
Abstract
Gray leaf spot (GLS) disease, caused by the Stemphylium (Sm) fungus, affects a wide range of horticultural crops including tomato. In this study, the South Korean Sm isolate (PNU-SM01) was identified, and the applicability of Sm-resistance gene (Sm)-linked DNA markers in marker-assisted selection was validated. ITS/gpd sequencing and morphological observation identified the isolate as S. lycopersici. A GLS disease assay was conducted on 80 tomato varieties, including commercial F1 hybrids and elite inbred lines. The reference line ‘Motelle’ was highly resistant (R, DSI = 4.0), whereas the ‘Moneymaker’ line was susceptible (S) and showed severe disease symptoms (DSI = 79.3). Among 30 commercial F1 hybrids, five were scored as S by DSI; whereas the rest, including all 17 hybrids claimed as Sm-resistant by the supplier, were R or moderately resistant (MR). Among 48 inbred lines, four were scored as S and 44, including five Lycopersicon accessions reported as R, were R or MR. Three known Sm-linked DNA markers (CT55, D5, and Sm-InDel) were genotyped in all 80 cultivars; the highest match (92.5%) to the GLS disease assay was observed for Sm-InDel. We converted the recessive marker CT55 into a codominant marker (CT55-Co) and developed dCAPS (Sm-SNP) and KASP (Sm-KASP) markers from a previously identified Sm-linked SNP. Sm-SNP and Sm-KASP showed a similar level of phenotype to marker genotype association as Sm-InDel, whereas CT55-Co displayed a high level of mismatch. To screen for the source of Sm resistance, 138 tomato accessions were genotyped with Sm-InDel and Sm-SNP: 15 were homozygous for the resistance allele of both markers. In this study, resistance to the Korean isolate of S. lycopersici in commercial F1 hybrids and breeding sources was characterized to reveal which DNA markers can be effectively applied for marker-assisted selection of Sm-resistance.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gray leaf spot (GLS) disease is caused by the Stemphylium (Sm) fungus in a wide range of horticultural crops, such as pepper (Capsicum annum L.), cotton (Gossypium hirsutum L.), spinach (Spinacia oleracea L.), and tomato (Solanum lycopersicum L.). GLS is distributed globally but affects particularly humid tropical and subtropical regions. Tomato plants infected by Sm develop small spots with a yellow halo that eventually become necrotic lesions with gray centers and dark brown borders. In severely affected plants, leaves become chlorotic with perforated centers on lesions, before ultimately falling off. A fungal spore is noticeable on the surface of lesions. The pathogen causing tomato GLS mainly include four species: Stemphylium solani, S. lycopersici (syn. S. floridanum), S. botryosum, and S. vesicarium (Weber 1930; Hannon and Weber 1955; Rotem et al. 1966; Porta-Puglia 1981). The occurrence of GLS by S. lycopersici and S. solani in tomato-cultivating areas in Korea has been reported previously (Min et al. 1995; Kim et al. 1999).
Planting tomato varieties resistant to GLS is the ideal approach to prevent loss by this disease. A resistance gene, named Sm, has been identified in the wild tomato species Solanum pimpinellifolium (Andrus et al. 1942). The Sm gene affords a broad spectrum of resistance to the aforementioned Sm species and is conveniently inherited by single incomplete dominance (Hendrix and Frazier 1949). Currently available commercial tomato cultivars resistant to GLS have been developed by introgression of Sm into elite lines through conventional hybridization breeding, in which resistant plants are selected based on phenotype.
Breeding of Sm-resistant cultivars can be facilitated by marker-assisted selection (MAS), whereby plants carrying the Sm trait are selected at seedling stage using DNA markers tightly linked to the Sm locus. In an early mapping approach, Behare et al. (1991) mapped Sm to Chr. 11 using an F2 population derived from crossing Sm-resistant ‘Motelle’ and Sm-susceptible ‘Moneymaker’ lines. Sm was flanked by two restriction fragment length polymorphism (RFLP) markers, TG110 and T10, and was found to be linked to a Fusarium race 1 resistance gene. Later, Ji and Scott (2009) produced a simple PCR-based cleaved amplified polymorphic sequence (CAPS) marker ‘CT55,’ which located between TG110 and T10 marker. Because CT55 is a recessive marker, heterozygous and homozygous susceptible plants shared the same PCR band pattern. CT55 has been used for pyramiding multiple disease resistance genes in tomato MAS breeding programs (Handson et al. 2016). Recently, Yang et al. (2017) used ‘Motelle’ as a source of Sm and mapped it to a genomic interval of 0.26 Mb containing 37 genes on Chr. 11 based on bulked segregant analysis combined with genome resequencing. Additionally, the D5 sequence characterized amplified region (SCAR) marker was developed from a gene, Solyc11g011880.1.1, that cosegregated with the resistance locus. More recently, Su et al. (2019) identified the Sm locus within a 185 kb interval between two SCAR markers, InDel343 and InDel-FT-32, by fine mapping the segregating populations derived from the ‘9706’ resistant line. A candidate gene, ORF9, was identified by virus-induced gene silencing (VIGS) of several open reading frames (ORFs) in the 185 kb interval, and a codominant marker, Sm-InDel, was developed based on vicinity to the candidate gene. However, sequence data for ORF9 and development of a marker directly from the gene were not described by Su et al. (2019).
GLS disease has been detected in greenhouse production in Korea, especially for cherry or grape tomato. Several commercial cultivars are claimed as Sm-resistant by breeding companies, but large-scale disease screening on a germplasm collection using the Korean Sm isolate has not been reported. In addition, applicability of known Sm-linked DNA markers has not been tested on Korean Sm isolates. The present study aimed to evaluate host reaction in tomato germplasm, including commercial F1 hybrids and elite breeding lines, and validate the use of Sm-linked markers for selecting cultivars resistant to the Korean isolate of S. lycopersici.
