotonic dystrophy [58]. CILP which encodes cartilage intermediate layer protein 1 has been identified as a marker of fibrosis potentially leading to heart failure. The study also found SNPs associated with the trait, LA active emptying fraction (LAAEF). A decrease in LAAEF has been associated with increased risk of AF [59]. CASQ2 encodes calsequestrin2 which is important for calcium handling and cardiac contraction [17]. Myosin-18B (MYO18B) was recently shown to support myofibril organization in differentiating cardiomyocytes and acts as an actin cross-linker and myosin-2 in cardiac sarcomere [28]. MYO18B was also proposed to be associated with nemaline myopathy and cardiomyopathy [60].
A gene-based association study on 660 patients with a history of either paroxysmal or persistent AF suggested the contribution of ZFHX3 to AF remodeling and response to catheter ablation, an established AF treatment. When single SNPs from GWAS studies do not exhibit a large effect in GWAS univariate analyses, gene-based tests (miniSNP, VEGAS-versatile gene-based association study) can detect genome-wide significant genes [61].
A study that investigated variants causal to AF from loci previously identified from GWAS and a meta-analysis of GWAS, found a rare loss-of-function variant c.105 + 1G > T in SYNPO2L to be associated with a significantly increased risk of developing AF (Odds Ratio, OR 3.5) [6, 62]. The largest meta-analysis of GWAS studies of combined ancestry included more than 65,000 cases and identified 97 loci implicating genes in cardiac development, electrophysiological, contractile, and structural pathways, highlighting the pleiotropic nature of AF associated genetic factors [6]. The locus upstream of PITX2 at 4q25 was found to be the most significant AF associated region in Europeans, Japanese and African Americans. The different loci identified in the meta-analyses were classified into four themes. The loci that are the primary targets of current antiarrhythmic therapy (SCN5A, KCNH2), loci associated with transcriptional regulation, a key feature of AF (TBX5, NKX2), genes whose altered expression levels underlie AF (PRRX1, KCNJ5), and finally loci that are implicated in Mendelian forms of arrhythmia syndromes (CASQ2, PKP2). Mutations in CASQ2 and PKP2 can lead to ventricular tachycardia and impaired cardiomyocyte communication and structural integrity respectively. A previous meta-analysis of the earlier GWAS studies identified PITX2, ZFHX3, and KCNN3, the three top of susceptibility loci and SNPs in or near potential candidate genes involved in cardiopulmonary development, pace-making activity and, signal transduction (PRRX1, CAV1, SYNE2, HCN4, SYNPO2L, and MYOZ1)[36].
In a large scale, multi-ethnic meta-analyses of common and rare variant association studies, Christophersen et al. included individuals in the Atrial Fibrillation Genetics (AFGen) Consortium from the analyses of GWAS, exome-wide association studies (ExWAS), and rare variant association studies (RVAS) in 33 studies, including 22,806 individuals with atrial fibrillation and 132,612 referents [7]. The study identified 12 novel genetic loci that exceeded genome-wide significance, implicating genes involved in cardiac electrical and structural remodeling. The comprehensive meta-analysis identified loci most frequently associated with AF (TTN, PLN, KCNN2, KCNJ5, SH3PXD2A, and SCN10A) and some uncommon AF linked genes (KIFAP3, ANXA4, CEP68, WNT8A, PCM1, and SOX5). KCNN2 encodes calcium-dependent potassium channel SK2 that forms heteromeric channel complexes with SK3 encoded by the previously implicated KCNN3 gene. KCNN2 along with KCNJ5 is known to be involved in the maintenance of the atrial cardiac action potential.
