
Abstract
Soybean cyst nematode (SCN) (Heterodera glycines Ichinohe) is a highly recalcitrant endoparasite of soybean roots, causing more yield loss than any other pest. To identify quantitative trait loci (QTL) controlling resistance to SCN (HG type 2.5.7, race 1), a genome-wide association study (GWAS) was performed. The association panel, consisting of 120 Chinese soybean cultivars, was genotyped with 7189 single nucleotide polymorphism (SNPs). A total of 6204 SNPs with minor allele frequency >0.05 were used to estimate linkage disequilibrium (LD) and population structure. The mean level of LD measured by r 2 declined very rapidly to half its maximum value (0.51) at 220 kb. The overall population structure was approximately coincident with geographic origin. The GWAS results identified 13 SNPs in 7 different genomic regions significantly associated with SCN resistance. Of these, three SNPs were localized in previously mapped QTL intervals, including rhg1 and Rhg4. The GWAS results also detected 10 SNPs in 5 different genomic regions associated with SCN resistance. The identified loci explained an average of 95.5% of the phenotypic variance. The proportion of phenotypic variance was due to additive genetic variance of the validated SNPs. The present study identified multiple new loci and refined chromosomal regions of known loci associated with SCN resistance. The loci and trait-associated SNPs identified in this study can be used for developing soybean cultivars with durable resistance against SCN.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Soybean cyst nematode (SCN) (Heterodera glycines Ichinohe) is a plant-parasitic nematode that attacks the roots of soybeans. The nematode is the most devastating soybean pest worldwide, causing more than US$1.5 billion in yield losses annually in the USA alone (Wrather and Koenning 2006). The use of resistant cultivars and cultural practices, such as crop rotation, are the principal strategies to limit SCN damage (Chen et al. 2001). Therefore, the development of elite cultivars with SCN resistance is a major goal of soybean breeding programs. Although soybean breeders have been successful in incorporating resistance genes from a few plant introductions (PIs), such as PI88788 and Peking (PI548402), into commercial soybean cultivars, soybean germplasm still has a narrow genetic base of SCN resistance (Concibido et al. 2004). Due to the overuse of the few resistance sources, SCN populations have emerged which show increasing reproductive capacity on previously resistant soybean cultivars (Colgrove and Niblack 2008; Mitchum et al. 2007). Therefore, it is urgent to screen and incorporate new sources of resistance to SCN in breeding programs for effective management of SCN globally.
Understanding the genetic basis of SCN resistance is critical for developing resistant cultivars. Mapping quantitative trait loci (QTL) for SCN resistance with molecular markers is a basic tool for dissection of the genetic basis of soybean resistance to SCN. Traditional bi-parental mapping studies have identified a total of 164 QTL (http://www.soybase.org/search/index.php?search=true&result=qtl&qtl=SCN). Among these, the QTLs on chromosomes (Chr.) 18 and 8 (rhg1 and Rhg4, respectively) have been well studied (Concibido et al. 1997; Matthews et al. 1998; Meksem et al. 2001; Guo et al. 2005, 2006; Kim et al. 2010) and are involved in resistance to SCN races 1 to 5. Recently, genes underlying the loci have been cloned (Cook et al. 2012; Liu et al. 2012). However, due to the limited coverage of resistance sources, inadequate statistical procedures, and small mapping populations, many QTLs underlying SCN resistance, therefore, have remained un-detected. With the availability of a soybean reference genome sequence and the development of high-throughput single nucleotide polymorphism (SNP) assay platforms, association mapping (AM) has been demonstrated to be a powerful approach to dissect various traits, such as disease resistance, yield, and quality-related traits in soybean (Wen et al. 2014, 2015; Hao et al. 2012a, b; Bastien et al. 2014; Hwang et al. 2014). Recent genome-wide association study (GWAS) on 282 accessions of soybean breeding lines have detected significant signals at two loci corresponding to rhg1 and FGAM1, plus a third locus located at the end (55.9 Mb) of Chr. 18 underlying resistance to SCN HG type 0 (Bao et al. 2014). By using a data set of 55,159 SNPs across 440 diverse soybean landraces and elite cultivars, Han et al. (2015) also identified 19 association loci significantly related to the resistance for two SCN HG types. Another GWAS was performed by using a diverse set of 553 soybean PIs belonging to maturity groups from III to V to detect QTL/genes associated with SCN resistance to HG type 0 (Vuong et al. 2015). They confirmed six QTL that were previously mapped using bi-parental populations and also identified eight novel QTL.