Materials and methods
Plant materials
A tomato cultivar collection containing 30 commercial F1 hybrids and 50 elite inbred lines was used in this study (Table 1). Seeds of commercial Fl hybrids and elite inbred lines were provided by private seed companies and the Plant Genetics and Breeding Research Center at Pusan National University (PGBRC at PNU), respectively. Inbred ‘Motelle’ (LA2823) and ‘Moneymaker’ (LA2706) reference lines were obtained from the Tomato Germplasm Research Center at the University of California-Davis (TGRC at UC-Davis).
Plants with the Sm-resistance marker genotype were screened using a tomato germplasm collection comprising 138 inbred lines, including 113 Lycopersicon accession (LA), 14 plant introduction (PI), and 11 PSL accessions, obtained from the TGRC, Germplasm Resources Information Network (GRIN), and PGBRC, respectively.
Pathogen maintenance and disease assay
The original Sm isolate (PNU-SM01) was obtained from Gangneung-Wonju National University and was maintained by placing the fungal mycelial agar plug onto a Potato Dextrose Agar (PDA; Difco, Detroit, MI, USA) and V8 juice agar (V8; HiMedia, Mumbai, India) and incubating at 25 °C on a 12-h light-dark cycle. For the disease assay, tomato plants were grown in plastic pots until the 3rd true leaves were fully expanded. Inocula were prepared from conidial suspensions as follows. Cultures were grown on V8 agar for 15 days at 25 °C under 12 h/12 h Near-Ultra Violet (NUV) light/darkness. Conidia were separated from the cultures by flooding with sterile distilled water and brushing gently, followed by filtering through a cheesecloth to remove mycelial fragments. The conidial suspension (1 × 105 conidia mL−1) was mixed with several drops of 0.1% Tween 20 and sprayed onto tomato plants with a hand sprayer. Inoculated plants were incubated at 25 °C with a 12-h photoperiod and 90% relative humidity in a controlled environment chamber for 15 days until disease symptoms were comparable to those of susceptible reference plants. The disease severity index (DSI) was assessed using a visual scale from 0 to 4, where 0: < 5% of diseased leaf tissue; 1: 5–25% diseased tissue; 2: above 25–50% diseased tissue; 3: above 50–75% diseased tissue; and 4: > 75% diseased tissue. The DSI was calculated as follows (Pandey et al. 2003):
The host was determined as resistant (R): DSI < 10%, moderately resistant (MR): DSI = 10–30%; and susceptible (S): DSI > 30%.
Pathogen identification
Fungal DNA extraction
Mycelial mats (hyphae, conidiophores, and conidia) were mixed with ddH2O and scrubbed off gently from the V8 agar with a sterile spatula. Collected mycelial mats were transferred to 1.5-mL tubes and centrifuged at 14,240×g for 2 min. Then, the supernatant was discarded; 600 μL of DNA extraction buffer (Kim et al. 2016) was added to each tube, and these were incubated at 65 °C for 45 min. The lysate was mixed with 200 μL of 7.5 M ammonium acetate, placed on ice for 15 min, and then centrifuged at 14,240×g for 10 min. The supernatant was transferred to a new 1.5-mL tube containing 2.5 μL glycogen solution (10 mg mL−1) and 600 μL isopropanol. After centrifugation at 14,240×g for 10 min, the liquid was carefully decanted, the DNA pellet washed with 300 μL of 70% ethanol, dried, and resuspended in 50 μL Tris-EDTA buffer. DNA quality and concentration were evaluated with a Nanodrop 1000 Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA).
Sequencing of internal transcribed spacer (ITS) region and gpd
The ITS region of nuclear rDNA and the glyceraldehyde-3-phosphate dehydrogenase gene (gpd) of the pathogen were amplified using the ITS1 (5′-TCCGTAGGTGAACCTGCGG-3′); ITS4 (5′-TCCTCCGCTTATTGATATGC-3′) (White et al. 1990) and gpd1 (5′-CAACGGCTTCGGTCGCATTG-3′); gpd2 (5′-GCCAAGCAGTTGGTTGTGC-3′) primer sets (Berbee et al. 1999), respectively. PCR amplifications were performed in a total volume of 20 μL containing 2 μL genomic DNA (20 ng μL−1), 2 μL 10× buffer Solg™ (SolGent, Daejeon, Korea), 0.4 μL dNTPs (10 mM, SolGent), 1 μL of each primer (10 pmol), and 0.2 μL eTaqSolg™ Taq polymerase (5 U μL−1, SolGent). Amplifications were conducted on a T100™ thermocycler (Bio-Rad, Hercules, CA, USA) as follows: denaturation at 94 °C for 10 min followed by 40 cycles of denaturation at 94 °C for 1 min, annealing at 55 °C for 1 min, and extension at 72 °C for 1 min. Amplification products were electrophoresed on a 2% agarose gel and purified using the Expin™ Gel SV gel extraction kit (GeneAll, Seoul, Korea). Purified PCR products were directly sequenced using the dye-terminator method by Genotech (Daejeon, Korea).
Sequence and phylogenetic analysis
Homologous genes for the ITS and gpd sequences of the PNU-SM01 isolate were retrieved from the National Center for Biotechnology Information (NCBI; https://www.ncbi.nlm.nih.gov/) database using BlastN. Sequences obtained from the Blast search were aligned using MEGA software version X (Kumar et al. 2018), inspected manually, and adjusted where necessary. The phylogenetic tree of Stemphylium spp. was constructed using the neighbor-joining method based on the combined sequence data set for the ITS and gpd. The distances were determined according to Jukes-Cantor model with 1000 bootstrap replicates. Alternaria alternaria (EGS 34-016) and Cochliobolus sativus (AF071329, AF081385) were used as an out-group.
