
Abstract
Mungbean germplasm characterization, evaluation and improvement are fundamentally based on morpho-agronomic traits. The lack of break-through in mungbean production has been due to non-availability of genetic variability for high yield potential. Forty-four genotypes of mungbean [Vigna radiata (L.)Wilczek] were subjected to random amplified polymorphic DNA (RAPD) analysis to assess the genetic diversity and relationships among the genotypes. Multilocus genotyping by twelve RAPD primers generated 166 markers and detected an average of intraspecific variation amounting to 82% polymorphism in banding patterns. Dendrogram obtained from cluster analysis delineated all the 44 genotypes into six clusters. Higher values of Nei’s gene diversity (h) and Shannon information index (i) and genetic distance analysis validate existence of wide genetic diversity among mungbean genotypes tested. Besides internal transcribed spacer (ITS) length variations, single nucleotide polymorphisms (SNPs) and insertions/deletions (INDELS) were detected at number of sites in nuclear rDNA region and the sequences of representatives of each sub-cluster and all distinct genotypes have been submitted to NCBI database and assigned Gen accession numbers HQ 148136-148147. Multiple sequence alignment revealed further lineages of distinct genotypes with main RAPD clusters. The measures of relative genetic distances among the genotypes of mungbean did not completely correlate the geographical places of their development. The homogeneous phenotypic markers proved insufficient in exhibiting genetic divergence among mungbean genotypes studied. RMG-62, RMG-976, and NDM-56 have been identified as potential source of parents for crop improvement. RAPD primers, OPA-9 and OPA-2 as polymorphic genetic markers and number of pods/plant and number of seeds/plant as dependable phenotypic markers have been identified for improving yield potentials. This genetic diversity will be of significance in developing intraspecific crosses in mungbean crop improvement programme.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Green gram [Vigna radiata (L.) Wilczek] an Asiatic species of pantropical genus Vigna, is an important legume crop, widely cultivated in tropics and sub-tropics for human consumption, animal feed, as a cover-crop and as supplemented crop in cereal based cropping system. It is thought to be originated in the Indian sub-continent [1–3] with maximum diversity in the upper Western Ghats and Deccan hills and has secondary centre of diversity in Indo-gangetic plains. The major constraints for achieving higher yields are inherently low yielding potential, poor harvest index and susceptibility to biotic and abiotic stresses. Jain [4] attributed that a large proportion of alleles of higher productivity have been lost in the present populations of mungbean due to overriding role of natural selection even long after the crop domestication.
Characterization and cataloguing of mungbean germplasm have been traditionally carried out by using morpho-agronomic traits [5, 6]. Whereas, these characters are often subjected to constraints from environmental interactions, epistatic interactions and the largely unknown genetic control of the traits [7]. The assessment by evaluation of morphological traits indicates that the germplasm is constrained by low level of diversity in the working gene pool of this crop [8]. Evaluation of morphological traits, pedigrees, geographic origins, isozymes and DNA markers have also been used for the assessments of Vigna species genetic diversity [9–13].
Random amplified polymorphic (RAPD) allows the analysis of individual and large number of markers in relatively short time, as only a few primers allow the generation of sufficient data to obtain a robust estimate of diversity index and have allowed the resolution of complex taxonomic relationships [14, 15]. It uses ten base primer to amplify the random portion of genomic DNA [16]. The data from RAPD analysis have indicated greater genetic diversity than allozymes in plant species [17, 18]. This technique has been used on a number of taxa including barley [19], brassica [20], wheat [21], pigeon pea [22], and chickpea [23]. In Vigna radiata a few previous studies have been carried out using RAPD profiles alone or in combination with ISSR profiles [24–29].
Currently, nuclear ribosomal internal transcribed spacer (ITS) is considered as one of the most useful phylogenetic marker due to less functional constrains and comparatively higher evolutionary changes [30]. It evolves relatively quickly and can be useful in determining inter-species [31, 32] and sometimes intra-species relationships [33]. The rate and patterns of ITS sequence mutation are typically appropriate for resolving relationships among species and genera [34, 35]. Although thousands of copies of the ITS exist in Angiosperm genomes, they are generally homogenized by concerted evolution, and thus can be treated as a single locus [36]. The nuclear ribosomal RNA (rRNA) gene complex is a tandem repeat unit of one to several thousand copies. This complex has several domains that evolve at varying rates [37] and thus have different phylogenetic utilities.