SCN HG type 2.5.7 (race 1) is the predominant race in the Huang-Huai region (between 32° N and 41° N latitude) of China (Lu et al. 2006). To deploy the major SCN resistance genes of this race in breeding germplasm, understanding the genetic basis of resistance to SCN in soybean is essential. To the best of our knowledge, GWAS has not yet been employed to study the resistance of this HG type in soybean. By selecting a representative collection of soybean cultivars, the objective of this study was to apply GWAS to dissect the genetic architecture of resistance to SCN in a panel of 120 diverse soybean varieties. To this end, the soybean accessions were genotyped with a high-quality custom-designed 7189 oligonucleotide genotyping array, and phenotyped for plant responses to SCN HG type 2.5.7 (race 1). The present research will aid efforts to dissect the genetic architecture of SCN resistance in soybean.
Materials and methods
Sampling and genotyping
One hundred twenty released cultivars representing maturity groups (MGs) II to IV were collected and used in this study (Supplementary Table S1). DNA was extracted from young leaves of each accession using standard CTAB method (Kisha et al. 1997). DNA sample from each accession was genotyped with SoySNP8k iSelect BeadChip (Illumina, San Diego, Calif., USA), which consists of 7189 SNPs. The quality of each SNP was checked manually as previously reported by Yan et al. (2010). The SNPs with >5% minor allele frequency (MAF) and a missing data rate <20% were retained. A total of 6204 SNPs markers passed the filters and were used in the subsequent analyses.
Phenotyping
A greenhouse assay of plant responses to SCN HG type 2.5.7 (race 1) was performed at the Shandong Academy of Agricultural Sciences in the summer of 2013 using established methods (Riggs and Schmitt 1988). The greenhouse experiment was run twice with 20 plants for each accession in first run and 10 plants for second run. Three seeds of each accession were planted in 120- by 150-mm plastic pot with 10 replications. After germination, the seedlings were thinned to two seeding per pot for first run and one seedling for second run. Thirty days after inoculation, the plant roots were washed and the cysts on the roots were counted. Resistance evaluations were conducted by counting the number of cysts (NC) on the roots and calculating the index of parasitism (PI) (Schmitt and Shannon 1992) using Jindou 21 as susceptible control line. The linear model for PI was y ij = u + g i + r j + (gr) ij + e ij , where y ij was the PI of each plant, u was the total mean, g i was the genetic effect of ith genotype, r j was the mean effect of jth run, (gr) ij was the interaction effect between the ith accession and jth run, and e ij was the residual. The heritability estimates were calculated using variance components obtained by the abovementioned linear model (Nyquist 1991).
Population genetic analyses
Principal component analysis and neighbor-joining tree were applied to infer population stratification. A pairwise distance matrix derived from a modified Euclidean distance for all polymorphic SNPs was calculated to construct neighbor-joining trees using the TASSEL 5.0 software (Bradbury et al. 2007). Principal component analysis was done using TASSEL 5.0 based on 4549 SNPs with minor allele frequency (MAF) >20% and physical distance >60 kb. Kinship matrixes were calculated using TASSEL 5.0 to determine relatedness among individuals based on the same sets of SNPs. TASSEL 5.0 was used to make all pairwise comparisons of alleles to calculate squared correlation coefficient (r 2) of alleles between markers. The extent of linkage disequilibrium (LD) decay was measured as the chromosomal distance at which the average pairwise correlation coefficient (r 2) dropped to half its maximum value.
Genome-wide association analysis
GWAS were performed with a mixed linear model (MLM, Zhang et al. 2010), to test the associations between the SNPs (MAF > 5%) and the resistance variations. The top five principal components were used to build up the P matrix for population structure correction. Analyses were performed by the software TASSEL 5.0. False discovery rate (FDR) ≤ 0.05 was used to identify significant associations. To select major QTLs among all the significant SNPs, these SNPs were clumped by using an LD block as a criterion (Gabriel et al. 2002), and the strongest association within each LD block was kept. In order to make sure that the present sample size has enough power to conduct GWAS, GWApower (Feng et al. 2011) was used to estimate the power of the present study with a SNP number, heritability score, and sample size with 6204, 0.81, and 120, respectively.