Improvement and validation of Sm-linked markers
Conversion of the CT55 recessive marker to the codominant marker CT55-Co
The insert sequence of the RFLP clone for the CT55 marker (Ji and Scott 2009) was obtained from the Sol Genomics Network (SGN) database by searching for the marker’s name. Genomic location and full-length gene sequence of the insert were searched by BlastN against the tomato reference genome assembly version SL3.0 from the SGN database. Genomic sequences of two gene homologs identified by the blast were aligned using CLUSTALW, and their nucleotide variations were compared. A primer pair for the CT55-Co marker was designed based on the nucleotide variant specific for the target gene homolog.
Development of derived CAPS (dCAPS) and kompetitive allele-specific PCR (KASP) markers
The genomic location of the Sm-linked single nucleotide polymorphism (SNP) (C/T, SL2.50 ch11_9,317,565 bp) identified in the genome-wide association study (GWAS) by Su et al. (2019) was searched against the tomato reference genome assembly version SL4.0 (iTAG 4.0) in the SGN database. A dCAPS marker (Sm-SNP) was designed using dCAPS finder 2.0 (http://helix.wustl.edu/dcaps/) and Primer3 program (http://bioinfo.ut.ee/primer3-0.4.0/) based on flanking sequences of the SNP.
The Sm-linked SNP was converted also to a KASP marker (Sm-KASP) for high-throughput automated genotyping based on real-time PCR. KASP™ Assay (Allele-1 forward, Allele-2 forward, and reverse primers) for Sm-KASP was designed and synthesized by LGC genomics (Teddington, England).
Genotyping of the Sm-linked DNA markers
For genotyping of Sm-linked DNA markers (CT55, CT55-Co, D5, Sm-InDel, Sm-SNP, and Sm-KASP) (Table 2), 1st to 2nd true leaves of tomato seedlings were collected, and genomic DNA was extracted with sodium dodecyl sulfate following the method of Kim et al. (2016) with minor modifications. Each sample was amplified in a total volume of 10 μL containing 1 μL genomic DNA template (20 ng μL−1), 0.2 μL dNTPs, 1 μL of 10× buffer, 0.5 μL of each primer (10 pmol), 0.1 μL Taq polymerase (SolGent), and 6.7 μL ddH2O. Amplifications were performed on a T100™ thermocycler (Bio-Rad) as follows: denaturation at 95 °C for 5 min followed by 35 cycles of denaturation at 95 °C for 30 s, annealing at Tm (°C, listed in Table 2 for each marker) for 30 s, and extension at 72 °C for 1 min. For CAPS and dCAPS markers, PCR products were digested with restriction enzymes for 90 min at 37 °C. PCR products were separated on 1.5% agarose gel at 180 V for 80 min, stained in ethidium bromide, and visualized using the Gel Image Analysis System (CoreBiosystem, Seoul, Korea).
KASP genotyping of each sample was performed in a total volume of 5 μL containing 2.5 μL DNA template (20 ng μL−1), 2.5 μL of 2× KASP Master mix containing the FRET cassette, Taq polymerase for allele-specific PCR and optimal buffer (LGC genomics), and 0.07 μL KASP Assay primer mix. The mixtures were amplified on a Lightcycler® 480 Instrument II (Roche, Basel, Switzerland) under the following conditions: hot-start activation at 94 °C for 15 min, 10 cycles of touchdown PCR (94 °C for 20 s; touchdown at 61 to 55 °C, − 0.6 °C per cycle for 1 min), and 26 cycles of further thermal cycling (94 °C for 20 s; 55 °C for 1 min). After plate reading at 37 °C for 1 min, end point genotyping data were analyzed on Lightcycler® software version 1.5.0 (Roche).
Characterization of the ORF9 sequence
Identification of the genomic location of ORF9
The forward and reverse primer sequences used by Su et al. (2019) to construct the cloning vector for VIGS were blasted against the tomato reference genome assembly version SL4.0. The structure of ORF9 was predicted based on the reference genome sequence using the FGENESH program (http://www.softberry.com/) with the S. lycopersicum generic tomato parameter. Protein function of ORF9 was confirmed by the NCBI BLASTP tool.
Cloning of the ORF9 region to detect polymorphisms
Three PCR primer sets were designed with Primer3 to clone the coding sequence (CDS) regions of ORF9 from ‘Moneymaker’ and ‘Motelle.’ Each sample was amplified in 50 μL of PCR reaction mixture containing 5 μL genomic DNA template (20 ng μL−1), 1 μL dNTPs, 5 μL of 10× buffer, 2.5 μL of each primer (10 pmol), 0.5 μL Taq polymerase (SolGent), and 33.5 μL ddH2O. Reactions were carried out in a T100™ thermocycler (Bio-Rad) under the following conditions: denaturation at 95 °C for 5 min followed by 35 cycles of denaturation at 95 °C for 30 s, annealing at 55 °C for 30 s, and extension at 72 °C for 1 min. PCR products were separated on a 1.5% agarose gel at 160 V for 30 min and purified using the Expin™ Gel SV extraction kit (GeneAll). Vector cloning of purified PCR products and cell transformation were carried out using the Dyne™ TA kit (Dynebio, Gyeonggi, Korea). Vector DNA was purified from competent cells using the Exprep™ Plasmid SV mini kit (GeneAll). All the procedures were conducted following the manufacturers’ instructions. Purified vector DNAs were sequenced using the dye-terminator method by Genotech (Daejeon, Korea), and the resulting insert sequences were aligned using the Multalin program (http://multalin.toulouse.inra.fr/multalin/).
Results
Identification of the Sm pathogen
The PNU-SM01 isolate formed dark olivaceous colonies with grayish aerial mycelia and secreted a yellowish to reddish-brown pigment when cultured on PDA and V8 agar plates (Fig. 1a, b). Microscopic examination revealed that PNU-SM01 conidia were morphologically similar to those of S. lycopersici as reported by Huang and Tsai (2017). Specifically, conidia were oblong, solitary, pale to mid brown, and 1–3 longitudinal septa and 3–4 transversal septa were observed in each conidium (Fig. 1c). Conidia of S. solani are distinguishable from those of S. lycopersici, which are longer and slenderer, with a symmetrically tapering core and a more pointed apex (Kim et al. 1999).