GWAS have demonstrated the polygenic nature of AF. Genome-based prediction models can be used to evaluate genetic risk scores. “An AF polygenic risk score summarizes the cumulative genetic risk” [63]. In 2014, Tada et al. evaluated the risk of incident AF with 12 SNPs associated with AF at a genome-wide significance level (P < 5 × 10–8). AF genotype-risk score (GRS) identified 20% of individuals at two-fold increased risk for incident AF and at 23% increased risk for ischemic stroke [64]. A recent study has shown that genome-wide polygenic risk scores (PRS) derived for common diseases can identify individuals at risk of the disease similar to monogenic mutations. The study employed genome-wide polygenic scores derived from more than 6 million variants from across the genome and identified 6.1% of the population at more than threefold risk for AF [65].
Integrative studies
Recent advances in the “omics” technologies have led to an integrated approach for the simultaneous study of genomics, epigenomics, transcriptomics and proteomics for a holistic understanding of the pathophysiological processes underlying complex human diseases [66]. A study set to unravel the genetic predisposition for AF by probing the non-coding regions, first prioritized candidate genes cross-referencing human and mouse transcriptomic, epigenomic, and chromatin conformation datasets, studied the impact of regulatory elements on putative target gene expression, and identified potential cardiac regulatory elements by Assay for Transposase Accessible Chromatin with high-throughput sequencing (ATAC-seq), a method for genome-wide mapping of chromatin accessibility [67], at the regions associated with AF and deployed EMERGE, an enhancer prediction tool [31]. The study identified BMP10, SMYD2, PITX2, MYOT, TBX5, GJA1, CAV1, HCN4, SPATS2L, and PLN genes to be likely affected by AF associated variants and the role of regulatory region variants in modulating the expression of the potential AF genes. BMP10 and SMYD2 exhibit highly enriched expression in the right atrium. Though the exact function of SPATS2L is not known, a previous study implicated this gene for QT duration [68]. PLN which is enriched in adult tissue showed the highest differential expression in cardiomyocytes in this study. PLN encodes a major substrate for the cAMP-dependent protein kinase in cardiac muscle and was previously found to be involved in the AF pathogenesis [32].
Wang et al. integrated information from three kinds of omics data from samples of European ancestry using computational approaches. GWAS data from the Atrial fibrillation genetics (AFGen) consortium 2017 were integrated with epigenome-wide association study (EWAS) and transcriptome-wide association study (TWAS) which analyzed whole blood DNA methylation markers and whole blood gene expression respectively. The study converted summary statistics from individual omics into gene-level associations and performed tissue-specific network analysis to identify 1931 potential AF-related genes which is much higher than the number of genes identified by GWAS (AFGen 2018) alone. Most of the genes identified in the study were involved in cardiac muscle structure [69].
Genes identified by high throughput sequencing
In recent years, whole genome or exome sequencing studies have identified variants with lesser frequency but that which have a greater effect on the disease phenotype. In lone AF, Olesen et al. found a higher prevalence of rare variations in previously implicated ion-channel proteins involved in cardiac depolarization (e.g. SCN5A and SCN1-3B), or cardiac repolarisation (e.g. KCNQ1, KCNA5, KCND3, KCNE1, KCNH2, and KCNJ2). The results compared with data from NHLBI Exome Variant Server (EVS) found that rare variations (minor allele frequency, MAF < 0.1) were significantly higher in early-onset lone AF patients with an odds ratio (OR) greater than ORs reported for common variations by GWAS [10]. Resequencing genes for cardiac potassium channels found rare variants in these genes to be significantly associated with a cohort of familial AF probands than in healthy controls [70].
Lieve et al. used a panel of genes associated with different cardiomyopathies and catecholaminergic polymorphic ventricular tachycardia to identify the functional genetic determinants in a Dutch family with ventricular arrhythmias and early-onset AF. The next-generation sequencing (NGS) analysis identified a gain–of–mutation in SCN5A in the proband. SCN5A encodes the alpha subunit of the main cardiac sodium channel. A novel missense mutation p.M1851V co-segregated with the clinical phenotype in the family and led to increased channel availability and increased window current [35].
