
Abstract
The stem color of young mung bean is a very useful tool in germplasm identification. Flowering time and plant height (PH) are known to be strongly correlated with crop adaption and yield. However, few studies have focused on elucidating the genetic mechanisms that regulate these five particular traits: young stem color (YSC), days to first flowering (DFF), days to maturity (DM), PH, and nodes on the main stem (NMS). In this study, a genetic linkage map for the F2 population was constructed using 129 InDel markers that were developed based on the sequence variations between parents. A total of 14 QTLs related to YSC, DFF, DM, PH, and NMS were detected. These QTLs were distributed on six chromosomes (1, 3, 4, 6, 7, and 10), which individually accounted for 1.32 to 90.07% of the total phenotypic variation. Using a short and high-density linkage map for the F3 population, six of the seven QTLs which clustered at two intervals on chromosomes 3 and 10 were detected again. Further analysis found that four QTLs between InDel markers R3-15 and R3-19 controlled DFF, DM, PH, and NMS, and each QTL accounted for a large percent of the total phenotypic variation. Analysis of two separated F2:3 lines also found that the phenotype was highly corresponded to its genotype which was between R3-15 and R3-19. Phenotype and genotype analysis for 30 mung bean accessions showed that the major effect QTL qDFF3 was a key regulator for DFF. Using a map-based cloning method, the major effect QTL qYSC4 for YSC was mapped in a 347 Kb interval on chromosome 4. Candidate gene analysis showed that sequence variations and expression level differences existed in the predicted candidate gene between the parents. These results provide a theoretical basis for cloning these QTLs and marker-assisted selection.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Mung bean [Vigna radiate (L.) Wilczek] is a fast-growing warm-season leguminous species, belonging to the subgenus Ceratotropis (genus Vigna), the tribe Phaseoleae, and the family Fabaceae. As a nutritional and healthy food with high-quality protein, folate, and iron, mung bean is mainly cultivated in Asia, Africa, and Australia; in certain developing countries, mung bean seed is regarded as a cheap source of carbohydrates (Kang et al. 2014; Kim et al. 2015). A benefit of mung bean is that its symbiosis with rhizobia can fix atmospheric nitrogen via biological nitrogen fixation, reducing the amount of chemical fertilizers needed while improving soil fertility and texture. Mung bean grown in rotation or intercropped with other cereals can increase the cereal grain and reduce pest incidence (Yaqub et al. 2010; Faria et al. 1989). For a long time, mung bean has been regarded as a minor grain, and thus, the basic research and genomics study for mung bean still lags far behind the other major crops such as rice, wheat, and corn.
InDels are an abundant form of genome variation, often more prevalent than SSRs on genomes and offering more genomic information than SNPs (Lv et al. 2016). Compared with other markers, InDel markers have special superiority in bi-allelic, co-dominant, high-density, and low-cost genotyping. Thus, this inexpensive and convenient marker has been widely used in evaluating genetic diversity and gene mapping as well as molecular-assisted selection (Lu et al. 2019; Yonemaru et al. 2015; Raman et al. 2006; Das et al. 2015). However, few studies have focused on the development of InDel markers in mung bean, not to mention using InDel markers in linkage map construction and QTL analysis, mainly because the high cost and low flux acquisition of insertion or deletion sequence information in the past. In recent years, high-throughput sequence information has become more available along with the rapid development of next-generation sequencing (NGS) technology. This progress has greatly facilitated and accelerated the discovery process for molecular markers. In mung bean genomics studies, QTL analysis has mainly been focused on insect and disease resistance, including bruchid (Mei et al. 2009; Wang et al. 2016), powdery mildew resistance (Chaitieng et al. 2002; Poolsawat et al. 2017), yellow mosaic virus resistance (Chen et al. 2013a; Sai et al. 2017), and cercospora leaf spot resistance (Chankaew et al. 2011), mainly because these issues are a great threat to mung bean yield. Among most of these QTL analysis studies, genetic linkage map construction usually used RFLP, RAPD, and SSR markers due to the lack of InDel markers. Flowering time and plant height (PH) are strongly correlated with crop yield. In addition, these two agronomic traits also determine the adaptability of mung bean cultivars at different light and temperature conditions. Thus, it is necessary to explore the molecular genetic mechanism controlling these traits in mung bean. Several studies focused on QTL detection for agronomic traits related to days to flowering time and maturity were completed. Isemura et al. constructed a mung bean linkage map by using 430 SSR markers, and detected 4 QTLs on linkage group (LG) 2, LG4, LG6, and LG11 controlled the number of days to first flowering (FLD) (Isemura et al. 2012). The QTL on LG2 had the largest effect (phenotypic variation explained = 32.9%). They also found 6 QTLs associated with the value for days to first pod maturity from first flowering on LG2, LG4, LG6, LG7, LG9, and LG11. As expected, the QTLs on LG2, LG4, LG6, and LG11 were located close to the 4 QTLs for FLD. Kajonphol et al. found 4, 3, and 3 QTLs related to FLD, days to first pod maturity (PDDM) and days to harvest (PDDH) by using 152 SSR markers (Kajonphol et al. 2012). These 10 QTLs were distributed on LG2, LG4, and LG11, and 6 QTLs (Fld2, Pddm2, and Pddh2 on LG2, Fld4.1, Pddm4.l, and Pddh4.1 on LG4) controlled these three traits were co-located at two chromosome intervals. Three QTLs for days to 50% flowering and 2 for days to 50% maturity were detected and mapped to LG4A, LG4B, and LG7 by using 101 SSR markers (Sompong et al. 2012). Somta et al. discovered 5 QTLs associated with days to flowering in two dry seasons and one rainy season by using 56 SSR markers (Somta et al. 2015). QDFL2.1 and qDFL2.2 could be detected in rainy season and one dry season, but the other three QTLs could only be identified in rainy season. They considered the difference in photoperiod regime between rainy season and dry seasons affected the detection for QTLs. In some of these studies, some QTLs for correlated traits were overlapped or co-located (Isemura et al. 2012; Kajonphol et al. 2012).