Pathogen identification of the Korean isolate of S. lycopersici (‘PNU-SM01’) based on morphological characteristics and DNA sequence. a Growth on PDA. b Growth on V8 agar. c Conidia and conidiophores; scale bar = 50 μm. d Pathogenicity test on ‘Moneymaker’ and gray leaf spot symptoms on infected leaves
The pathogenicity of the PNU-SM01 isolate was tested with reference to Sm-susceptible ‘Moneymaker’ plants (Behare et al. 1991; Yang et al. 2017). Development of disease symptoms typical for GLS (irregular black spots mostly surrounded by a yellowish halo) was observed on leaves 5 days post inoculation, indicating that PNU-SM01 was pathogenic to ‘Moneymaker’ (Fig. 1d).
The amplified sequences of the ITS region and gpd fragment of the fungal isolate were compared by BLAST analysis against the sequences deposited in GenBank. The ITS sequence of the isolate revealed 99.0% identity with S. lycopersici and 98.1% identity with S. solani; whereas the gpd sequence showed 99.0% and 94.2% identity, respectively. Phylogenetic analysis using a combined dataset of the ITS-5.8S rDNA and gpd regions showed that PNU-SM01 was clustered in the sub-group comprising S. lycopersici and differentiated clearly from other Stemphylium species used in this study (Fig. 2).

Molecular identification of ‘PNU-SM01 (red bold highlight)’ based on the ITS region and gpd sequence
GLS disease assay
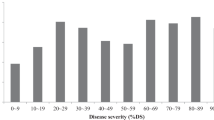
A total of 80 tomato cultivars were evaluated for host reaction to the PNU-SM01 isolate (Table 1). ‘Motelle’ (DSI = 4.0) and ‘Moneymaker’ (DSI = 79.3) were employed as R and S reference lines. The DSI of most cultivars was within the R or MR range (Fig. 3). Among 30 commercial F1 hybrids developed in Korea, Japan, and Europe, five were scored as S; whereas the remaining 25, including all 17 hybrids claimed as Sm-resistant (commercial information marked as bracketed R in Table 1) by the supplier, were either R or MR. Among the 48 inbred lines, four were scored as S and 44, including five LA accessions reported as R (TGRC information marked as bracketed R in Table 1), were either R or MR.

Disease severity index (DSI) of gray leaf spot in 80 tomato cultivars inoculated with the Korean isolate of S. lycopersici (PNU-SM01). Cultivars with DSI < 10, DSI = 10–30, and DSI > 30 were scored as resistant (R), moderately resistant (MR), and susceptible (S), respectively. Motelle and Moneymaker are marked with arrows
Validation of Sm-linked DNA markers
To assess applicability in MAS of Sm-resistance, three previously reported Sm-linked DNA markers, CT55 (Ji and Scott 2009), D5 (Yang et al. 2017), and Sm-InDel (Su et al. 2019), were genotyped in the 80 cultivars (Table 1). The genomic locations of the three markers are depicted in a diagram (Fig. 4).

Physical genomic locations of five Sm-linked DNA markers (CT55, CT55-Co, D5, Sm-InDel, and Sm-SNP) on chromosome 11
According to Ji and Scott (2009), CT55 is a recessive marker and hence heterozygous and susceptible homozygous plants cannot be distinguished because they share the same PCR banding pattern. A PCR fragment of 404 bp was generated for both Sm-resistant (allele for ‘Motelle’) and Sm-susceptible (allele for ‘Moneymaker’) genotypes. After digesting the PCR product with DdeI, the Sm-resistant genotypes produced three fragments of 194 bp, 134 bp, and 76 bp, while four bands of 328 bp, 194 bp, 134 bp, and 76 bp were generated from both Sm-susceptible and heterozygous genotypes (Fig. 5a). Therefore, Sm-resistant heterozygous plants and Sm-susceptible homozygous plants presented all four bands, whereas Sm-resistant homozygous plants produced only the three lower bands. Thus, in our CT55 marker genotyping, cultivars showing four bands were scored as homozygous susceptible (S) or heterozygous (H), while cultivars showing three bands were scored as homozygous resistant (R). Only cultivars that exhibited a clear R marker genotype were considered for the host reaction comparison. In our experiment, ‘Motelle’ and ‘Moneymaker’ displayed R and S/H CT55 marker genotype, respectively (Table 1, Fig. 5a). For 53 cultivars that showed R genotype, five cultivars were scored as susceptible by their host reaction; whereas for 27 cultivars with S/H marker genotype, five were phenotypically S and 22 were R or MR. For the D5 marker, no polymorphism was observed between ‘Motelle’ and ‘Moneymaker,’ and all cultivars were monomorphic (Table 1, Fig. 5c). Yang et al. (2017) reported that the D5 marker amplified a fragment of 876 bp from ‘Motelle’ and 820 bp from ‘Moneymaker,’ which cosegregated in the F2 population. In our experiment, all cultivars amplified a fragment of 876 bp. Sm-InDel is a SCAR located 15 kb downstream from the candidate gene (ORF9) of the Sm locus (Su et al. 2019), and only 4.4 Mb apart from D5 on Chr. 11 (Fig. 4). Sm-InDel marker validation in 80 cultivars revealed a good match between marker genotype and host reaction; only six cultivars (four F1 hybrids and two inbred lines) that were Sm-susceptibility in the GLS assay showed R or H marker genotype (Table 1, Fig. 5d).