Tucker et al. sequenced ~ 158 kilobases (kb) including PRRX1 in 962 individuals of European descent with or without AF and identified rs577676 within an enhancer that interacts with the PRRX1 promoter to potentially alter its expression in the left atrium. Functional analyses found that suppression of PRRX1 could shorten atrial action potential duration considered to be the hallmark of AF [71]. In a previous WES study on the affected members of a family presenting with a complex phenotype of AF, atrial, and ventricular septal defects, Tucker et al. identified four rare mutations with a gain-of-function effect in the GATA6 gene that encodes a transcription factor that regulates the expression of genes required to maintain electrical stability in the atrium [20]. They also studied a large cohort of early-onset AF using a combination of high-resolution melting and Sanger sequencing to screen GATA6 exons. GATA6 has been implicated in the maintenance of cardiac precursor cells in an undifferentiated proliferative state during cardiac development [72].
Gudbjartsson et al. performed WGS on 2636 Icelanders with AF. A recessive frameshift deletion in MYL4 (c.234delC, p.Cys78Trpfs*29) was identified for the first time. MYL4 encodes myosin essential light chain, the key sarcomeric component found in both embryonic muscle and adult atria [9]. Another study that performed WES on a family presenting with a syndrome characterized by early-onset AF (age < 35 years), conduction disease, and signs of atrial myopathy identified a novel p.Glu111Lys mutation in MYL4 in all its affected members. A primary atrial-specific sarcomeric defect was found to cause the rare AF subtype in this family [27].
A large-scale case–control study sought to identify genes associated with early-onset AF defined as AF onset < 66 years of age. The authors performed WGS with a mean genome coverage of 99.1% and found a loss of function variant in the TTN gene [73]. Interestingly, an excess of loss of function (LOF) variants has been reported in TTN when WES was performed on 24 families. These titin-truncating variants (TTNtv) were also found in an independent cohort of early-onset AF patients. TTNtvs could disrupt the assembly of sarcomeres in both atria and ventricles [74].
A high-throughput sequencing study in 20 parent–offspring trios identified 5 novel rare variants that included a variant in the 5′ regulatory region of PITX2 that downregulated PITX2c in atrial myocytes and four exonic nonsense mutations in SYNE2, ZFXH3, and KCNN3 [75]. A novel mutation (p.Trp498Ter) in the LMNA gene was found to co-segregate with AF-affected members in a four-generation family from the north of China by exome-sequencing [76]. LMNA belongs to the family of intermediate filaments type V lamins. Loss of function mutations similar to the novel mutation identified in this study has been found to underlie the mechanism for LMNA associated dilated cardiomyopathy [77]. Lubitz et al. examined WES data of 1734 AF cases and 9423 controls of predominantly European ancestry. The study did not find any significant association between coding region variants and AF and neither did they observe any significant variant enrichment in the previously reported loci in AF cases suggesting that variants of coding regions may not be the predominant factors to lead to common forms of AF [78].
A cohort study by Yoneda et al. in 2021 performed whole-genome sequencing for 1293 patients with early-onset AF for the analysis of genes included in the commercial gene panels available for arrhythmias and cardiomyopathies. The study found a greater overlap between AF-associated variants and loci linked with inherited cardiomyopathies. The study warrants genetic testing for early-onset AF with the results of the study demonstrating positive genetic test results for up to 16.8% patients diagnosed with AF before 30 years of age [79].
Conclusions
The recent findings point to the genetic complexity of AF with both common and rare variants adding to the heterogeneity. Most AF cases are multifactorial, following the traits of a common complex disorder. Polygenic risk scores that take into consideration the contributions of common variants from relevant pathways can better predict genetic susceptibility. The recent studies have identified loci linked with myocardial structural components, transcription factors and atrial development, cardiomyocyte contractility and electrophysiology to contribute to AF pathogenesis. The major challenge lies in the translation of the genetic data into clinically useful information and clinical decision-making.