Results
Analysis of phenotypic variation
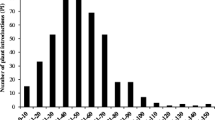
Averaged over two runs, a large variation in SCN resistance was observed across assayed soybean accessions. NC on the root had a mean of 30.5 with a range from 1 to 83, whereas index of parasitism (IP) ranged from 1.8 to 120 with a mean of 43 (Fig. 1). ANOVA for the IP indicated that the effect of accession, run, and accession by run had significant effects (Table 1, Supplementary Fig. S1). The broad-sense heritability estimates for NC and IP were 0.74 and 0.81, respectively, not only indicating that the number of cysts per plant within inbred lines was relatively consistent but also suggesting that there was a major genetic component conditioning SCN resistance in this population. Having high heritability and expecting few genetic loci contributing to the phenotypic variation, the population size of 120 lines should be adequate to detect the QTL.

Number of cyst nematode (NC, a) and index of parasitism (IP, b) for 120 soybean accessions. R resistant (IP < 10), red, MR moderately resistant (10 < IP < 30), blue, MS moderately susceptible (30 < IP < 60), green, S susceptible (IP > 60), brown (color figure online)
Totally, 70% of the tested accessions were moderately susceptible to completely susceptible to SCN HG type 2.5.7 (race 1), with only six accessions being highly resistant (IP < 10) and 30 accessions being moderately resistant (10 < IP < 30; Fig. 1). We checked the pedigree information of the six accessions with high resistance level to SCN, and then found that four of them have Peking as an ancestral resistance source.
Patterns of linkage disequilibrium and profile of population structure
A total of 6204 SNPs distributed across the soybean genome with minor allele frequency > 0.05 and missing data of less than 20% were used for the estimation of LD level in the association panel. These SNP markers spanned 934 Mbp, which represents approximately 84.9% of the 1100-Mbp soybean genome, resulting in an average SNP density of 1 SNP every 162 kilobase (kb). The mean level of LD measured by r 2 declined very rapidly to half its maximum value (0.51) at 220 kb (Supplementary Fig. S2). Therefore, the estimate of LD decay rate is approximately 220 kb in our diversity panel.
Since population structure can result in spurious association, it has constrained the use of association studies in human and plant genetics (Yu et al. 2006). To understand the geographic structure of genetic diversity and population stratification in the diversity panel, neighbor-joining (NJ) tree plots and principal components analysis (PCA) were applied to determine the relatedness among the sampled accessions (Fig. 2). The resulting NJ trees and PCA plots showed that the landraces had four subgroups. Based on the analysis of origin of each accession for every subgroup, we found that overall population structure was approximately coincident with geographic origin (Supplementary Table S2). For example, accessions from Sichuan province, developed in the southwest of China, dominate subgroup 1; subgroup 3 contained 73 accessions, of which 49 were from Huang-Huai river region of China. The chi-squared test was used to test whether the four SNP data-based subgroups were associated with geographic origin (Supplementary Table S2). The results showed very significant association (P < 0.0001) between the two grouping factors. These results show population structuring along geographic origin, confirming previous analyses (Li et al. 2010; Hao et al. 2012a, b) and providing additional insights into the fine-scale patterns of ancestry resulting from regional soybean breeding efforts. Taken together, these results highlight the need to account for population structure when performing association analyses in soybean.

Population structures of soybean cultivars in the association panel. a Neighbor-joining tree of 120 accessions. b PCA plots of the first two components of 120 accessions. The four subgroups identified from the tree are color coded in (a) and (b) (color figure online)
GWAS for SCN resistance
GWAS was conducted by using the mean value of IP over two runs in a MLM, which accounts for both population structure (top five PCs) and familial relatedness (K). A total of 13 SNPs significantly associated with IP were identified (Table 2 and Fig. 3a). Since the distribution of observed −log10 P values from MLM did not depart from the expected distribution under a model of no association with significant inflation of nominal P values (Fig. 3b), our MLM model showed good control of type I error rate. Moreover, after running GWApower (Feng et al. 2011) with three parameters specific to present study (SNP number, heritability, and sample size of 6204, 0.81, and 120, respectively), the results showed that the theoretical sample size is 126 to reach the power = 1.0. Given that the sample size of the present study is 120, we believe that the present study has enough power to detect significant SNP associated with SCN resistance.