Genotyping results of various Sm-linked markers: a CT55. b CT55-Co. c D5. d Sm-InDel. e Sm-SNP (Agarose gel lane 1, ‘Moneymaker’; lane 2, ‘Motelle’; lane 3, ‘Bacchus’; lane 4, ‘Doterang Dia’; lane 5, ‘Doterang Master’; lane 6, ‘Escort’; lane 7, ‘Galaxy’; lane 8, ‘Qupirang’; lane 9, ‘SV0052TC’; lane 10, ‘AV017-4’; lane 11, ‘TSS3-395’; M, 100 bp DNA size marker). f Sm-KASP [Green, cultivars with homozygous resistance (TT); Red, heterozygous cultivars (CT); Blue, cultivars with homozygous susceptibility (CC); Gray, No-Template-Control]
Conversion of the recessive CT55 marker to a codominant marker
To ensure MAS is efficient, a heterozygous genotype needs to be distinguishable from homozygous genotypes for a specific trait. To this end, here, we converted CT55 to a codominant marker that allowed discrimination between all three genotypes. While exploring the reason for the recessive nature of CT55, we blasted the mRNA sequence (NM_001247089.2) of the cDNA clone from which CT55 was originated (Ji and Scott 2009) against the tomato reference genome. This yielded two tandemly repeated homologous gene sequences, Solyc11g062130 and Solyc11g062190, located 74 kb apart on Chr. 11. The length of each gene was about 4 kb and included four exons. Alignment revealed sequence variations comprising 34 InDels and 74 nucleotide mismatches (SNPs). Primer sites for CT55 were conserved between the two genes, indicating that both gene sequences would be amplified simultaneously from a PCR run. The DdeI enzyme recognition site for the susceptible allele (A, uncut, 328/76 bp) was found in Solyc11g062130. However, in Solyc11g062190, its corresponding site was C, which resulted in R allele-specific bands (cut, 194/135/76 bp) in addition to the S allele band (328 bp) (Fig. 6). We believe that this observation explains why S and H produced the same banding pattern with CT55.

Schematic diagram comparing two gene homologs (Solyc11g062130 and Solyc11g062190) harboring the CT55 sequence
We designed a new set of primers for CT55-Co with annealing sequences specific to Solyc11g062130 by altering slightly the original binding site of the forward and reverse primers of CT55. CT55-Co could amplify fragments only from Solyc11g062130 (Fig. 6). After DdeI digestion of the PCR products amplified by the CT55-Co primer set, a banding pattern specific for the S allele (418/300 bp) appeared with ‘Moneymaker’ DNA, and a banding pattern specific for the heterozygous genotype (418/300/284/134 bp) appeared the DNA mixture of ‘Moneymaker’ and ‘Motelle,’ indicating that the recessive marker had been successfully converted to a codominant one (Table 2, Fig. 5b). The CT55-Co marker allowed all cultivars that showed S/H marker genotype with CT55 to be scored unequivocally as either S or H (Table 1). In the case of CT55-Co, only 17 cultivars exhibited a mismatch between genotype and host reaction.
Development of dCAPS and KASP markers
In Su et al. (2019), a genome-wide association study identified a SNP (C/T) significantly linked to Sm. We converted this SNP to a dCAPS marker and evaluated its applicability for MAS of Sm-resistance by genotyping 80 cultivars (Fig. 5e). The dCAPS marker (named Sm-SNP) genotypes matched perfectly the Sm-InDel genotypes in all cultivars. To avoid any additional time-consuming steps when running dCAPS, such as restriction enzyme digestion and gel electrophoresis, we converted Sm-SNP to a KASP marker (Sm-KASP) suitable for high-throughput automated genotyping. The Sm-KASP genotypes matched perfectly the Sm-SNP genotypes in all 80 cultivars (Fig. 5f).
Screening of tomato accessions with markers for Sm-resistance
A total of 138 tomato accessions were genotyped using Sm-InDel and Sm-SNP markers (Table S1). For Sm-InDel, 16 accessions were genotyped as homozygous for the resistant allele of ‘Motelle’ (122 bp), whereas 90 accessions were homozygous for the susceptible allele of ‘Moneymaker’ (140 bp), and 32 accessions showed genotypes (160 bp and 180 bp) different from either ‘Motelle’ or ‘Moneymaker.’ For Sm-SNP, 20 accessions were homozygous for the resistant allele of ‘Motelle’ (TT), 12 were heterozygous (CT), and 106 were homozygous for the susceptible allele of ‘Moneymaker’ (CC). Fifteen accessions were homozygous for resistance using both Sm-InDel and Sm-SNP (122 bp/TT).
Characterization of the ORF9 sequence
In Su et al. (2019), the VIGS of 33 ORF sequences in the genomic region (82,790 bp, SL2.50 chr11: 9,198,580–9,281,370 bp) carrying the Sm locus identified ORF9 as a candidate gene responsible for resistance to GLS (Fig. 4). Here, we aimed to identify and characterize the full sequence of ORF9 and develop a marker based on the polymorphisms between ORF9 of ‘Motelle’ and ‘Moneymaker.’
By blasting the primer sequences of ORF9 used for vector cloning in VIGS (Su et al. 2019), we identified a 277-bp region on Chr. 11 (SL4.0 chr11: 9,295,938–9,296,214 bp) flanked by the two primers. To obtain the full-length sequence of ORF9, an 80-kb sequence (SL4.0 chr11: 9,281,433–9,361,726 bp) flanking the 277 bp region was obtained from the reference genome assembly SL4.0 and tested for potential gene structures using FGENESH (Fig. S1). Twelve potential genes were predicted, and the 277 bp region of ORF9 was found in the 3rd gene, which was 15.7 kb long (SL4.0 chr11: 9,292,885–9,308,544 bp) and contained 15 exons (5571 bp CDS, 1856 amino acids). The CDS of this gene encodes seven protein domains (Retrotrans_gag, zf-CCHC, retropepsin_like, RVP_2, RT-LTR, RVT_1, and RNase_HI_RT_Ty3) reported by Su et al. (2019) and an additional three protein domains (PTZ00368, ZnF_C2HC, and CD_POL_like). The result indicated that this putative gene corresponded to the full-length sequence of ORF9.