The region of 5.8S rRNA gene was shown to contain considerable phylogenetic information [38]. The ITS polymorphism might occur at a genus, species or individual levels, making it useful for phylogenetic, evolutionary and bio-geographical diversity studies [39]. ITS sequences have been widely used in several species [40, 41] for phylogenetic inferences.
The present study was undertaken to assess the extent of genetic diversity and relationship among different populations of [Vigna radiata (L) Wilczek] based on nuclear rDNA and RAPD polymorphism to select genetically diversed parents for crop improvement.
Materials and methods
Field experiment
Forty-four promising genotypes of mungbean including few check varieties were procured from All India Coordinated Research Project, CAZRI, Jodhpur. Field experiments were laid out in randomized block design with three replications at Central Arid Zone Research Institute, Jodhpur during rainy (Kharif) season in the years 2009 and 2010. Each plot was accommodating three rows of 4 m length with 30 cm and 10 cm distance from rows and plants, respectively. The recommended cultural practices were followed. The data of ten morphological characters namely plant height (cm), number of primary branches, number of secondary branches, number of pods/plant, number of seeds/pod, pod length (cm), number of cluster/plant, days to 50% flowering, 100 seed weight (g) and seed yield (kg/ha) were recorded. The phenotypic correlation of all the morpho-agronomic traits studied with seed yield was calculated to identify possible phenotypic markers. The details of the genotypes are given in Table 1.
DNA extraction and quantification
The genomic DNA was extracted from 100 mg of fresh leaves of each of the 44 genotypes of V. radiata, crushed with pestle and mortar in liquid nitrogen. The plant genomic DNA purification spin kit “Hi Pura” of Hi-media Company and protocols suggested by Birren and Lai [42] and Sambrook et al. [43] were followed for genomic DNA isolation. Finally, the genomic DNA was eluted with 200 μl of Tris–EDTA buffer (TE) for DNA fingerprinting. DNA was quantified with UV/VIS spectrophotometer by measuring OD260 and OD280. The quantified DNA samples were diluted in TE buffer to make a final concentration of 50 ng/μl for PCR reactions.
RAPD analysis
A set of 26 decamer random primers of OPA, OPB, and OPP series (Operon Technologies) were used for initial screening of 44 genotypes of V. radiata. Multilocus genotyping by RAPD was performed using 12 decamer arbitrary primers supplied by Operon Technologies. Each amplification was performed in a total reaction mixture of 25 μl. Each reaction mixture contained: decamer primer, 2 μl (50 pmol/μl); dNTP mix, 2 μl (2 mM each of dATP, dGTP, dCTP, and dTTP from MBI, Fermentas); MgCl2, 1 μl (25 mM, MBI, Fermentas); Taq DNA polymerase, 0.4 μl (5 U/μl, Sigma chem); 10× PCR buffer, 2.5 μl (100 mM, Tris–HCl, pH 8.3, 15 mM MgCl2, 250 mM KCl), 12.9 μl of dH2O, and 4 μl of genomic DNA (50 ng/μl). RAPD-PCR amplification were performed in a gradient thermal cycler (Corbett Research, USA) with lid heating option at 110°C with initial denaturation step of 94°C for 3 min followed by 38 amplification cycles of 94°C for 40 s, 50°C for 40 s, and 72°C for 2 min and final elongation at 72°C for 7 min.
Amplicons were separated on a 1.5% agarose gel pre-stained with ethidium bromide solution using 1× TAE buffer. The gels were run for 4 h at 45 V and the RAPD amplicons profiles were recorded using Syngene Gel Documentation System with Genesnap software. The size of the amplified fragments was determined using 100 bp plus ladder (MBI Fermentas). All RAPD reactions were performed twice to test the reproducibility of the amplicons profile.
ITS amplification
The genomic DNA of 12 mungbean genotypes namely, ML-818, Ganga-8, AKM-9911, COGG-934, PUSA-0771, PM-047, NDM-56, RMG-976, MH-418, MH-429, NVL-1 and RMG-62 representing different RAPD clusters and all distinct/out group genotypes were used for amplification and sequencing of 5.8S gene region. The polymerase chain reaction (PCR) primer ITS-1 (19 bp) and ITS-4 (20 bp) developed by White et al. [44] were used to amplify the ITS of ribosomal DNA, which encompasses the 5.8S gene and both ITS-1 and ITS-2 regions. Amplification by PCR was performed in a total volume of 50 μl containing: 1 U Taq DNA polymerase (Promega), 2.5 mM MgCl2, 160 μM dNTP mix (MBI, Fermentas), 50 pmol of each ITS-1 and ITS-4 primers, 50 ng genomic DNA in dH2O. The reactions were performed in the gradient thermal cycler (Corbett Research, USA) with following conditions. 1 min denaturation at 95°C, 30 s annealing at 50°C, 1 min 20 s elongation at 72°C, for 36 cycles with a final elongation step of 72°C for 7 min. The PCR products were visualized on 1.4% agarose gel in 1× TAE buffer at 60 V for 100 min. Agarose gels were stained with ethidium bromide and photographed under UV light for amplified ITS products.