References
Chugh SS, Havmoeller R, Narayanan K et al (2014) Worldwide epidemiology of atrial fibrillation: a global burden of disease 2010 study. Circulation 129:837–847. https://doi.org/10.1161/CIRCULATIONAHA.113.005119
Camm AJ, Kirchhof P, Lip GYH et al (2010) Guidelines for the management of atrial fibrillation. Eur Heart J 31:2369–2429. https://doi.org/10.1093/eurheartj/ehq278
Vermond RA, Geelhoed B, Verweij N et al (2015) Incidence of atrial fibrillation and relationship with cardiovascular events, heart failure, and mortality a community-based study from the Netherlands. J Am Coll Cardiol 66:1000–1007. https://doi.org/10.1016/j.jacc.2015.06.1314
Fox CS, Parise H, D’Agostino RB Sr et al (2004) Parental atrial fibrillation as a risk factor for atrial fibrillation in offspring. JAMA 291:2851–2855. https://doi.org/10.1001/jama.291.23.2851
Lubitz SA, Yin X, Fontes JD et al (2010) Association between familial atrial fibrillation and risk of new-onset atrial fibrillation. JAMA 304:2263–2269. https://doi.org/10.1001/jama.2010.1690
Roselli C, Chaffin MD, Weng LC et al (2018) Multi-ethnic genome-wide association study for atrial fibrillation. Nat Genet 50:1225–1233. https://doi.org/10.1038/s41588-018-0133-9
Christophersen IE, Rienstra M, Roselli C et al (2017) Large-scale analyses of common and rare variants identify 12 new loci associated with atrial fibrillation. Nat Genet 49:946–952. https://doi.org/10.1038/ng.3843
Tucker NR, Ellinor PT (2014) Emerging directions in the genetics of atrial fibrillation. Circ Res 114:1469–1482. https://doi.org/10.1161/CIRCRESAHA.114.302225
Gudbjartsson DF, Helgason H, Gudjonsson SA et al (2015) Large-scale whole-genome sequencing of the Icelandic population. Nat Genet 47:435–444. https://doi.org/10.1038/ng.3247
Olesen MS, Andreasen L, Jabbari J et al (2014) Very early-onset lone atrial fibrillation patients have a high prevalence of rare variants in genes previously associated with atrial fibrillation. Heart Rhythm 11:246–251. https://doi.org/10.1016/j.hrthm.2013.10.034
Roberts JD, Gollob MH (2010) Impact of genetic discoveries on the classification of lone atrial fibrillation. J Am Coll Cardiol 55:705–712. https://doi.org/10.1016/j.jacc.2009.12.005
Roberts JD, Gollob MH (2014) A contemporary review on the genetic basis of atrial fibrillation. Methodist Debakey Cardiovasc J 10:18–24. https://doi.org/10.14797/mdcj-10-1-18
Mahida S, Lubitz SA, Rienstra M et al (2011) Monogenic atrial fibrillation as pathophysiological paradigms. Cardiovasc Res 89:692–700. https://doi.org/10.1093/cvr/cvq381
Kalstø SM, Siland JE, Rienstra M, Christophersen IE (2019) Atrial fibrillation genetics update: toward clinical implementation. Front Cardiovasc Med 6:1–16. https://doi.org/10.3389/fcvm.2019.00127
Sinner MF, Tucker NR, Lunetta KL et al (2014) Integrating genetic, transcriptional, and functional analyses to identify 5 novel genes for atrial fibrillation. Circulation 130:1225–1235. https://doi.org/10.1161/CIRCULATIONAHA.114.009892
Aoki T, Okada N, Ishida M et al (1999) TIP120B: a novel TIP120-family protein that is expressed specifically in muscle tissues. Biochem Biophys Res Commun 261:911–916. https://doi.org/10.1006/bbrc.1999.1147
Ahlberg G, Andreasen L, Ghouse J et al (2021) Genome-wide association study identifies 18 novel loci associated with left atrial volume and function. Eur Heart J 42:4523–4534. https://doi.org/10.1093/eurheartj/ehab466