Genome-wide association study of SCN in the association panel. a Manhattan plots of the MLM model for IP in the association panel. The −log10 P values from a genome-wide scan are plotted against the position on each of the 20 chromosomes. The horizontal red line indicates the genome-wide significance threshold (FDR < 0.05). b Quantile-quantile (QQ) plot of simple model for IP in the association panel. c Number of resistance allele possessed by soybean cultivars and corresponding average IP (color figure online)
After the clumping of SNPs within same LD block, we identified seven significant loci associated with SCN race 3 resistance on the Chr. 8, 11, 13, 17, and 18 (Table 2). The peak SNPs at the identified loci explained approximately 90% of the total phenotypic variance with FDR < 0.05. Since the variance explained by the validated SNPs is similar with the heritability, therefore, there is no failure of the validated SNP associations to explain the estimated heritability like the previous GWAS result (Han et al. 2015). Note that the known loci rhg1 on Chr. 18 and Rhg4 on Chr. 8 were identified in this study (locus no. 1 and locus no. 6). Moreover, locus no. 3 (Gm11-18083338), on Chr. 11, fell within the interval of previously described QTL associated with resistance to SCN race 1 in a bi-parental mapping population derived from a PI 89772 × Hamilton cross (Yue et al. 2001). Locus no. 7 (Gm18-5112839) was not only within the previously reported interval (Winter et al. 2007) but also 4 Mb away form an SNP marker (BARC-019001-03050) tagging a previously described locus associated with resistance to SCN HG type 0 (Bao et al. 2014). Apart from these overlaps, our GWAS identified 3 novel QTL, including QTL on Chr. 8 (locus no. 2), 13 (locus no. 4), and 17 (locus no. 5), which have not been previously mapped by using a bi-parental population. The gene that is nearest to the peak SNP in each locus was listed in Table 2 as the most likely candidate gene underlying SCN resistance. Three candidate gene identified in the novel GWAS loci are Glyma.08g303900, Glyma.13g175200, and Glyma.17g010200. Among them, Glyma.08g303900 codes for a sterol regulatory element-binding protein (SREBP). A homologous gene in Arabidopsis plays a role in the regulation of endodermal plastid size and number. These plasmids are involved in ethylene-dependent gravitropism of light-grown hypocotyls (Guo et al. 2008). Glyma.13g175200, a GTP-binding related gene, is believed to be associated with resistance genes (R-genes) and regulates the activity of an NADPH oxidase complex in plants (Grant and Loake 2000). Glyma.17g010200 is annotated as homologous to homeobox transcription factor genes previously shown to be involved in response to aphid feeding (Smith and Boyko 2006).
To facilitate molecular breeding, it is necessary to determine which allele is the resistant allele. Therefore, average IP value of the soybean accessions that possessed each allele of significant SNPs was calculated. The resistance alleles of each significant SNP were identified (Table 2). The relationships between number of resistance allele possessed by soybean cultivars and corresponding average IP was showed in Fig. 3c. It clearly showed that the proportion of phenotypic variance is due to additive genetic variance of the validated SNPs. Since our GWAS model is still based on single-locus tests, we cannot exclude the possibility that structural variation, such as copy number variation, or other compound gene interaction may have functional importance.
Discussion
During the past few years, GWAS has been now well established in soybean with the availability of high-throughput genotyping platforms such as SNP array or genotyping by sequencing (GBS). The inbreeding nature of soybean has resulted in a long-range LD decay. The long extent of LD decay estimate (220 kb) is very similar with previously published values (Wen et al. 2014). The estimates of LD decay herein suggests that at least 5000 markers will be needed for whole genome scanning in soybean, as the soybean genome is known to extend over 1000 Mb. Since the number of polymorphic marker in this panel is 6204, which ensures the coverage of most LD block and reasonable power to identify common variants of large effect associated with SCN resistance. Note that LD decay varies across different chromosomes and even within heterochromatic and euchromatic chromosome regions. Sonah et al. (2015) has shown LD for each of the chromosomes separately and observed range from 250 kb to 2.5 Mb. Hwang et al. (2014) identified that the LD decay rate in heterochromatic and euchromatic chromosome regions was 360 and 9600 kb, respectively. On one hand, population with long extent of LD is amenable to coarse mapping with fewer markers (Abdurakhmonov and Abdukarimov 2008). On the other hand, LD decay rate is also the primary factor limiting the mapping resolution in GWAS for soybean. Therefore, the low rate of LD decay in the present study also may lead to resolution limitations for the following association mapping.