Sequencing of ITS region
PCR product of ITS amplified region containing ITS-1, 5.8S rDNA and ITS-II were directly sequenced using ITS-1 (forward primer) and ITS-4 (reverse primer) by Big dye terminator method in ABI prism DNA sequencer. The sequenced data obtained from ITS-4 reverse primer was inversed using Gene doc software and clubbed with sequence data of ITS-1 to obtain complete sequences of amplified ITS product. Nucleotide sequence comparisons were performed by using basic local alignment search tool (BLAST) network services against the National Centre for Biotechnology Information (NCBI), USA database. The molecular characterization of the genotypes was done based on similarity with the best-aligned sequence of BLAST search.
Molecular analysis of RAPD
The RAPD amplification products were scored as present (1) or absent (0) for each primers genotype combination. Molecular data were entered into a binomial matrix and were used to determine Jaccard’s similarity coefficient with NTSYS-pc software [45, 46]. To perform molecular analysis, the 44 genotypes were divided into five regions of the country (North, South, East, West, and Central) and considered as 11 populations based on the state they represent in All India Coordinated Pulse Improvement Programme. Principal Coordinate Analysis via covariance matrix was calculated using GenALEx 6 software [47]. Whereas, diversity in the frequency of fragment size of RAPD patterns was apportioned within and among mungbean genotypes using Shannon’s information index (i) [48] and gene diversity index (h) following Nei [49] using PopGen 32 programme.
Sequence analysis
Nucleotide sequence comparisons were performed by using the basic local alignment search tool (BLAST) network services against the National Centre for Biotechnology Information databases. The multiple sequence alignment of ITS region (ITS-1, 5.8S r-RNA gene and ITS-2) of all the ten representative mungbean genotypes was performed using CLUSTAL X 1.83 software to detect SNPs. The phylogenetic relationships among the genotypes were estimated after the construction of a phylogram based on multiple sequence alignment of rDNA ITS sequences with the Tree View software [50].
Results
The perusal of phenotypic characterization data of 44 mungbean genotypes showed significant variations in most of the characters (Table 1). The plant height varied from 38 (NVL-1) to 50 cm (NDM-7-31); number of primary branches/plant from six (six genotypes) to nine (RMG-62); number of secondary branches/plant from six (nine genotypes) to 12 (OBGG-11-1 and RMG-976); number of pods/plant from ten (COGG-934 and NVL-1) to 24 (RMG-62); number of seeds/pod from seven (NVL-1) to 13 (RMG-62); pod length from six (NVL-1) to 10 cm (RMG-62 and RMG-976); number of clusters/plant from four (11 genotypes) to seven (three genotypes), days to 50% flowering from 29 (MH-530) to 51 (PM-0427) and 1,000 seed weight from 31 (NVL-1) to 56 g (RMG-976). The maximum seed yield of 768 kg/ha was recorded from RMG-62 followed by the yield of 741 kg/ha (RMG-976) and 724 kg/ha (NDM-56) which were statistically at par with RMG-62 and the minimum yield of 261 kg/ha was harvested from NVL-1.
The mean values of morpho-agronomic characters exhibited phenotypic diversity among and within the populations. The population three exhibited the maximum phenotypic diversity within the population, i.e., NVL-1 was recorded with the minimum seed yield of 261 kg/ha with minimum values of most of the yield attributing phenotypic traits. Whereas, within this population NDM-5-6 was recorded with the maximum seed yield of 724 kg/ha and the maximum values of the most of the phenotypic traits studied. While, populations, ten followed by eight and seven resulted in relatively higher seed yield as compared to others. The phenotypic correlation of morpho-agronomic traits with seed yield (Table 1) resulted in highly significant and positive correlation with number of pods/plant (98%), number of seeds/plant (90%), pod length (84%) and 1,000 seed weight (81%).