Volonte D, McTiernan CF, Drab M et al (2008) Caveolin-1 and caveolin-3 form heterooligomeric complexes in atrial cardiac myocytes that are required for doxorubicin-induced apoptosis. Am J Physiol 294:392–401. https://doi.org/10.1152/ajpheart.01039.2007
Oka T, Maillet M, Watt AJ et al (2006) Cardiac-specific deletion of gata4 reveals its requirement for hypertrophy, compensation, and myocyte viability. Circ Res 98:837–845. https://doi.org/10.1161/01.RES.0000215985.18538.c4
Tucker NR, Mahida S, Ye J et al (2017) Gain-of-function mutations in GATA6 lead to atrial fibrillation. Heart Rhythm 14:284–291. https://doi.org/10.1016/j.hrthm.2016.10.014
Salameh A, Haunschild J, Bräuchle P et al (2014) On the role of the gap junction protein Cx43 (GJA1) in human cardiac malformations with fallot-pathology. A study on paediatric cardiac specimen. PLoS ONE 9:1–11. https://doi.org/10.1371/journal.pone.0095344
Song K, Nam YJ, Luo X et al (2012) Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature 485:599–604. https://doi.org/10.1038/nature11139
Schulze-Bahr E, Neu A, Friederich P et al (2003) Pacemaker channel dysfunction in a patient with sinus node disease. J Clin Invest 111:1537–1545. https://doi.org/10.1172/JCI200316387
Olesen MS, Refsgaard L, Holst AG et al (2013) A novel KCND3 gain-of-function mutation associated with early-onset of persistent lone atrial fibrillation. Cardiovasc Res 98:488–495. https://doi.org/10.1093/cvr/cvt028
Lin J, Lin S, Choy PC et al (2008) The regulation of the cardiac potassium channel (HERG) by caveolin-1. Biochem Cell Biol 86:405–415. https://doi.org/10.1139/O08-118
Ellinor PT, Lunetta KL, Glazer NL et al (2010) Common variants in KCNN3 are associated with lone atrial fibrillation. Nat Genet 42:240–244. https://doi.org/10.1038/ng.537
Orr N, Arnaout R, Gula LJ et al (2016) A mutation in the atrial-specific myosin light chain gene (MYL4) causes familial atrial fibrillation. Nat Commun 7:1–8. https://doi.org/10.1038/ncomms11303
Latham SL, Weiß N, Schwanke K et al (2020) Myosin-18B regulates higher-order organization of the cardiac sarcomere through thin filament cross-linking and thick filament dynamics. Cell Rep 32:108090. https://doi.org/10.1016/j.celrep.2020.108090
Perrot A, Tomasov P, Villard E et al (2016) Mutations in NEBL encoding the cardiac Z-disk protein nebulette are associated with various cardiomyopathies. Arch Med Sci 12:263–278. https://doi.org/10.5114/aoms.2016.59250
Gudbjartsson DF, Arnar DO, Helgadottir A et al (2007) Variants conferring risk of atrial fibrillation on chromosome 4q25. Nature 448:353–357. https://doi.org/10.1038/nature06007
van Ouwerkerk AF, Bosada FM, van Duijvenboden K et al (2019) Identification of atrial fibrillation associated genes and functional non-coding variants. Nat Commun 10:1–14. https://doi.org/10.1038/s41467-019-12721-5
Kayvanpour E, Sedaghat-Hamedani F, Amr A et al (2017) Genotype-phenotype associations in dilated cardiomyopathy: meta-analysis on more than 8000 individuals. Clin Res Cardiol 106:127–139. https://doi.org/10.1007/s00392-016-1033-6
Asperti C, Astro V, Totaro A et al (2009) Liprin-α1 promotes cell spreading on the extracellular matrix by affecting the distribution of activated integrins. J Cell Sci 122:3225–3232. https://doi.org/10.1242/jcs.054155
Ihida-Stansbury K, McKean DM, Gebb SA et al (2004) Paired-related homeobox gene Prx1 is required for pulmonary vascular development. Circ Res 94:1507–1514. https://doi.org/10.1161/01.RES.0000130656.72424.20