In soybean, QTL on Chr. 18 (rhg1) and 8 (Rhg4) have been consistently mapped in a bi-parental population and a variety of soybean germplasm (Meksem et al. 2001; Kim et al. 2010; Liu et al. 2012; Han et al. 2015; Vuong et al. 2015). Two loci, rhg1 and Rhg4, represent the major sources of resistance in soybean cultivars (Concibido et al. 2004). Our association mapping results for SCN resistances were compared with the QTL and GWAS hit loci that were previously reported in SoyBase (http://www.soybase.org/search/index.php?search=true&result=qtl&qtl=SCN) and in literature (Supplementary Fig. S3). Of the seven loci identified in present study, four overlapped with previously identified genes (or QTLs) (Table 2). Notably, one significant SNP on Chr. 18 (Gm18-1979161) associated with IP was found to be located at 290 kb upstream of three key genes (Glyma.18g022400, Glyma.18g022500, and Glyma.18g022700) in rhg1, which consistently contributes much more effective SCN resistance than any other known loci (Cregan et al. 1999). The distance from marker to causal genes (rhg1) in our study was shorter than that (350 kb) reported by Vuong et al. (2015), but longer than that (5 kb) reported by Han et al.(2015). Note that the two functional SNP markers (GSM381 and GSM383) have been developed from Glyma.18g022500 for the selection of the rhg1 resistance (Shi et al. 2015). Another significant SNP on Chr. 8 (Gm08-8281564) associated with IP was found to be located within an exon of one putative rhg4 gene (Glyma.08g107700, Lightfoot and Meksem 2002), but 110 kb away from another gene (Glyma.08g108900) identified by Liu et al. (2012). Moreover, Gm18-1979161and Gm08-8281564 also overlapped with the same loci associated with resistance to SCN HG type 0 (Han et al. 2015), confirming that they can be used as molecular markers to accelerate the development of soybean varieties with the resistance to both SCN HG type 0 and HG type 2.5.7. Previous studies have identified that Peking needs both rhg1 and Rhg4 resistance alleles for a resistant phenotype (Liu et al. 2012; Meksem et al. 2001). As mentioned earlier, a few resistance sources in this panel derive resistance from Peking. The identification of significant SNPs in both rhg1 and Rhg4 regions is what we expected and reasonable. Apart from the two loci, we also identified two significant SNPs, Gm18-51128392 and Gm11-18083338 on Chr. 11 and 18, respectively. The two loci explained about 30% of the phenotypic variation, which were also located at the interval of previously identified QTL underlying resistance to SCN race 1 (Yue et al. 2001). In the present study, Gm08-41568898 on Chr. 8 (LG A2), Gm13-27796524 on Chr. 13 (LG F), and Gm17-835879 on Chr. 17 (LG B2) were the novel loci that have not been previously mapped by using bi-parental populations. However, Gm08-41568898 was approximately 400 kb away from an SNP marker (rs41643371) tagging a previously GWAS hit loci associated with resistance to SCN HG type 0 (Han et al. 2015). They have the potential to be used in MAS for developing soybean cultivars with more durable resistance against SCN HG type 2.5.7.
Since marker-assisted selection using DNA markers is currently cost-effective compared with greenhouse screening, it is useful to determine the beneficial allele of each peak SNP associated with SCN resistance. Although no accession has all 13 resistance alleles, however, the average IP values of accessions with resistant alleles gradually decrease with the increasing of resistance allele possessed by soybean accessions. In other words, the more resistance alleles one accession possesses, the higher resistance level the accession has. Pedigree information of the six accessions with high resistance level to SCN showed that two of them do not have Peking or PI 88788 as ancestral resistance source. Moreover, there are 30 accessions which exhibited moderate resistance (10 < IP < 30). We believe that the use of these germplasm with different genes for resistance will broaden genetic diversity and stabilize yield for soybean. Undoubtedly, the loci and trait-associated SNPs identified in this study will facilitate developing soybean cultivars with durable resistance against SCN. Those resistant alleles identified in the present study could be useful for marker-assistant selection (MAS) of SCN resistance. In the past, the development of the cultivars carrying SCN resistance genes is labor intensive and costly. The advantage of MAS over conventional SCN screening was obvious. Mudge et al. (1997) showed that with using two SSR markers, Satt038 and Satt130, they were 98% accurate in identifying resistant lines. Recently, three robust Kompetitive Allele Specific PCR (KASP) SNP marker assays (GSM381, GSM383, and GSM191) were developed and proved to be high throughput and efficient for MAS in identifying plants with resistance of SCN race 3 (Shi et al. 2015). These SNP markers have been validated, and a strong correlation was observed between the SNP genotypes and the reactions to SCN race 3 using a panel of 153 soybean lines, as well as a bi-parental population, F5-derived recombinant inbred lines (RILs) from G00-3213 × LG04-6000 (Shi et al. 2015).