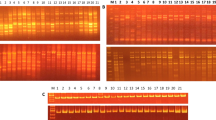
Out of 26 decamer random primers tested, 12 primers detected intraspecific variations generating scorable amplicons reproducible patterns and generated 166 marker bands in the range of 175 bp to 4 Kb (Table 2). Among these, 141 markers were polymorphic amounting to 84.93% polymorphism and exhibited 60–100% polymorphism in banding pattern. The number of PCR amplified products formed ranged from nine (OPP-07) to 22 (OPP-02) with an average of about 14 bands per primer. The primer OPA-09 was most informative primer which exhibited 100% polymorphism in RAPD banding patterns. The RAPD profiles generated by two informative primers (OPA-09 and OPA-02) are shown vide Figs. 1a, b and 2a, b. The dendrogram obtained from cumulative cluster analysis of twelve primers scorable fragments varied from 0.72 to 0.92 and clearly delineated all 44 genotypes of V. radiata. The combined dendrogram of the 12 random primer data matrixes delineated 44 genotypes into six main clusters (Fig. 3).

a RAPD profiles of 1–22 genotypes of Vigna radiata amplified by OPA-9 primer. b RAPD profiles of 23–44 genotypes of Vigna radiata amplified by OPA-9 primer

a RAPD profiles of 1–22 genotypes of Vigna radiata amplified by OPA-2 primer. b RAPD profiles of 23–44 genotypes of Vigna radiata amplified by OPA-2 primer

Dendrogram of 44 genotypes of Vigna radiata based on 12 RAPD informative primers
Cluster I had two sub-clusters, the first sub-cluster contained genotypes ML-818, KM-2262, GM-0213, BGS-9 and ML-1405, while the second sub-cluster contained genotypes PUSA-0771, SKAM-300, PUSA-0772 and MH-530. Cluster II comprised of two sub-clusters containing genotypes Ganga-8, MH-521 and RMG-977 in first sub-cluster while, genotypes ML-1333, NDM-734, PM-0427, GS-6999 and COGG-934 in second sub-cluster. Cluster III consisted of two sub-clusters in which OBGG-111 and IPM-0214 belonged to first sub-cluster while the genotypes PM-047 and TMB-26 were catalogued in the second sub-cluster along with AKM-9911 as a distinct genotype in the III cluster. Cluster IV comprised of IPM-0219, NDM-56, NDM-731, and COGG-936 along with GM-9924, PUSA-0672 and MH-418 as distinct genotypes of IV cluster. Cluster V comprised of the genotypes S-8, K-851, and RMG-62. Cluster VI comprised of four sub-clusters with ML-1349 and MH-429 in first sub-cluster, KM-2260, RMG-976 and HUM-23 in second sub-cluster, PUSA-0671, PUSA-0571, GM-9925 and RMG-34 in third sub-cluster, KM-2241 and RMG-267 in fourth sub-cluster along with NVL-1 as a distinct genotype in VI cluster.
On the basis of delineation one representative genotype from each RAPD sub-cluster and all distinct genotypes were selected for nuclear rDNA ITS region DNA sequencing. All the 12 selected genotypes of mungbean generated a single band which included partial sequence 18S gene, complete sequence (ITS-1, 5.8S gene, ITS-2) and partial sequence 28S gene upon direct sequencing using ITS-1 and ITS-4 primers (Table 3).
All the gene sequences have been submitted to NCBI databases and assigned Gen Accession numbers HQ148136-148147. The conserved 5.8 S r-DNA region was recorded with uniform nucleotide length of 167 base pairs in all the genotypes. Whereas, ITS-1 region exhibited base pair length diversity ranging from 203 to 212 bp and ITS-2 region from 218 to 224 bp.
We recorded not only single nucleotide polymorphisms (SNPs) at number of places but also small insertions and deletions (INDELS) in both the ITS regions. Whereas, 5.8S rDNA conserved region was recorded with SNPs by way of replacement of single nucleotide at number of places. The phylogram generated using Tree view software programme delineated all the genotypes representing all clusters and distinct genotypes and revealed further lineages among representative genotypes of different RAPD sub-clusters, i.e., the distinct genotypes NVL-1, RMG-62, COGG-934, and MH-418 have shown close lineages with the representatives of other clusters upon multiple sequence alignment of rDNA regions (Fig. 4).