Lieve KV, Verkerk AO, Podliesna S et al (2017) Gain-of-function mutation in SCN5A causes ventricular arrhythmias and early onset atrial fibrillation. Int J Cardiol 236:187–193. https://doi.org/10.1016/j.ijcard.2017.01.113
Ellinor PT, Lunetta KL, Albert CM et al (2012) Meta-analysis identifies six new susceptibility loci for atrial fibrillation. Nat Genet 44:670–675. https://doi.org/10.1038/ng.2261
Peng J, Raddatz K, Molkentin JD et al (2007) Cardiac hypertrophy and reduced contractility in hearts deficient in the titin kinase region. Circulation 115:743–751. https://doi.org/10.1161/CIRCULATIONAHA.106.645499
Jiang Q, Ni B, Shi J et al (2014) Down-regulation of ATBF1 activates STAT3 signaling via PIAS3 in pacing-induced HL-1 atrial myocytes. Biochem Biophys Res Commun 449:278–283. https://doi.org/10.1016/j.bbrc.2014.05.041
Lee JY, Kim TH, Yang PS et al (2017) Korean atrial fibrillation network genome-wide association study for early-onset atrial fibrillation identifies novel susceptibility loci. Eur Heart J 38:2586–2594. https://doi.org/10.1093/eurheartj/ehx213
Lubitz SA, Lunetta KL, Lin H et al (2014) Novel genetic markers associate with atrial fibrillation risk in Europeans and Japanese. J Am Coll Cardiol 63:1200–1210. https://doi.org/10.1016/j.jacc.2013.12.015
Haïssaguerre M, Jaïs P, Shah DC et al (1998) Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N Engl J Med 339:659–666. https://doi.org/10.1056/NEJM199809033391003
Kirchhof P, Kahr PC, Kaese S et al (2011) PITX2c is expressed in the adult left atrium, and reducing Pitx2c expression promotes atrial fibrillation inducibility and complex changes in gene expression. Circ Cardiovasc Genet 4:123–133. https://doi.org/10.1161/CIRCGENETICS.110.958058
Collins MM, Ahlberg G, Hansen CV et al (2019) Early sarcomere and metabolic defects in a zebrafish pitx2c cardiac arrhythmia model. Proc Natl Acad Sci USA 116:24115–24121. https://doi.org/10.1073/pnas.1913905116
Ma JF, Yang F, Mahida SN et al (2016) TBX5 mutations contribute to early-onset atrial fibrillation in Chinese and Caucasians. Cardiovasc Res 109:442–450. https://doi.org/10.1093/cvr/cvw003
Nadadur RD, Broman MT, Boukens B et al (2016) Pitx2 modulates a Tbx5-dependent gene regulatory network to maintain atrial rhythm. Sci Transl Med. https://doi.org/10.1126/scitranslmed.aaf4891
Low SK, Takahashi A, Ebana Y et al (2017) Identification of six new genetic loci associated with atrial fibrillation in the Japanese population. Nat Genet 49:953–958. https://doi.org/10.1038/ng.3842
Gutierrez A, Chung MK (2016) Genomics of atrial fibrillation. Curr Cardiol Rep. https://doi.org/10.1007/s11886-016-0735-8
Nica AC, Dermitzakis ET (2013) Expression quantitative trait loci: present and future. Philos Trans R Soc B Biol Sci. https://doi.org/10.1098/rstb.2012.0362
Herrmann S, Layh B, Ludwig A (2011) Novel insights into the distribution of cardiac HCN channels: an expression study in the mouse heart. J Mol Cell Cardiol 51:997–1006. https://doi.org/10.1016/j.yjmcc.2011.09.005
Milanesi R, Baruscotti M, Gnecchi-Ruscone T, DiFrancesco D (2006) Familial sinus bradycardia associated with a mutation in the cardiac pacemaker channel. N Engl J Med 354:151–157. https://doi.org/10.1056/nejmoa052475