To find polymorphisms in ORF9, a genomic region covering 2.6 kb of the gene was PCR-cloned and sequenced from ‘Motelle’ and ‘Moneymaker.’ Two colonies per tomato line were picked, and inserts on their plasmid vectors were sequenced. Sequence alignment and comparison revealed polymorphisms including 184 SNPs and two InDels (1 bp, 15 bp) between the two GLS resistant and susceptible lines (Fig. S2). Furthermore, the PCR fragments from the same line did not match completely and showed several sequence variants, supporting the existence of ORF9 homologs.
A SCAR marker was designed from the 15 bp InDel and genotyped in ‘Moneymaker’ and ‘Motelle.’ Two PCR bands specific for the 15 bp insertion and 15 bp deletion were observed from both cultivars, and no polymorphism was detected, supporting the existence of ORF9 homologs. The sequence amplified by the SCAR marker (about 1 kb) was blasted against the SGN SL4.0 assembly and, as expected, sequences with high similarity (> 93.9%) were retrieved from all 12 chromosomes.
Discussion
Various molecular markers for selecting disease resistance in tomato plants are publicly available. Tomato diseases with known resistance markers for MAS include viral tomato mosaic virus (ToMV) and tomato yellow leaf curl virus (TYLCV); Bacterial wilt and Bacterial spot; fungal Fusarium crown root rot, Fusarium wilt, and Verticillium wilt; and nematode (Lee et al. 2015; Hanson et al. 2016). Availability of these markers for MAS has accelerated breeding programs for pyramiding R genes and developing new cultivars with multiple disease resistances. Nevertheless, continuous marker development is needed even for the same disease, because, at times, resistance to different isolates of the same pathogen may be conferred by different genes, and existing resistance genes can be easily overcome by new virulent races evolved in a natural environment (Andersen et al. 2018; Stam and McDonald 2018).
Four different Sm species are known to cause GLS in tomato (Weber 1930; Hannon and Weber 1955; Rotem et al. 1966; Porta-Puglia 1981). In South Korea, appearance of two Sm species, S. lycopersici and S. solani, has been reported in tomato-cultivating areas (Min et al. 1995; Kim et al. 1999). The Sm gene conferring resistance to all four Sm species (Andrus et al. 1942; Behare et al. 1991) enables Sm-resistance breeding, which is facilitated also by the simple genetic inheritance of Sm via single incomplete dominance and long-term resistance (Behare et al. 1991). When a new pathogenic strain evolves rather slowly, a resistance gene can remain effective for a long time without being overcome by the pathogen. Similar simple inheritance and durable resistance have been shown for resistance to Fusarium in cabbage (Walker 1930), leaf rust in barley (Cebada Capa gene) (Jin et al. 1993), stem rust in wheat (Sr26 gene) (Singh et al. 2006), and Alternaria stem canker in tomato (ASC gene) (Witsenboer et al. 1989).
Conventional field breeding for Sm-resistance in tomatoes has a long history, and currently diverse cultivars exhibiting reliable Sm-resistance are available worldwide. Large pink fresh fruit cultivars typical in Japan such as the ‘Doterang’ series were introduced in South Korea more than two decades ago. These cultivars were Sm-resistance at the time they were introduced and were used for breeding by seed companies. However, the source of the original Sm-resistance gene carried by these commercial cultivars has remained unknown. The results of our study imply that most resistant cultivars grown in South Korea likely carry the Sm gene originated from S. pimpinellifolium, because Sm-resistant cultivars showed marker genotype for ‘Motelle,’ which was developed by introgression of the Sm locus from this red-fruited wild species (Behare et al. 1991).
In our GLS disease bioassay, 70 of 80 cultivars showed R or MR response to the Korean isolate (PNU-SM01) of S. lycopersici. A high percentage of Sm-resistant cultivars stems from the fact that we collected the F1 cultivars claimed as Sm-resistant by their suppliers and selected inbred lines that showed R marker genotype (‘Motelle’) by screening with CT55. Among F1 hybrids evaluated in this study, four (Alround-Daemok, AS6, Fighting-Daemok, and Ultra-Daemok) displayed severe susceptibility to PNU-SM01, even if their marker genotypes were R (‘Motelle’) or H. Notably, these four hybrids are rootstock cultivars used in grafting of tomato plants, whereas all other hybrids with a perfect match between phenotype and marker genotype are cultivars used as scions. In breeding of rootstock cultivars, wild Solanum species, such as S. pimpinellifolium or S. habrochaites, are generally used as breeding sources with the intent of improving resistance to soil-borne pathogens and plant vigor required for long-term cultivation and high yields (Grieneisen et al. 2018). Thereby, it is possible that these rootstock hybrids were derived from S. pimpinellifolium plants that were susceptible to Sm but carried a Sm-linked marker genotype for resistance. This circumstance emphasizes the importance of developing functional gene-based markers that discern R alleles from the Sm locus in both wild and cultivated species, thus enabling widespread use in MAS.
In this study, six Sm-linked DNA markers, CT55, CT55-Co, D5, Sm-InDel, Sm-SNP and Sm-KASP, were validated for their applicability in MAS of Sm-resistance. CT55 was the first PCR-based Sm-linked marker and was reported in 2009 by Ji and Scott. However, we found that linkage of CT55 to Sm was not strong enough for MAS. Nevertheless, CT55 was tightly linked to Sm in RFLP mapping of Behare et al. (1991), possibly due to lack of markers located on the genomic region or suppression of recombination in their segregation population. Similar phenomena were reported for the Frl locus conferring resistance to Fusarium crown root rot (FCRR) in tomato (Kim et al. 2016; Devran et al. 2018). Being a recessive marker, CT55 poses another limitation in MAS as the susceptible and heterozygous marker genotypes are indistinguishable from each other. Here, we successfully converted CT55 to a codominant marker that detected dominant and recessive homozygosity and heterozygosity, and consequently enabled better evaluation of marker-phenotype association. Nevertheless, mismatches in the CT55-Co marker genotype and the host reaction exhibited for the 17 cultivars in this study indicated that chromosomal recombination and linkage breaks between the marker and Sm occur to a considerable extent and, thus, limit the use of the CT55-Co marker for the MAS.