The stem color of young mung bean is a very useful phenotype marker in germplasm identification. This trait is mainly determined by the kinds and content of anthocyanins. Anthocyanins are important water-soluble pigments and are widely distributed in berries and cellular fluids of leaves, stems, flowers, and fruit, attracting pollinators such as insects. Previous studies found that MYB-type transcription factor plays a regulatory role in anthocyanin biosynthesis (Jaakola 2013; Schwinn et al. 2006). In fact, anthocyanins not only play a decisive role in the appearance color of most plants but also protect them from various biotic and anti-biotic stresses depending on the anti-oxidation and anti-mutation ability (Wang et al. 2019; Petrussa et al. 2013). In this study, QTL analysis for young stem color (YSC), days to first flowering (DFF), days to maturity (DM), PH, and nodes on main stem (NMS) was performed using populations derived from the cross between Sulu16-10 and Weilu11. InDel marker development was based on sequence insertion or deletion between the parents. One hundred twenty-nine polymorphic markers that were uniformly distributed on 11 chromosomes were selected for genetic linkage map construction. The availability of markers linked with the major QTL controlled DFF for assist selection was verified. Fine mapping and candidate gene analysis for the major QTL associated with YSC were also performed. These newly developed InDel markers in this study can enrich the current molecular markers resources and can be used in mung bean genomic research. Dissection of QTLs will advance our knowledge on underlying genetic mechanisms of these traits in mung bean. In addition, markers linked with major- and stable-expressed QTLs could be used in marker-assisted selection and facilitated the breeding of elite breeding varieties.
Materials and methods
Plant materials and agronomic trait measurement
The F2 and F3 population consisted of 136 and 134 individuals, respectively. They were developed from a cross between Sulu16-10 and Weilu11. Sulu16-10 is a new mung bean variety with green YSC, high PH and late DFF; it was cultivated by the Jiangsu Academy of Agricultural Sciences, Jiangsu, China. Weilu11 has a purple YSC, dwarf PH, and early DFF; it was bred by the Weifang Academy of Agricultural Sciences, Shandong, China. The F2 population and the two parental lines were grown in 2019 in Hefei (31.88° North/117.25° East), China. The F3 population, F2:3 lines, and 30 mung bean accessions were planted in 2020 in Hefei, China. Agronomic traits investigation criteria were as follows: DFF, the number of days from sowing to first flowering; DM, the number of days from sowing to maturity; PH, the distance from the cotyledon node to the top of the plant; NMS, the number of nodes from the cotyledon node to the last compound leaf node at the top of the main stem of the plant. The phenotypic data were analyzed for distribution and significance of trait variation using the Excel 2007 and SPSS V22.0 software.
Anthocyanidin profiling and data analysis
Young stem samples from each of the parents were collected 10 days after germination. The samples were frozen in liquid nitrogen for further use. Each replicate was from 15–20 plants, and three biological replications were conducted. The sample preparation, extraction, anthocyanidin identification, and quantification were performed following Wang (Wang et al. 2017a) and Chen (Chen et al. 2013b). Sample data were collected using an LC–ESI–MS/MS system (HPLC, Shim-pack UFLC SHIMADZU CBM30A system; MS, Applied Biosystems 6500 Q TRAP). Data analysis also followed the methods described by Wang (Wang et al. 2017a) and Zhuang (Zhuang et al. 2019). Metabolites with significant differences in content were set with a threshold of fold change of ≥ 2 or ≤ 0.5.
Illumina sequencing and data filtering
Homozygous parental lines were obtained by self-pollinating for several generations. Genomic DNA was extracted from fresh leaves of parents using a DNeasy Plant Mini Kit (Qiagen, Hilden, Germany). Sequencing libraries with 400-bp insertion were constructed and sequenced on Illumina HiSeq2000 sequencing system with paired-end sequencing strategy. After the production of raw sequences, all adapter sequences, short reads (length < 50 bp), and low-quality reads were deleted. Duplicates (PCR duplicates and optical duplicates) produced in the sequencing process and DNA library amplification was removed by the Picard software (http://www.psc.edu/index.php/user-resources/software/picard).
Primer design and PCR reaction
To find sequence differences between the two parents and reference genome sequence, all of the high-quality sequencing data were aligned to the mung bean reference genome (https://www.ncbi.nlm.nih.gov/genome/664) using the Bwa package (Li and Durbin 2009). If the insertion/deletion sequence between the parents and the reference genome was not inconsistent at the same position, this sequence difference could regard as an InDel between parents. InDels between 5 and 50 bp were selected for primer design using Primer3 V2.3.6 (Untergasser et al. 2012). The polymorphism of markers was validated using Sulu16-10 and Weilu11 by PCR reaction. The 10 μL PCR mixture contained 1.0 μL template DNA (50 ng·μL−1), 5.0 μL10 × PCR MIX (TSINGKE, Beijing, China), 10 μmol·L−1of each forward and reverse primer 0.5 μL, and ddH2O 3.0 μL. Thermal Cycler (Dongshenglong, Beijing, China) was used to perform PCR amplification with the following procedures: first denaturation step at 94 °C for 4 min, followed by 35 cycles of 94 °C for 30 s, annealing (temperature set was based on primers, usually 57 °C) for 30 s, 72 °C for 30 s, final extension step at 72 °C for 10 min and storage at 8 °C. PCR products were separated with 6% non-denaturing polyacrylamide gel and visualized by silver staining.