Phylogram generated using Tree View of multiple sequence aligned rDNA region of 12 Vigna radiata genotypes
Genetic analyses of mungbean populations
Eleven V. radiata populations were subjected to principal coordinate analysis (PCA) using GenALEx software programme (Fig. 5). The first three principal coordinates accounted for 35.01, 18.50, and 13.24%, respectively, amounting to a total of 66.75% of total variance. The Eigen vector analysis indicated that the contributions of the first three factors were 20.49, 10.83, and 7.75, respectively, (explaining a total of 39.07 of total variability). A perusal of distribution of populations across principal coordinates clearly showed that populations 2, 3, and 7 are widely distributed across all the three principal coordinates as compared to others. The results validate the existence of higher genetic diversity among mungbean genotypes studied. A summary of mean genetic variation statistics of all the eleven populations and mean of all loci is presented in Table 4.

The principal coordinates analysis (PCA) of all the six clusters (11 populations) using GenALEx software [47] for 44 genotypes of Vigna radiata
The mean values of all the eleven populations together for Nei’s gene diversity (h) was 0.2079 and Shannon information index (i) was 0.3287. Results showed that the genetic diversity of mungbean genotypes of Pop 3 was the richest among all the eleven populations. Nei’s unbiased measure of genetic distance was employed to further elucidate the gene differentiation among populations (Table 5). The Nei’s genetic distance ranged from 0.0155 to 0.2673 and genetic identity ranged from 0.7655 to 0.9846. The largest genetic distance (0.2673) occurred between population 11 and 6 and the least (0.0155) between populations 7 and 3 and vice versa for genetic identity.
Discussion
Under present investigation morphological markers vis-à-vis genetic markers, i.e., RAPD analysis indicated that there is no consistency in grouping mungbean genotypes with similar morphological traits as phenotypically similar genotypes were genetically catalogued into different clusters of RAPD dendrogram. For example the fact that mungbean genotypes belonging to different agro climatic regions fall into same phylogenetic cluster, i.e., ML-818 (Punjab), KM-2262 (UP), GM-0213 (Gujarat) and BGS-9 (Bihar). While, the different genotypes belonging to states of Haryana, Rajasthan Punjab, Gujarat, Delhi, Orissa, Uttar Pradesh, Tamil Nadu, Maharashtra, Bihar, and Jammu and Kashmir grouped under all the six different clusters. It suggests that V. radiata genetic diversity has wide genetic distribution across agro-climatic regions of the country.
The measures of relative genetic distances among populations did not completely correlate the geographical distances of places of their origins. For instance, studies on other plant species including (Phaseolus vulgaris) [51] (Cyamopsis tetragonoloba) [15] and (Trigonella foenum-graecum) [33] have also been reported with low correlation between geographic distribution and genetic distances measured by DNA markers. Such clustering of the genotypes of different locations ignored the influence of geographic variations within the genetic diversity of mungbean. Such a lack of correlation between geographic diversity and genetic diversity in mungbean has also been reported by Bisht et al. [5], Manivannan et al. [6] and Lavanya et al. [28].
Under present investigation RAPD marker system showed high levels of polymorphism among the mungbean genotypes (84.93%), revealing rich genetic diversity among the genotypes analysed and indicating its effectiveness for evaluating genetic diversity in mungbean. Earlier, low to moderate polymorphism was observed while analyzing Indian mungbean cultivars using RAPD primers [24, 52–54]. All the 44 cultivars were delineated into six clusters indicating variability among the genotypes. Earlier, Afzal et al. [25] obtained five clusters in RAPD analysis of selected mungbean cultivars and predicted reasonable variability that may be exploited for yield improvement.
A uniform base pair length of 167 bp in all the 12 sequenced genotypes validates the conserved nature of 5.8S gene region. Whereas, length polymorphism in intervening intron ITS regions indicate non-coding nature of the region. In contrast to highly conserved ribosomal RNA genes, the rDNA spacers such as the ITS and the intergenic spacer present high variability and faster rates of evolution [55, 56]. The presence of SNPs and small insertions and deletions (INDELS) validate the existence of genetic variability that enabled us not only in delineating the sub-clusters but also in estimating genetic distances and lineages with other genotypes of RAPD sub-clusters as shown in the phylogram (Fig. 4). The genotype Ganga-8 (check variety) is shown as the most distinct genotype in the phylogram because of the maximum number of SNPs and INDELS in the intervening ITS regions.
The ITS length variants and polymorphism have been reported for several plant species [38, 41, 55]. Saini et al. [56] reported intra-individual heterogeneity in nuclear rDNA ITS region in V. radiata which did not cause any phylogenetic errors at species level. Barkley et al. [57] employed digested mixed accessions of V. radiata variety radiata with CEL-1 and observed SNPs and suggested EcoTILLING as a powerful genetic analysis tool for rapid identification of naturally occurring variations.