Beqqali A, Monshouwer-Kloots J, Monteiro R et al (2010) CHAP is a newly identified Z-disc protein essential for heart and skeletal muscle function. J Cell Sci 123:1141–1150. https://doi.org/10.1242/jcs.063859
Bang ML, Chen J (2015) Roles of nebulin family members in the heart. Circ J 79:2081–2087. https://doi.org/10.1253/circj.CJ-15-0854
Santiago-Medina M, Gregus KA, Nichol RH et al (2015) Regulation of ECM degradation and axon guidance by growth cone invadosomes. Development 142:486–496. https://doi.org/10.1242/dev.108266
Nielsen JB, Fritsche LG, Zhou W et al (2018) Genome-wide study of atrial fibrillation identifies seven risk loci and highlights biological pathways and regulatory elements involved in cardiac development. Am J Hum Genet 102:103–115. https://doi.org/10.1016/j.ajhg.2017.12.003
Nielsen JB, Thorolfsdottir RB, Fritsche LG et al (2018) Biobank-driven genomic discovery yields new insight into atrial fibrillation biology. Nat Genet 50:1234–1239. https://doi.org/10.1038/s41588-018-0171-3
Norgett EE, Hatsell SJ, Carvajal-Huerta L et al (2000) Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum Mol Genet 9:2761–2766. https://doi.org/10.1093/hmg/9.18.2761
Norman M, Simpson M, Mogensen J et al (2005) Novel mutation in desmoplakin causes arrhythmogenic left ventricular cardiomyopathy. Circulation 112:636–642. https://doi.org/10.1161/CIRCULATIONAHA.104.532234
Thornton CA (2014) Myotonic dystrophy. Neurol Clin 32:705–719. https://doi.org/10.1016/j.ncl.2014.04.011
Bertelsen L, Diederichsen SZ, Haugan KJ et al (2020) Left atrial volume and function assessed by cardiac magnetic resonance imaging are markers of subclinical atrial fibrillation as detected by continuous monitoring. Europace 22:724–731. https://doi.org/10.1093/europace/euaa035
Malfatti E, Böhm J, Lacène E et al (2015) A premature stop codon in MYO18B is associated with severe nemaline myopathy with cardiomyopathy. J Neuromuscul Dis 2:219–227. https://doi.org/10.3233/JND-150085
Husser D, Büttner P, Ueberham L et al (2017) Association of atrial fibrillation susceptibility genes, atrial fibrillation phenotypes and response to catheter ablation: a gene-based analysis of GWAS data. J Transl Med 15:4–9. https://doi.org/10.1186/s12967-017-1170-3
Clausen AG, Vad OB, Andersen JH, Olesen MS (2021) Loss-of-function variants in the SYNPO2L gene are associated with atrial fibrillation. Front Cardiovasc Med. https://doi.org/10.3389/fcvm.2021.650667
Roselli C, Roselli C, Rienstra M et al (2020) Genetics of atrial fibrillation in 2020: GWAS, genome sequencing, polygenic risk, and beyond. Circ Res. https://doi.org/10.1161/CIRCRESAHA.120.316575
Tada H, Shiffman D, Smith JG et al (2014) Twelve-single nucleotide polymorphism genetic risk score identifies individuals at increased risk for future atrial fibrillation and stroke. Stroke 45:2856–2862. https://doi.org/10.1161/STROKEAHA.114.006072
Khera AV, Chaffin M, Aragam KG et al (2018) Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet 50:1219–1224. https://doi.org/10.1038/s41588-018-0183-z
Sun YV, Hu YJ (2016) Integrative analysis of multi-omics data for discovery and functional studies of complex human diseases. Elsevier Ltd, Amsterdam
Buenrostro JD, Wu B, Chang HY, Greenleaf WJ (2015) ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr Protoc Mol Biol 2015(2129):1–21299. https://doi.org/10.1002/0471142727.mb2129s109