Linkage map construction and QTL analysis
Polymorphic InDel markers were chosen to analyze the genotype of individuals in the F2 population. The segregation of each marker was analyzed by a chi-square test for goodness of fit for the expected Mendelian segregation ratio (1:2:1). Markers showed segregation distortion were excluded from genetic linkage map construction. All markers used for genetic map construction are listed in Table S1. For the F3 population, markers on chromosomes 3 and 10, additional with other seven polymorphic markers, were used to construct a short but high-density genetic map. QTL detection was performed with the QTL IciMapping software V4.1 (Meng et al. 2015; Li et al. 2008) using inclusive composite interval mapping of additive QTL ICIM-ADD modules. The scanning step was set at 1.0 cM. The LOD thresholds for each dataset were determined by permutation test (1000 permutations, P < 0.05). QTLs were declared when their LOD scores were larger than 2.0. QTLs were named according to McCouch (McCouch et al. 1997).
Fine mapping for qYSC4 and expression level analysis
New polymorphic markers used for fine mapping qYSC4 were designed by Primer Premier 5.0 software based on sequence differences between parents. PCR-based molecular markers used to identify the genotypes of 256 F2 individuals with green stem were listed in Table S1. Genotyping for SNP markers PCR productions were performed by the sequencing method. Young stem samples collected at 10 days after germination were used for RNA extraction with the OmniPlant RNA extraction Kit (DNase I). At this growth stage, the hypocotyl of Weilu11 was purple, and the epicotyl was green. Both the hypocotyl and epicotyl of Sulu16-10 were green. RNA was treated with DNase I and the first-strand cDNA was synthesized using cDNA Synthesis SuperMix (TransGen). QRT-PCR was conducted on LightCycler 96. Each 20 μL reaction system contained 2 μL of template cDNA, 10 μL 2 × SYBR Premix EX-Taq Mix, 10 μmol·L−1 of each forward and reverse primer 0.5 μL, and 7 μL RNase free ddH2O. The relative expression level of each transcript was obtained by comparing with the expression of the Vrtubulin gene. QRT-PCR primers for candidate genes and Vrtubulin are listed in Table S2. The 2−ΔΔCT method (Livak and Schmittgen 2001) was used to analyze gene relative expression level.
Results
Phenotypic variation and correlations between traits
The phenotypic differences between the parents and the agronomic trait variation in the F2 and F3 populations are summarized in Table 1. For YSC, Sulu16-10 exhibited green, whereas Weilu11 showed purple. Metabolomic analysis detected 11 types of anthocyanins in the mung bean young stem. Compared with Sulu16-10, the comparative content of four types of anthocyanins (cyanidin 3-O-glucoside, delphinidin 3-O-glucoside, delphinidin 3-O-rutinoside, and cyanidin 3-O-galactoside) in Weilu11 was increased (Fig. S1). The stems of all of the F1 plants showed purple, meaning purple was dominant to green. The ratio of individuals with purple to green (99 to 37) was fit to 3:1 by a chi-square test in the F2 population (χ2 = 1.3 < χ20.05 = 3.84), indicating the YSC was controlled by a single gene or a major effect QTL with a large contribution rate. The other four traits, DFF, DM, PH, and NMS, showed significant differences between the parents. Sulu16-10 showed higher PH and later DFF when compared with Weilu11. DFF, DM, PH, and NMS were exhibited continuous distribution in the F2 population (Table 1). DFF, DM, and PH were normally distributed, whereas the distribution of NMS was skewed and toward increased nodes. Transgressive segregation was observed for all traits. Therefore, we concluded that these traits were quantitatively inherited and controlled by multiple genes. These four traits were also observed to be continuous in the F3 population. The mean values of DFF and DM in the F3 population were larger than those in the F2 population, whereas the mean values of PH and NMS were less than those in the F2 population. Correlation analysis (Table 2) showed that all these four traits were significantly positive correlated with each other at two years. The YSC only correlated with PH at significance level in 2020.
Sequencing and data filtering of Illumina paired-end reads
Whole-genome sequencing for Sulu16-10 and Weilu11 was conducted using the Illumina next-generation sequencing platform. In total, 104.0 and 106.9 million paired-end raw reads were obtained, containing 15.7 and 16.1 Gb of sequencing data with guanine and cytosine (GC) content of 34.5% and 35.6% for Sulu16-10 and Weilu11, respectively. After removal of adapter sequences and low-quality reads, 88.9 and 92.9 million clean reads remained, encompassing 12.7 and 13.3 Gb for Sulu16-10 and Weilu11, respectively.
InDel marker development
All of the clean reads were mapped to the mung bean reference genome, and the mapping ratio (mapped reads/total reads) for parents was both greater than 99.0%. Finally, a total of 17, 280 InDels that ranged from 5 to 150 bp were identified between the parents based on the sequence differences. Most of these InDels were between 5 and 50 bp, and these InDels were chosen to design primers. Based on the flanking sequences of these InDels, PCR primer pairs were successfully designed for 14,509 loci. Two hundred nine markers that were uniformly distributed on 11 chromosomes were selected to distinguish the genotype of the parents. Of these, 205 (98.1%) markers were successfully amplified clear PCR products of the expected sizes. Of these successfully amplifying markers, 180 produced polymorphic bands between the parents, accounting for 86.1% of the total tested markers.
Genetic map construction
Considering the polymorphic and position, 145 InDel markers that were uniformly distributed on the 11 chromosomes were chosen for linkage map construction. According to chi-square test and linkage analysis results, 12 markers showed serious segregation distortion and 4 markers were not mapped on chromosomes. Therefore, these 16 markers were excluded from the linkage map. Finally, 129 InDel markers could be anchored on the 11 chromosomes covering a total of 707.43 cM (Table 3, Fig. S2). The number of markers per chromosome was between 6 (chromosome 10) to 20 (chromosome 7) with a mean of 11.73. The length of the chromosomes ranged from 23.66 cM (chromosome 10) to 158.08 cM (chromosome 7) with an average of 64.31 cM. The average distance between the adjacent markers varied from 3.56 cM (chromosome 2) to 10.15 cM (chromosome 8) at an average marker density of 6.00 cM.