The depiction of principal coordinate analysis of RAPD data clearly delineated all the 44 genotypes of V. radiata and showed that first three principal coordinates contributed as much as 66.75% of total variance. The maximum phenotypic diversity in morpho-agronomic characters of population 3 is validated by the molecular analysis of RAPD data as population 3 is shown as the most diverse population across all the three principal coordinates in Fig. 5. The analysis validates the existence of higher genetic diversity within population than amongst population with higher (h and i values) because of rich genetic diversity in mungbean varieties.
Based on obtained results, the elite mungbean genotypes RMG-62, RMG-976 and NDM-56 with higher seed yields of 768, 741 and 724 kg/ha, respectively, showing significant phylogenetic distances in both RAPD dendrogram and phylogram based on ITS diversity have been identified as potential source of parents for Vigna radiata crop improvement. Present study resulted in identification of RAPD primers OPA-9 and OPA-2 showing 100 and 95% polymorphism in banding patterns, respectively, as polymorphic genetic markers. Whereas, the number of pods/plant and number of seeds/plant as dependable phenotypic markers for improving yield potentials and mungbean improvement due to highly significant and positive correlation with seed yield. We report existence of reasonable genetic variability in mungbean genotypes that may be useful in crop improvement.
References
De Candolle A (1886) Origin of cultivated plants. Hafner Publishing, New York
Vavilov NI (1926) The origin, variation, immunity and breeding of cultivated plants (translated by K Starr, Chester). Chronica Bot 13:1–6
Zukovskij PM (1962) Cultivated plants and their wild relatives. Commonwealth Agriculture Bureau, London
Jain HK (1994) Pulses—the wonder plants of world agriculture. In: Ali M, Asthana AN, Mehta SL (eds) Twenty five years of pulses research in India. Indian Institute of Pulses Research, Kanpur, pp 1–4
Bisht IS, Mahajan RK, Kawalkar TG (1998) Diversity in green gram [Vigna radiata (L.) Wilczek] germplasm collections and its potential use in crop improvement. Ann Appl Biol 132:301–312
Manivannan N, Murugan E, Viswanathan PL, Dhankodi CV (1998) Genetic divergence in green gram. Legume Res 21:131–133
Smith JSC, Smith OS (1989) The description and assessment of distances between inbred lines of Maize-the utility of morphological, biochemical and genetic descriptors and a scheme for testing of distinctiveness between inbred lines. Maydica 34:151–161
Ramanujam S (1981) Biometrical basis for yield improvement in mungbean. In: Cowell R (ed) Proceedings 1st international mungbean symposium, AVRDC, Shanhua, p 210
Fatokun CA, Danesh D, Young ND, Stewart EL (1993) Molecular taxonomic relationships in the genus Vigna based on RFLP analysis. Theor Appl Genet 86:97–104
Kaga A, Tamooka N, Egawa Y, Hosaka K, Kamijima O (1996) Species relationships in subgenus Ceratotropis (genus Vigna) as revealed by RAPD analysis. Euphytica 88:17–24
Ehlers JD, Hall AE (1997) Cowpea (Vigna unguiculata L. Walp.). Field Crop Res 53:187–204
Tosti N, Negri V (2002) Efficiency of three PCR based markers in assessing genetic variation among cowpea (Vigna unguiculata spp. unguiculata) landraces. Genome 45:268–275
Bisht IS, Bhat KV, Lakhanpaul S, Latha M, Jayan PK, Biswas BK, Singh AK (2005) Diversity and genetic resources of wild Vigna species in India. Genet Resour Crop Evol 52:53–68
Ruangsuttapha S, Eimert K, Schroder MB, Silayoi B, Denduangboripant J, Kanchanapoom K (2007) Molecular phylogeny of banana cultivars from Thailand based on HAT-RAPD markers. Genet Resour Crop Evol. doi:10.1007/s10722-006-9169-2
Pathak R, Singh SK, Singh M, Henry A (2010) Molecular assessment of genetic diversity in cluster bean (Cyamopsis tetragonoloba) genotypes. J Genet 89:243–246