The marker genotypes Sm-InDel and Sm-SNP (Sm-KASP) matched in all 80 cultivars. When 138 tomato accessions were examined using these markers, the linkage between the markers was retained well in most S. lycopersici accessions, whereas a marker genotype mismatch (or recombination between the markers) was observed in several accessions of the wild tomato species. Similar results were also reported by Su et al. (2019) in their test of 289 accessions using these two markers. In addition, PCR band sizes that were different from those of ‘Motelle’ or ‘Moneymaker’ were observed, mostly from the wild species accessions, which indicates the existence of high levels of DNA variation in these marker regions in the wild tomato species. Therefore, further work is required to develop a functional gene-based marker for Sm-resistance for universal applicability to the MAS. Fifteen accessions that were homozygous for the resistance allele in both Sm-InDel and Sm-SNP could be useful breeding sources for Sm-resistance; however, a disease assay is required to confirm their host reaction to Sm and the marker-phenotype association.
The Sm-linked D5 marker (Yang et al. 2017) failed to distinguish between Sm-resistant and Sm-susceptible alleles, showing instead a monomorphic PCR banding pattern for all tomato cultivars tested. A similar observation was reported by Su et al. (2019), confirming poor applicability of D5 in MAS. Our validation of Sm-linked markers pointed to Sm-InDel and Sm-SNP as the markers with the best applicability to MAS. Both markers matched genotype and host reaction to Sm in 92.5% of cases across the 80 tomato cultivars tested. In addition, genotyping of 138 accessions revealed 15 accessions (three from S. pimpinellifolium and 12 from S. lycopersicum) that were homozygous resistance (122 bp/TT) for both markers. These 15 accessions may be good candidates for bioassays aimed at future validation of their Sm-resistant status.
In Su et al. (2019), the ORF9 sequence located 15 kb upstream of Sm-InDel was suggested as a candidate gene for Sm based on gene knockout using VIGS. The ‘9706’ resistant line exhibited a high level of susceptibility after ORF gene silencing. We aimed to develop a polymorphic marker from ORF9 using its full-length sequence, but it failed due to extensive duplications of the sequence throughout the tomato genome. Multiple homologous regions with high sequence similarity made it difficult to discriminate polymorphisms specific to ORF9 between ‘Motelle’ and ‘Moneymaker,’ which is essential for the development of an ORF9 gene-based DNA marker.
In conclusion, for the first time, the host reaction of the tomato germplasm to the Korean isolate of S. lycopersici was evaluated, which provides useful information on the commercial F1 hybrids and breeding sources with resistance to GLS. Our validation of Sm-linked markers based on the GLS disease assay indicated that Sm-InDel and Sm-SNP are currently the most suitable markers for the MAS of Sm-resistance. The MAS, using these markers, will significantly improve the breeding process for new Sm-resistant cultivars. However, further work is required to develop a functional gene-based marker for Sm-resistance.
Abbreviations
- CAPS:
-
Cleaved amplified polymorphic sequence
- dCAPS:
-
derived CAPS
- DSI:
-
Disease severity index
- GLS:
-
Gray leaf spot
- ITS:
-
Internal transcribed spacer
- KASP:
-
Kompetitive allele-specific PCR
- MAS:
-
Marker-assisted selection
- NCBI:
-
National Center for Biotechnology Information
- PGBRC:
-
Plant Genetics and Breeding Research Center
- RFLP:
-
Restriction fragment length polymorphism
- SCAR:
-
Sequence characterized amplified region
- SGN:
-
Sol Genomics Network
- Sm:
-
Stemphylium
- Sm :
-
Sm-resistance gene
- SNP:
-
Single nucleotide polymorphism
- LA:
-
Lycopersicon accession
- TGRC:
-
Tomato Germplasm Research Center
- VIGS:
-
Virus-induced gene silencing
References
Andersen EJ, Ali S, Byamukama E, Yen Y, Nepal MP (2018) Disease resistance mechanisms in plants. Genes 9:339. https://doi.org/10.3390/genes9070339
Andrus CF, Reynard GB, Wade BL (1942) Relative resistance of tomato varieties, selections, and crosses to defoliation by Alternaria and Stemphylium. US Department of Agriculture Circular 652
Behare J, Laterrot H, Sarfatti M, Zamir D (1991) Restriction fragment length polymorphism mapping of the Stemphylium resistance gene in tomato. Mol Plant-Microbe Interact 4:489–492
Berbee M, Pirseyedi M, Hubbard S (1999) Cochliobolus phylogenetics and the origin of known, highly virulent pathogens, inferred from ITS and glyceraldehyde-3-phosphate dehydrogenase gene sequences. Mycologia 91:964–977. https://doi.org/10.1080/00275514.1999.12061106
Devran Z, Kahveci E, Hong Y, Studholme DJ, Tör M (2018) Identifying molecular markers suitable for Frl selection in tomato breeding. Theor Appl Genet 131:2099–2105. https://doi.org/10.1007/s00122-018-3136-0
Grieneisen ML, Aegerter BJ, Stoddard CS, Zhang M (2018) Yield and fruit quality of grafted tomatoes, and their potential for soil fumigant use reduction. A meta-analysis. Agron Sustain Dev 38:29. https://doi.org/10.1007/s13593-018-0507-5