QTL analysis for agronomic traits
In total, 14 QTLs distributed on chromosomes 1, 3, 4, 6, 7, and 10 were found to be associated with YSC, DFF, DM, PH, and NMS (Table 4 and Fig. S2). LOD scores of these QTLs ranged from 2.06 to 97.45, and the phenotypic variance explained (PVE) were between 1.32 and 90.07% for each QTL. Three QTLs distributed on chromosomes 1, 4, and 6 were detected for YSC. They collectively accounted for 94.18% of the phenotypic variation. QYSC4, the major effect QTL for YSC, could explain 90.07% of the total phenotypic variation, and the positive allele came from Weilu11. Four QTLs located on chromosomes 3, 6, and 10 were related to DFF and DM. Seven QTLs located on chromosomes 1, 3, 7, and 10 controlled PH and NMS. Among these QTLs, 7 were clustered at two intervals on chromosome 3 (qDFF3, qDM3, qPH3.1, and qNMS3) and chromosome 10 (qDFF10, qPH10, and qNMS10).
Confirmation for the two QTL hot spots
To confirm the two QTL hot spots detected in 2019. Markers on chromosomes 3 and 10 were used to construct a short linkage map. Another seven polymorphic markers on chromosome 3 were developed to narrow the distance between markers. Among these newly developed markers, two (R3-9 and R3-12) were between ID3-5 and ID3-6, and the other five (R3-5, R3-13, R3-15, R3-19, and R3-20) were between ID3-6 and ID3-7. QTL analysis found 8 QTLs for DFF, DM, PH, and NMS on these two chromosomes (Table 5). Six of these QTLs were detected in 2019. Based on the QTL analysis results, four QTLs between R3-15 and R3-19 controlled all of these four traits, and the PVE was ranged from 62.04 to 84.72%. Chromosome region between ID10-5 and ID10-6 hold QTLs for DFF, DM, and NMS (Table 5). Further analysis found 49 individuals with Sulu16-10 genotype within R3-15 and R3-19 region showed late DFF and high PH (DFF = 59.0 ± 4.1 days, PH = 89.3 ± 13.2 cm). The phenotypes of 30 heterozygotes were similar to these plants with homozygous Sulu16-10 genotype (DFF = 57.9 ± 2.3 days, PH = 84.2 ± 7.9 cm). Meanwhile, 49 plants with homozygous Weilu11 genotype showed early DFF and dwarf PH (DFF = 42.0 ± 2.8 days, PH = 54.1 ± 8.9 cm). Ten plants with three different genotypes from the F2 population were selected to confirm the existence of these major QTLs between R3-15 and R3-19. All the descendants of the two F2 plants with homozygous Sulu16-10 genotype showed late DFF and high PH. The F2:3 lines generated from the two F2 plants with homozygous Weilu11 genotype showed early DFF and dwarf PH. The phenotypes of descendants generated from these six heterozygotes were segregated. Two segregated F2:3 lines were selected to analyze the consistency between genotype and phenotype. The genotypes of plants in these two lines are listed in Table S3. All the plants with Sulu16-10 or heterozygote genotype showed later DFF, later DM, higher PH, and more NMS when compared with plants hold Weilu11 genotype (Table 6), and the differences reached a significant level. Consideration of combined phenotype and genotype of these two segregated F2:3 lines, these QTLs were limited between R3-5 and ID3-7. These results indicated that QTLs with large PVE conferring DFF, DM, PH, and NMS existed in this chromosome region.
Molecular marker assistant selection
Since marker R3-15 and R3-19 were flanked with qDFF3, these two markers could be used in marker assistant selection. To verify the accuracy rate of marker assistant selection, 30 mung bean varieties with different DFF were divided into two groups that is early DFF group (EF) and late DFF group (LF). Generally speaking, DFF was consistent with the genotype identified by marker R3-15 and R3-19 (Table 7, Fig. S3). Correlation analysis indicated the correlation coefficients between DFF with R3-15 and R3-19 were 0.78** and 0.86**, respectively. Among these tested mung bean varieties, 15 were showed Weilu11 genotype at qDFF3 region, and 14 of them showed early DFF. Eleven varieties were showed Sulu16-10 genotype, and all of them showed late DFF. The accuracy rate was 96.2%. All of these results demonstrated that InDel markers R3-15 and R3-19 could be used as a very useful tool in mung bean DFF prediction and identification.
Fine mapping for the YSC gene qYSC4
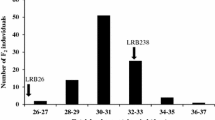
Among these three QTLs that controlled YSC, qYSC4 can be regarded as a major gene because it could explain 90.07% of the phenotypic variation. For fine mapping qYSC4, four InDel and two SNP markers were used to screen the genotypes of 256 green stem individuals in the F2 population. QYSC4 was finally mapped within a 347 Kb region between InDel marker ID4-14.9 and SNP marker 771 K (Fig. 1a). Based on the mung bean genome annotation information, there are 38 open reading frames (ORFs) with different predicted biological functions in this candidate region. Among these ORFs, LOC106758035 and LOC106758748 encoded MYB transcription factor were found in this target interval. Sequencing results showed there were no sequence variations in the LOC106758035 gene between parents. The gene LOC106758748 in Weilu11 had two synonymous SNPs in the second exon, four SNPs, and an AT box (35 AT repeats) deletion in introns (Fig. 1b). QRT-PCR results showed there was no significantly different expression level for LOC106758035 in hypocotyl between parents. The expression level of LOC106758748 was strongly up-regulated in the hypocotyl of Weilu11 when compared with Sulu16-10 (Fig. 1c). Further analysis found the expression level of LOC106758748 in hypocotyl was higher than that in epicotyl of Weilu11 (Fig. 1d). This result was consistent with their phenotype since the hypocotyl of Weilu11 was purple, while the epicotyl was green at this growth stage. Besides, the expression levels of LOC106758748 in the hypocotyl of three F2:3 lines with purple YSC were higher than those in three F2:3 lines with green YSC (Fig. 1d). Based on these results, LOC106758748 was selected candidate gene for qYSC4.