Williams JGK, Kubelik AR, Livak KJ, Rafalski JA, Tingey SV (1990) DNA polymorphisms amplified by arbitrary primers are useful as genetic markers. Nucl Acids Res 18:6231–6235
Esselman EJ, Crawford D, Brauner S, Stussy IF, Anderson GJ, Silva MO (2000) RAPD marker diversity within and divergence among species of Dendroseris (Asteraceae: Lactuaceae). Am J Bot 87:591–596
Li YC, Fahima T, Krugman T, Beiles A, Röder MS, Korol AB, Nevo E (2000) Parallel microgeographic patterns of genetic diversity and divergence revealed by allozyme, RAPD, and microsatellites in Triticum dicoccoides at Ammiad, Israel. Conserv Genet 1:191–207
Yu Z, Li QL, Huan L, Jie B, Man YY, Chen M, Ying FC, Xiao LQ, Fang C (2002) RAPD markers in diversity detection and variety identification of Tibetan hulless barley. Pol Mol Biol 20:369–377
Ghosh KK, Haque ME, Parvin S, Akhter F, Rahim MM (2009) Genetic diversity analysis in brassica varieties through RAPD markers. Bangladesh J Agric Res 34(3):493–503
Thomas G, Mohapatra T, Rao AR, Sharma RP (2006) Distinguishing Indian commercial wheat varieties using RAPD based DNA fingerprints. Indian J Biotechnol 5:200–206
Malviya N, Yadav D (2010) RAPD Analysis among Pigeon pea [Cajanus cajan (L.) Mill sp.] Cultivars for their Genetic Diversity. Genet Eng Biotechnol J 2010:1–9
Talebi R, Fayaz F, Mardi M, Pirsyedi SM, Naji AM (2008) Genetic relationships among chickpea (Cicer arietinum) elite lines based on RAPD and agronomic markers. Int J Agric Biol 10:301–305
Lakhanpaul S, Chadha S, Bhat KV, Chadha S (2000) Random amplified polymorphic DNA analysis in Indian mungbean [Vigna radiata (L.) Wilczek] cultivars. Genetica 109:227–234
Afzal MA, Haque MM, Shanmugasundaram S (2004) Random amplified polymorphic DNA (RAPD) analysis of selected mungbean [Vigna radiata (L) Wilczek] cultivars. Asian J Pol Sci 3(1):20–24
Saini A, Reddy SK, Jawali N (2004) Evaluation of long primers for AP-PCR analysis of mungbean genetic relationships and fingerprinting of some genotypes. Indian J Biotechnol 3(4):511–518
Chattopadhyay K, Ali MN, Sarkar HK, Mandal N, Bhattacharyya S (2005) Diversity analysis by RAPD and ISSR markers among the selected mungbean [Vigna radiata (L.) Wilczek] genotypes. Indian J Genet Pol Breed 65(3):173–175
Lavanya GR, Srivastava J, Ranade SA (2008) Molecular assessment of genetic diversity in mungbean germplasm. J Genet 87(1):65–74
Tantasawat P, Trongchuen J, Prajongjai T, Thongpae T, Petkhum C, Seehalak W, Machikowa T (2010) Variety identification and genetic relationships of mungbean and black gram in Thailand based on morphological characters and ISSR analysis. Afr J Biotechnol 9(27):4452–4464
Alvarez I, Wendel JF (2003) Ribosomal ITS sequences and plant phylogenetic inference. Mol Phylogenet Evol 29:435–455
Singh SK, Tiwari M, Kamal S, Yadav MC (2005) Morel phylogeny and diagnostics based on restriction fragment length polymorphism analysis of ITS region of 5.8S ribosomal DNA. J Biochem Biotechnol 14:179–183
Vogler DR, Bruns TD (1998) Phylogenetic relationships among the pine stem rust fungi (Cronartium and Peridermium spp.). Mycologia 90:244–257
Kakani RK, Singh SK, Pancholy A, Meena RS, Pathak R, Raturi A (2011) Assessment of genetic diversity in Trigonella foenum-graecum based on nuclear ribosomal DNA, internal transcribed spacer and RAPD analysis. Pol Mol Bio Rep 29:315–323
Hershkovitz MA, Zimmer EA, Hahn WJ (1999) Ribosomal DNA sequences and angiosperm systematics. In: Hollingsworth PM, Bateman RM, Gornall RJ (eds) Molecular systematics and plant evolution. Taylor and Francis, London, pp 268–326
Saini A, Reddy SK, Jawali N (2008) Intra individual and intra species heterogeneity in nuclear rDNA ITS region of Vigna species from subgenus Ceratotropis. Genet Res 90:299–316
Baldwin BG, Sanderson MJ, Porter JM, Wojciechowski MF, Campbell CS, Donoghue MJ (1995) The ITS region of nuclear ribosomal DNA: a valuable source of evidence on angiosperm phylogeny. Ann Mo Bot Gard 82:247–277