Hannon C, Weber G (1955) A leaf spot of tomato caused by Stemphylium floridanum sp. nov. Phytopathology 45
Hanson P, Lu S-F, Wang J-F, Chen W, Kenyon L, Tan C-W, Tee KL, Wang Y-Y, Hsu Y-C, Schafleitner R, Ledesma D, Yang RY (2016) Conventional and molecular marker-assisted selection and pyramiding of genes for multiple disease resistance in tomato. Sci Hortic 201:346–354. https://doi.org/10.1016/j.scienta.2016.02.020
Hendrix JW, Frazier WA (1949) Studies on the inheritance of Stemphylium resistance in tomatoes. Hawaii Agricultural Experiment Station, technical bulletin no. 8, pp 24. http://hdl.handle.net/10125/35662
Huang C-J, Tsai W-S (2017) Occurrence and identification of Stemphylium lycopersici causing Stemphylium leaf spot disease on tomato in Taiwan. Eur J Plant Pathol 148:35–44. https://doi.org/10.1007/s10658-016-1066-8
Ji Y, Scott J (2009) A CAPS marker linked to the tomato gray leaf spot (Stemphylium sp.) resistance gene Sm. TGC Rep 59:29–31
Jin Y, Statler G, Franckowiak J, Steffenson B (1993) Linkage between leaf rust resistance genes and morphological markers in barley. Phytopathology 83:230–233
Kim B-S, Cho H-J, Hwang H-S, Cha Y-S (1999) Gray leaf spot of tomato caused by Stephylium solani. Plant Pathol J 15:348–350
Kim B, Kim N, Kim JY, Kim BS, Jung H-J, Hwang I, Noua I-S, Sim S-C, Park Y (2016) Development of a high-resolution melting marker for selecting Fusarium crown and root rot resistance in tomato. Genome 59:173–183. https://doi.org/10.1139/gen-2015-0115
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549. https://doi.org/10.1093/molbev/msy096
Lee JM, Oh C-S, Yeam I (2015) Molecular markers for selecting diverse disease resistances in tomato breeding programs. Plant Breeding and Biotechnology 3:308–322. https://doi.org/10.9787/PBB.2015.3.4.308
Min J, Kim B, Cho K, Yu S (1995) Grey leaf spot caused by Stemphylium lycopersici on tomato plants. Korean J Plant Pathol (Korea Republic)
Pandey KK, Pandey PK, Kalloo G, Banerjee MK (2003) Resistance to early blight of tomato with respect to various parameters of disease epidemics. J Gen Plant Pathol 69:364–371. https://doi.org/10.1007/s10327-003-0074-7
Porta-Puglia A (1981) Stemphylium vesicarium (Wallr.) Simmons on tomato in the marches. Ann Istit Sper Patol Veg Roma 7:39–46
Rotem J, Cohen Y, Wahl I (1966) A new tomato foliage disease in Israel caused by Stemphylium botryosum. Can J Plant Sci 46:265–270
Singh RP, Hodson DP, Jin Y, Huerta-Espino J, Kinyua MG, Wanyera R, Njau P, Ward RW (2006) Current status, likely migration and strategies to mitigate the threat to wheat production from race Ug99 (TTKS) of stem rust pathogen. CAB Reviews 1:504. https://doi.org/10.1079/PAVSNNR20061054
Stam R, McDonald BA (2018) When resistance gene pyramids are not durable—the role of pathogen diversity. Mol Plant Pathol 19:521–524. https://doi.org/10.1111/mpp.12636
Su X, Zhu G, Huang Z, Wang X, Guo Y, Li B, Du Y, Yang W, Gao J (2019) Fine mapping and molecular marker development of the Sm gene conferring resistance to gray leaf spot (Stemphylium spp.) in tomato. Theor Appl Genet 132:871–882. https://doi.org/10.1007/s00122-018-3242-z
Walker J (1930) Inheritance of Fusarium resistance in cabbage. J Agric Res 40:721–745
Weber GF (1930) Gray leaf spot of tomato caused by Stemphylium solani, sp. nov. Phytopathology 20
White TJ, Bruns T, Lee S, Taylor J (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR protocols: a guide to methods and applications. 18:315–322
Witsenboer H, Van de Griend E, Tiersma J, Nijkamp H, Hille J (1989) Tomato resistance to Alternaria stem canker: localization in host genotypes and functional expression compared to non-host resistance. Theor Appl Genet 78:457–462. https://doi.org/10.1007/BF00290828
Yang H, Zhao T, Jiang J, Wang S, Wang A, Li J, Xu X (2017) Mapping and screening of the tomato Stemphylium lycopersici resistance gene, Sm, based on bulked segregant analysis in combination with genome resequencing. BMC Plant Biol 17:266. https://doi.org/10.1186/s12870-017-1215-z
Funding
This research was supported by the Golden Seed Project (Center for Horticultural Seed Development, No. 2013007-05-1-SBF10), Ministry of Agriculture, Food and Rural Affairs (MAFRA), Ministry of Oceans and Fisheries (MOF), Rural Development Administration (RDA), and Korea Forest Service (KFS).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest/competing interests
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Table S1
Genotyping results of 138 tomato accessions using Sm-InDel and Sm-SNP. (XLSX 20 kb)
Fig. S1
Prediction of potential genes in the 80,294 bp genomic DNA of Solanum lycopersicum harboring the ORF9 sequence. (PDF 289 kb)
Fig. S2
Sequence alignment of ORF9 cloned from ‘Motelle’ and ‘Moneymaker’. (PDF 421 kb)
Rights and permissions
About this article

Cite this article
Park, J., Kwon, S., Park, G. et al. Host reaction of tomato varieties and applicability of Sm-linked DNA markers to Stemphylium lycopersici. Mol Breeding 40, 106 (2020). https://doi.org/10.1007/s11032-020-01188-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-020-01188-8