Fine mapping and candidate gene analysis for qYSC4. a Fine mapping for qYSC4. There are 38 ORFs between marker ID4-14.9 and 771 K. Green arrow and red arrow represent LOC106758035 and LOC106758748, respectively. b Sequence differences of LOC106758748 between Sulu16-10 and Weilu11. Black boxes indicate exons, and lines between black boxes indicate introns. c Relative expression levels of LOC106758035 and LOC106758748 in hypotocyl between the parents. Each bar indicates the mean ± SD of three independent replicates. Student’s t test was used for statistical analysis. **Represents significant difference at 0.01 level. d Relative expression levels of LOC106758748 in the parents and six F2:3 lines. 19, 35, and 36 were three F2:3 lines with purple stem; 7, 9, and 15 were three F2:3 lines with green stem.W–H, S–H, 19-H, 35-H, 36-H, 7-H, 9-H, and 15-H represent the hypocotyl of Weilu11, Sulu16-10, 19, 35, 36, 7, 9, and 15, respectively. W-E and S-E mean the epicotyl of Weilu11 and Sulu16-10, respectively
Discussion
Molecular marker development in mung bean had been performed in previous studies, and most of these studies were focused on SSR markers (Wang et al. 2015; Chen et al. 2015; Liu et al. 2016). In this study, high-throughput development of InDel markers could be achieved by directly compare the sequence differences between parents, and the rate of polymorphic InDel markers reached 86.1%, higher than the SSR markers (Wang et al. 2015; Chen et al. 2015; Liu et al. 2016). Several studies focused on genetic linkage map construction and QTL analysis for mung bean agronomic traits has been conducted by using SSR markers (Isemura et al. 2012; Kajonphol et al. 2012; Sompong et al. 2012; Somta et al. 2015). However, the positions of these QTLs were signed with genetic distance, and the markers linked with QTLs were not anchored on mung bean chromosomes. Therefore, it is difficult to compare the positions and contribute rate among these QTLs. In this study, all the markers used in linkage map construction were anchored on mung bean chromosomes. Thus, it will facilitate fine mapping or cloning for these QTLs, and also using in marker-assistant selection in the future. A genetic linkage map consisted of 129 developed InDel markers was constructed. The map totally covered 707.43 cM (Table 3), slightly shorter than map described by Wang et al. (Wang et al. 2016). For the purpose of genetic analysis and QTL mapping, varieties with significant phenotypic differences were usually chosen for segregation population construction. In this study, all the agronomic traits showed significant differences between the two the parents, Sulu16-10 and Weilu11.
A total of 11 QTLs were found to be responsible for DFF, DM, PH, and NMS, and 7 of them were clustered at the two chromosome regions (Table 4). It was note worthy that QTLs for different traits were co-located at ID3-6 and ID3-7 and the positive alleles all from Sulu16-10. These results supported the significant correlation between these four traits. QTLs controlled correlated traits were clustered together also found in other studies (Mei et al. 2009; Isemura et al. 2012; Kajonphol et al. 2012; Sompong et al. 2012). The reasons for this phenomenon were that either a pleiotropic existed that controlled these traits or QTLs related to these traits were closely linked. In rice, Ghd7 and Ghd8 were pleiotropic genes, which controlled flowering time and PH at the same time (Gao et al. 2014; Yan et al. 2011). Thus, these four QTLs between ID3-6 and ID3-7 were considered the same loci. To distinguish chromosome regions whether contained one QTL with pleiotropy effect or closely linked QTLs, near isogenic lines need to be constructed, and then, large segregation populations need to be generated to break the link relationship. QTL detection is usually conducted at different conditions, and these stable expressed QTLs have great significance in elite cultivars breeding. However, most loci in plants of F2 population were not homozygous, and these heterozygous loci will be segregated in the progeny. Thus, the linkage map which constructed for the F2 population could not be used in the progeny populations. Using the F3 population and markers on chromosomes 3 and 10 additional with other seven polymorphic InDel markers, another short and high-density linkage map was constructed. All these four QTLs located between ID3-6 and ID3-7 can be detected again, and the QTL region was narrowed at a 170 Kb region between R3-15 and R3-19 (Table 5). In addition, phenotype and genotype analysis for two segregate F2:3 lines also verified the regulator role of qDFF3 (Table 6). BLAST analysis for sequences of markers that closely linked with QTLs controlled flowering time and growth period (Isemura et al. 2012; Kajonphol et al. 2012; Sompong et al. 2012; Somta et al. 2015; Hwang et al. 2017); qDFF3 was corresponding to Dff 3–1, and near to Fld5.4.1. Comparative analyzing the candidate genomic region with soybean, a putative phytochrome A gene related to flowering time was found, which was an orthologue of the soybean E3/E4 (Liu et al. 2008; Watanabe et al. 2009). Besides, qDM3 was corresponding to Pddm5.4.1 and Df3-1 (Isemura et al. 2012; Hwang et al. 2017); qDFF10 was corresponding to qDFL5.1 (Somta et al. 2015); qDM6 was near to the region of Pddm5.7.1 (Isemura et al. 2012). Some new loci such as qYSC1, qYSC6, qPH1, qPH7, qNMS10 were detected in this study. Two minor QTLs (qDFF10 and qNMS10) located at ID10-4 and ID10-6 could also be detected in 2020 (Table 5). Since DFF and PH were two positively correlated traits, they should share the similar change tendency in different populations or conditions. Strangely, even though the average DFF and DM in F3 population was larger than the average value in F2 population, the average PH and NMS in F3 population was obviously decreased when compared with that in F2 population (Table 1). The main reason was that the mung bean phenotype was strongly influenced by the weather since mung bean growth period in 2019 was hot and drought, but cool and rainy in 2020. The average temperature and total rainfall among mung bean growth stage were 27.2 °C and 190.2 mm in 2019. Meanwhile, the data were 26.7 °C and 748.5 mm in 2020. Especially in July, the difference in the average temperature and total rainfall between 2019 (28.1 °C and 21.3 mm) and 2020 (25.1 °C and 317.9 mm) was more obvious. Thus, these repeatedly detected QTLs were considered stable expressed at different conditions. Genotype analysis found 14 of 15 varieties showed homozygous Weilu11 genotype in the early DFF group, and 11 of 15 varieties showed homozygous Sulu16-10 genotype in the late DFF group (Table 7, Fig. S3). This result showed qDFF3 was the key regulator loci for DFF in mung bean germplasm. The accuracy rate of linked markers in DFF identification reached 96.2%. Thus, InDel markers R3-15 and R3-19 were very useful molecular marker in mung bean DFF prediction and identification. Future work will focused on fine mapping population construction for these major effect QTLs and depth analysis of candidate genes.