Jorgenson RD, Cluster PD (1988) Modes and tempos in the evolution of nuclear ribosomal DNA: New characters of evolutionary studies and new markers for genetic and population studies. Ann Mo Bot Gard 75:1238–1247
Mir BA, Koul S, Kumar A, Koul MK, Soodan AS, Raina SN (2010) Intraspecific variation in the internal transcribed spacer (ITS) regions of rDNA in Withania somnifera (Linn.) Dunal. Indian J Biotechnol 9:325–328
Carvalho A, Guedes-Pinto H, Lima BJ (2009) Genetic diversity among old Portuguese bread wheat cultivars and botanical varieties evaluated by ITS rDNA PCR-RFLP markers. J Genet 88(3):363–367
Andrew M, Kohn Linda M (2009) Single nucleotide polymorphism based diagnostic system for crop-associated Sclerotinia species. Appl Environ Microbiol 75:5600–5606
Xiao LQ, Möller M, Zhu H (2010) High nrDNA ITS polymorphism in the ancient extant seed plant cycas: incomplete concerted evolution and the origin of pseudogenes. Mol Phylogenet Evol 55:168–177
Birren B, Lai E (1993) Pulsed field gel electrophoresis: a practical guide. Academic Press, San Diego, p 253
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, Plainview
White TJ, Bruns SL, Taylor J (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Geltand DH, Sninsky TT, White TJ (eds) PCR protocols: a guide to methods and applications, Academic press, San Diego, p 315
Rohlf FJ (1997) NTSYS pc: Numerical taxonomy and multivariate analysis system version 2.02 h. Exeter Software, New York
Sneath PHA, Sokal RR (1973) Numerical taxonomy. W. H. Freeman, San Francisco
Peakall R, Smouse PE (2006) GENALEX 6: genetic analysis in excel. Population software for teaching and research. Mol Ecol Notes 6:288–295
Lewontin RC (1972) The apportionment of human diversity. Evol Biol 6:391–398
Nei M (1973) Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci USA 70:3321–3323
Page RDM (1996) Tree View: an application to display phylogenetic on personal computers. Comp Appl Biol Sci 12:357–358
Martins SR, Vences FJ, Saenz de Miera LE, Barrosa MR, Carnide V (2006) RAPD analysis of genetic diversity among and within Portuguese landraces of common white bean (Phaseolus vulgaris L.). Sci Hortic 108:133–142
Gaffor AS, Ahmed AZ, Zahid MA, Rabbani MA (2001) Genetic diversity in black gram [Vigna mungo (L.) Hepper]. Field Crop Res 69:183–190
Bhat KV, Lakhanpaul S, Chadha S (2004) Amplified fragment length polymorphism (AFLP) analysis of genetic diversity in Indian mungbean [Vigna radiata (L.) Wilczek] cultivars. Indian J Biotechnol 4:56–64
Souframanien J, Gopalakrishnan T (2004) A comparative analysis of genetic diversity in black gram genotypes using RAPD and ISSR markers. Theor Appl Genet 109:1687–1693
Andreev IO, Spiridonova KV, Solovyan VT, Kunakh VA (2005) Variability of ribosomal RNA genes in Rauwolfia species: parallelism between tissue culture-induced rearrangements and interspecies polymorphism. Cell Biol Int 29(1):21–27
Saini A, Reddy SK, Jawali N (2008) Intra-individual and intra-species heterogeneity in nuclear rDNA ITS region of Vigna species from subgenus Ceratotropis. Genet Res 90(4):299–316
Barkley NA, Wang ML, Gillaspie AG, Dean RE, Pederson GA, Jenkins TM (2008) Discovering and verifying DNA polymorphisms in a mungbean [V. radiata (L.) R Wilczek] collection by EcoTILLING and sequencing. BMC Res Notes 1:28
Acknowledgments
The authors are thankful to Dr. M. M. Roy, Director, Central Arid Zone Research Institute, Jodhpur for providing necessary laboratory and field facilities and CSIR for providing financial assistance to carry out this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Raturi, A., Singh, S.K., Sharma, V. et al. Molecular characterization of Vigna radiata (L.) Wilczek genotypes based on nuclear ribosomal DNA and RAPD polymorphism. Mol Biol Rep 39, 2455–2465 (2012). https://doi.org/10.1007/s11033-011-0996-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-011-0996-7