Arking DE, Pulit SL, Crotti L et al (2014) Genetic association study of QT interval highlights role for calcium signaling pathways in myocardial repolarization. Nat Genet 46:826–836. https://doi.org/10.1038/ng.3014
Wang B, Lunetta KL, Dupuis J et al (2020) Integrative omics approach to identifying genes associated with atrial fibrillation. Circ Res. https://doi.org/10.1161/CIRCRESAHA.119.315179
Mann SA, Otway R, Guo G et al (2012) Epistatic effects of potassium channel variation on cardiac repolarization and atrial fibrillation risk. J Am Coll Cardiol 59:1017–1025. https://doi.org/10.1016/j.jacc.2011.11.039
Tucker NR, Dolmatova EV, Lin H et al (2017) Diminished PRRX1 expression is associated with increased risk of atrial fibrillation and shortening of the cardiac action potential. Circ Cardiovasc Genet 10:1–12. https://doi.org/10.1161/CIRCGENETICS.117.001902
Gove C, Walmsley M, Nijjar S et al (1997) Over-expression of GATA-6 in Xenopus embryos blocks differentiation of heart precursors. EMBO J 16:355–368. https://doi.org/10.1093/emboj/16.2.355
Choi SH, Weng LC, Roselli C et al (2018) Association between Titin loss-of-function variants and early-onset atrial fibrillation. JAMA 320:2354–2364. https://doi.org/10.1001/jama.2018.18179
Ahlberg G, Refsgaard L, Lundegaard PR et al (2018) Rare truncating variants in the sarcomeric protein titin associate with familial and early-onset atrial fibrillation. Nat Commun 9:1–11. https://doi.org/10.1038/s41467-018-06618-y
Tsai CT, Hsieh CS, Chang SN et al (2015) Next-generation sequencing of nine atrial fibrillation candidate genes identified novel de novo mutations in patients with extreme trait of atrial fibrillation. J Med Genet 52:28–36. https://doi.org/10.1136/jmedgenet-2014-102618
Zhao J, Yao H, Li Z et al (2016) A novel nonsense mutation in LMNA gene identified by exome sequencing in an atrial fibrillation family. Eur J Med Genet 59:396–400. https://doi.org/10.1016/j.ejmg.2016.06.006
Gupta P, Bilinska ZT, Sylvius N et al (2010) Genetic and ultrastructural studies in dilated cardiomyopathy patients: a large deletion in the lamin A/C gene is associated with cardiomyocyte nuclear envelope disruption. Basic Res Cardiol 105:365–377. https://doi.org/10.1007/s00395-010-0085-4
Lubitz SA, Brody JA, Bihlmeyer NA et al (2016) Whole exome sequencing in atrial fibrillation. PLoS Genet 12:1–12. https://doi.org/10.1371/journal.pgen.1006284
Yoneda ZT, Anderson KC, Quintana JA et al (2021) Early-onset atrial fibrillation and the prevalence of rare variants in cardiomyopathy and arrhythmia genes. JAMA Cardiol 6:1371–1379. https://doi.org/10.1001/jamacardio.2021.3370
Acknowledgements
Not applicable.
Funding
The work was supported by ICMR Project: No.5/4-5/1-6/2020-NCD-II.
Author information
Authors and Affiliations
Contributions
AM conceptualized, initiated, performed the literature search, and wrote the original draft. RS and VB supervised, reviewed, and revised the review critically for important intellectual content.
Corresponding author
Ethics declarations
Conflict of interest
Authors declare no conflict of interest.
Research involving human participants
This article does not contain any studies with human participants performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Manoharan, A., Sambandam, R. & Ballambattu, V.B. Genetics of atrial fibrillation—an update of recent findings. Mol Biol Rep 49, 8121–8129 (2022). https://doi.org/10.1007/s11033-022-07420-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-022-07420-2