YSC is a very useful tool for varietal purity identification and assistant breeding. Previous studies showed MYB-type transcription factor was the key regulator for synthesis of anthocyanins (Zhou et al. 2016; Cho et al. 2016). In soybean, soluble pigment in stems was considered had relationship with white mold resistance (Li et al. 2010; Wegulo et al. 1998). Thus, it is necessary to research the relationship between disease resistance and YSC in mung bean. Previous studies showed YSC was controlled by a single gene (Wang et al. 2013) or multi-genes (Wang et al. 2017b). Chi-square test found the number of plants with purple stem to green was fit to 3:1 in F2 population, indicating there was a single gene or major QTL conferring this trait. QTL detection results showed that YSC was controlled by one major QTL qYSC4 plus two minor QTLs: qYSC1 and qYSC6 (Table 4). Actually, we have identified an YSC mutant by using EMS mutagenesis strategy, and the mutation site did not allele to qYSC4. These results demonstrated that YSC was controlled by multi-genes. According to our phenotype investigation results, qYSC1 and qYSC6 could not affect the phenotype judge for YSC because alleles from Sulu16-10 could increase the effect while this parent showed green stem. Thus, the phenotype of YSC in F2 population and parents were completely determined by qYSC4, which could explain 90.07% of the total phenotype variation. Map-based cloning limited the qYSC4 within a 347 Kb region which contained two predicted MYB-type transcription factor, LOC106758035 and LOC106758748 (Fig. 1a). Homology analysis showed LOC106758748 was orthologous to soybean R gene, which controlled seed coat color (Yan et al. 2015). Sequence variations were existed in LOC106758748 and may affect the expression level in parents and hybrid progeny (Fig. 1b–d). Expression level analysis also found LOC106758748 was highly expressed in the hypocotyl of purple stem at young growth stage. In summary, sequence variation and expression analysis revealed LOC106758748 was the preferred candidate gene for qYSC4.
Conclusions
High-throughput development of InDel marker could be performed based on the sequence variation between parents’ genome sequence obtained from NGS. The ratio of polymorphic InDel marker could reach 86.1%. A genetic linkage map which covered 707.43 cM for a F2 population developed by the cross between Sulu16-10 and Weilu11 was constructed. QTL analysis detected 14 QTLs for YSC, DFF, DM, PH, and NMS. The phenotypic variance explained by each QTL was between 1.32 and 90.07%. Seven of these QTLs were clustered at two intervals on chromosomes 3 and 10. Six of these co-located QTLs were detected again in the F3 population by using a short and high-density linkage map. Phenotype and genotype analysis for 30 mung bean accessions found that the qDFF3 was the key regulation factor for DFF and the linked markers could be used in marker-assisted selection. Map-based cloning limited the qYSC4, the major gene for YSC within a 347 Kb region. Sequence variation and expression level analyses confirm that the MYB transcription factor LOC106758748 was the preferred candidate gene for qYSC4. These results provide a foundation for cloning these QTLs and clarifying the molecular regulation mechanisms for these traits.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Code availability
Not applicable.
References
Chaitieng B, Kaga A, Han OK, Wang XW, Wongkaew S, Laosuwan P, Tomooka N, Vaughan DA (2002) Mapping a new source of resistance to powdery mildew in mungbean. Plant Breed 121(6):521–525
Chankaew S, Somta P, Sorajjapinun W (2011) Quantitative trait loci mapping of Cercospora leaf spot resistance in mungbean, Vigna radiata (L) Wilczek. Mol Breed 28(2):255–264
Chen HM, Ku HM, Schafleitner R, Bains TS, Kuo CG, Liu CA, Nair RM (2013a) The major quantitative trait locus for mungbean yellow mosaic Indian virus resistance is tightly linked in repulsion phase to the major bruchid resistance locus in a cross between mungbean [Vigna radiata (L.) Wilczek] and its wild relative Vigna radiata ssp. sublobata. Euphytica 192(2):205–216
Chen W, Gong L, Guo Z, Wang W, Zhang H, Liu X, Yu S, Xiong L, Luo J (2013b) A novel integrated method for large-scale detection, identification, and quantification of widely targeted metabolites: application in the study of rice metabolomics. Mol Plant 6(6):1769–1780
Chen H, Wang L, Wang S, Liu C, Blair MW, Cheng X (2015) Transcriptome sequencing of mung bean (Vigna radiate L.) genes and the identification of EST-SSR markers. PLoS One 10(4):e0120273
Cho K, Cho KS, Sohn HB, Ha IJ, Hong SY, Lee H, Kim YM, Nam MH (2016) Network analysis of the metabolome and transcriptome reveals novel regulation of potato pigmentation. J Exp Bot 67(5):1519–1533
Das S, Upadhyaya HD, Srivastava R, Bajaj D, Gowda CL, Sharma S, Singh S, Tyagi AK, Parida SK (2015) Genome-wide insertion-deletion (InDel) marker discovery and genotyping for genomics-assisted breeding applications in chickpea. DNA Res 22(5):377–386
De Faria SM, Lewis GP, Sprent JI, Sutherland JM (1989) Occurrence of nodulation in the Leguminosae. New Phytol 111:607–619
Gao H, Jin M, Zheng XM, Chen J, Yuan D, Xin Y, Wang M, Huang D, Zhang Z, Zhou K, Sheng P, Ma J, Ma W, Deng H, Jiang L, Liu S, Wang H, Wu C, Yuan L, Wan J (2014) Days to heading 7, a major quantitative locus determining photoperiod sensitivity and regional adaptation in rice. Pro Natl Acad Sci U S A 111(46):16337–16342
Hwang WJ, Ha J, Lee T, Jeong H, Kim MY, Kim SK, LeeYH JJW, Lee SH (2017) A candidate flowering gene in mungbean is homologous to a soybean Phytochrome A gene. Euphytica 213:79
Isemura T, Kaga A, Tabata S, Somta P, Srinives P, Shimizu T, Jo U, Vaughan DA, Tomooka N (2012) Construction of a genetic linkage map and genetic analysis of domestication related traits in mungbean (Vigna radiata). PLoS One 7(8):e41304
Jaakola L (2013) New insights into the regulation of anthocyanin biosynthesis in fruits. Trends Plant Sci 18(9):477–483
Kajonphol T, Sangsiri C, Somta P, Toojinda T, Srinives P (2012) SSR map construction and quantitative trait loci (QTL) identification of major agronomic traits in mungbean (Vigna radiata (L.) Wilczek). SABRAO J Breed Genet 44(1):71–86
Kang YJ, Kim SK, Kim MY, Lestari P, Kim KH, Ha BK, Jun TH, Hwang WJ, Lee T, Lee J, Shim S, Yoon MY, Jang YE, Han KS, Taeprayoon P, Yoon N, Somta P, Tanya P, Kim KS, Gwag JG, Moon JK, Lee YH, Park BS, Bombarely A, Doyle JJ, Jackson SA, Schafleitner R, Srinives P, Varshney RK, Lee SH (2014) Genome sequence of mungbean and insights into evolution within Vigna species. Nat Commun 5:5443
Kim SK, Nair RM, Lee J, Lee SH (2015) Genomic resources in mungbean for future breeding programs. Front Plant Sci 6:6626
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25(14):1754–1760
Li H, Ribaut JM, Li Z, Wang J (2008) Inclusive composite interval mapping (ICIM) for digenic epistasis of quantitative traits in biparental populations. Theor Appl Genet 116(2):243–260
Li D, Sun M, Han Y, Teng W, Li W (2010) Identification of QTL underlying soluble pigment content in soybean stems related to resistance to soybean white mold (Sclerotinia sclerotiorum). Euphytica 172(1):49–57
Liu B, Kanazawa A, Matsumura H, Takahashi R, Harada K, Abe J (2008) Genetic redundancy in soybean photoresponses associated with duplication of the phytochrome A Gene. Genetics 180(2):995–1007
Liu C, Fan B, Cao Z, Su Q, Wang Y, Zhang Z, Wu J, Tian J (2016) A deep sequencing analysis of transcriptomes and the development of EST-SSR markers in mungbean (Vigna radiata). J Genet 95(3):527–535
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25(4):402–408
Lu N, Zhang M, Xiao Y, Han D, Liu Y, Zhang Y, Yi F, Zhu T, Ma W, Fan E, Qu G, Wang J (2019) Construction of a high-density genetic map and QTL mapping of leaf traits and plant growth in an interspecific F1 population of Catalpa bungei × Catalpa duclouxii Dode. BMC Plant Biol 19(1):596
Lv Y, Liu Y, Zhao H (2016) mInDel: a high-throughput and efficient pipeline for genome-wide InDel marker development. BMC Genomics 17(1):290
McCouch SR, Cho YG, Yano M, Paul E, Blinstrub M, Morishima H, Kinoshita T (1997) Report on QTL nomenclature. Rice Genet Newsl 14(11):11–13
Mei L, Cheng XZ, Wang SH, Wang LX, Liu CY, Sun L, Xu N, Humphry ME, Lambrides CJ, Li HB, Liu CJ (2009) Relationship between bruchid resistance and seed mass in mungbean based on QTL analysis. Genome 52(7):589–596
Meng L, Li HH, Zhang LY, Wang JK (2015) QTL IciMapping: integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop J 3:269–283
Petrussa E, Braidot E, Zancani M, Peresson C, Bertolini A, Patui S, Vianello A (2013) Plant flavonoids-biosynthesis, transport and involvement in stress responses. Int J Mol Sci 14(7):14950–14973
Poolsawat O, Kativat C, Arsakit K, Tantasawat PA (2017) Identification of quantitative trait loci associated with powdery mildew resistance in mungbean using ISSR and ISSR-RGA markers. Mol Breed 37(12):150
Raman H, Raman R, Wood R, Martin P (2006) Repetitive indel markers within the ALMT1 gene conditioning aluminium tolerance in wheat (Triticum aestivum L.). Mol Breeding 18(2):171–183
Sai BC, Nagarajan P, Raveendran M, Rabindran R, Bapu JRK, Senthil N (2017) Understanding the inheritance of mungbean yellow mosaic virus (MYMV) resistance in mungbean (Vigna radiata L. Wilczek). Mol Breed 37(5):63
Schwinn K, Venail J, Shang Y, Mackay S, Alm V, Butelli E, Oyama R, Bailey P, Davies K, Martin C (2006) A small family of MYB-gegulatory genes controls floral pigmentation intensity and patterning in the genus Antirrhinum. Plant Cell 18(4):831–851
Sompong U, Somta P, Raboy V, Srinives P (2012) Mapping of quantitative trait loci for phytic acid and phosphorus contents in seed and seedling of mungbean (Vigna radiata (L.) Wilczek). Breed Sci 62(1):87–92
Somta P, Chankaew S, Kongjaimun A, Srinives P (2015) QTLs controlling seed weight and days to flowering in mungbean [Vigna radiata (L.) Wilczek], their conservation in azuki bean [V angularis (Ohwi) Ohwi & Ohashi] and rice bean [V. umbellata (Thunb.) Ohwi & Ohashi]. Agrivita 37(2):159–168
Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG (2012) Primer3—new capabilities and interface. Nucleic Acids Res 40(15):e115
Wang LX, Cheng XZ, Wang SH, Liu Y (2013) Inheritance of several traits in mungbean (Vigna radiata). Acta Agron Sin 39(7):1172–1178. (Chinese with English abstract)
Wang LX, Elbaidouri M, Abernathy B, Chen HL, Wang SH, Lee SH, Jackson SA, Cheng XZ (2015) Distribution and analysis of SSR in mung bean (Vigna radiata L.) genome based on an SSR-enriched library. Mol Breed 35(1):1–10
Wang LX, Wu CS, Zhong M, Zhao D, Mei L, Chen HL, Wang SH, Liu CJ, Cheng XZ (2016) Construction of an integrated map and location of a bruchid resistance gene in mung bean. Crop J 4(5):360–366
Wang Z, Cui Y, Vainstein A, Chen S, Ma H (2017a) Regulation of fig (Ficus carica L.) fruit color: metabolomic and transcriptomic analyses of the flavonoid biosynthetic pathway. Front Plant Sci 8:1990
Wang JH, Zhang YW, Cheng XZ, Wang LX (2017) Construction of genetic map and identification of QTLs related to agronomic traits in mung bean. Acta Agron Sin 43(7):1096–1102 (Chinese with English abstract)
Wang F, Xu Z, Fan X, Zhou Q, Cao J, Ji G, Jing S, Feng B, Wang T (2019) Transcriptome analysis reveals complex molecular mechanisms underlying UV tolerance of wheat (Triticum aestivum, L.). J Agric Food Chem 67(2):563–577
Watanabe S, Hideshima R, Xia Z, Tsubokura Y, Sato S, Nakamoto Y, Yamanaka N, Takahashi R, Ishimoto M, Anai T, Tabata S, Harada K (2009) Map-based cloning of the gene associated with the soybean maturity locus E3. Genetics 182(4):1251–1262
Wegulo SN, Yang XB, Martinson CA (1998) Soybean cultivar responses to Sclerotinia sclerotiorum in field and controlled environment studies. Plant Dis 82(11):1264–1270
Yaqub M, Mahmood T, Akhtar M, Iqbal MM, Ali S (2010) Induction of mungbean [Vigna radiata (L.) Wilczek] as a grain legume in the annual rice-wheat double cropping system. Pak J Bot 42(5):3125–3135
Yan F, Di S, Takahashi R (2015) CACTA-superfamily transposable element is inserted in MYB transcription factor gene of soybean line producing variegated seeds. Genome 58(8):365–374
Yan WH, Wang P, Chen HX, Zhou HJ, Li QP, Wang CR, Ding ZH, Zhang YS, Yu SB, Xing YZ, Zhang QF (2011) A Major QTL, Ghd8, plays pleiotropic roles in regulating grain productivity, plant height, and heading date in rice. Mol Plant 4(2):319–330
Yonemaru JI, Choi SH, Sakai H, Ando T, Shomura A, Yano M, Wu J, Fukuoka S (2015) Genome-wide indel markers shared by diverse Asian rice cultivars compared to Japanese rice cultivar ‘Koshihikari.’ Breed Sci 65(3):249–256
Zhou H, Peng Q, Zhao J, Owiti A, Ren F, Liao L, Wang L, Deng X, Jiang Q, Han Y (2016) Multiple R2R3-MYB transcription factors involved in the regulation of anthocyanin accumulation in peach flower. Front Plant Sci 7:1557
Zhuang H, Lou Q, Liu H, Han H, Wang Q, Tang Z, Ma Y, Wang H (2019) Differential regulation of anthocyanins in green and purple turnips revealed by combined de novo transcriptome and metabolome analysis. Int J Mol Sci 20(18):4387
Funding
This work was supported by grants from the Anhui Academy of Agricultural Sciences (Grant No. 2020YL012), the National Key Research and Development Program of China (2016YFE0203800), China Agriculture Research System (CARS-08-Z11).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. The experiments were designed by Weijun Ye, Liya Zhang, Lei Zhang, and Bin Zhou. Material preparation, data collection, and analysis were performed by Weijun Ye, Yong Yang, Peiran Wang, Yin Zhang, Dongfeng Tian, Lingling Zhang, and Bin Zhou. The first draft of the manuscript was written by Weijun Ye and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ye, W., Yang, Y., Wang, P. et al. InDel marker development and QTL analysis of agronomic traits in mung bean [Vigna radiate (L.) Wilczek]. Mol Breeding 41, 66 (2021). https://doi.org/10.1007/s11032-021-01233-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-021-01233-0