Taken together, the results of this work have provided SNPs for the application for high-throughput selection of SCN resistance. GWAS was successfully used to examine the genetic architecture of resistance to SCN HG type 2.5.7 in multiple genetic backgrounds. A total of seven loci were obtained for SCN resistance to this HG type and will expand the diversity for SCN resistance genes in soybean cultivars. The multiple beneficial alleles showed additive genetic variance and may be useful to breed varieties with improved resistance to SCN.
References
Abdurakhmonov I, Abdukarimov A (2008) Application of association mapping to understanding the genetic diversity of plant germplasm resources. Int J Plant Genom Vol 2008:1–18
Bao Y, Vuong T, Meinhardt C, Tiffin P, Denny R, Chen S, Nguyen HT, Orf JH, Young ND (2014) Potential of association mapping and genomic selection to explore PI 88788 derived soybean cyst nematode resistance. The Plant Genome 7(3):1–13. doi:10.3835/plantgenome2013.11.0039
Bastien M, Sonah H, Belzile F (2014) Genome wide association mapping of Sclerotinia sclerotiorum resistance in soybean with a genotyping-by-sequencing approach. Plant Genome 7(1):1–13
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635
Chen S, Porter PM, Reese CD, Stienstra WC (2001) Crop sequence effects on soybean cyst nematode and soybean and corn yields. Crop Sci 41:1843–1849. doi:10.2135/cropsci2001.1843
Colgrove AL, Niblack TL (2008) Correlation of female indices from virulence assays on inbred lines and field populations of Heterodera glycines. J Nematol 40:39–45
Concibido VC, Lange D, Denny RL, Orf J, Young N (1997) Genome mapping on soybean cyst nematode resistance genes in ‘Peking’, PI 90763, and PI 88788 using DNA markers. Crop Sci 37:258–264
Concibido VC, Diers BW, Arelli PR (2004) A decade of QTL mapping for cyst nematode resistance in soybean. Crop Sci 44:1121–1131
Cook DE, Lee TG, Guo X, Melito S, Wang K, Bayless AM, Wang J, Hughes TJ, Willis DK, Clemente TE, Diers BW, Jiang J, Hudson ME, Bent AF (2012) Copy number variation of multiple genes at Rhg1 mediates nematode resistance in soybean. Science 338(6111):1206–1209
Cregan PB, Mudge J, Fickus EW, Danesh D, Denny R, Young ND (1999) Two simple sequence repeat markers to select for soybean cyst nematode resistance conditioned by the rhg1 locus. Theor Appl Genet 99:811–818
Feng S, Wang S, Chen CC, Lan L (2011) GWAPower: a statistical power calculation software for genome-wide association studies with quantitative traits. BMC Genet 12:12
Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, Higgins J, DeFelice M, Lochner A, Faggart M, Liu-Cordero SN, Rotimi C, Adeyemo A, Cooper R, Ward R, Lander ES, Daly MJ, Altshuler D (2002) The structure of haplotype blocks in the human genome. Science 296:2225–2229
Grant J, Loake GJ (2000) Role of reactive oxygen intermediates and cognate redox signaling in disease resistance. Plant Physiol 124(1):21–30
Guo B, Sleper DA, Arelli PR, Shannon JG, Nguyen HT (2005) Identification of QTLs associated with resistance to soybean cyst nematode races 2, 3 and 5 in soybean PI 90763. Theor Appl Genet 111:965–971
Guo B, Sleper DA, Lu P, Shannon JG, Nguyen HT, Arelli PR (2006) QTLs associated with resistance to soybean cyst nematode in soybean: meta-analysis of QTL locations. Crop Sci 46:595–604
Guo D, Gao XR, Li H, Zhang T, Chen G, Huang PB, An LJ, Li N (2008) EGY1 plays a role in regulation of endodermal plastid size and number that are involved in ethylene-dependent gravitropism of light-grown Arabidopsis hypocotyls. Plant Mol Biol 66(4):345–360
Han Y, Zhao X, Cao G, Wang Y, Li Y, Liu D, Teng W, Zhang Z, Li D, Qiu L, Zheng H, Li W (2015) Genetic characteristics of soybean resistance to HG type 0 and HG type 1.2.3.5.7 of the cyst nematode analyzed by genome-wide association mapping. BMC Genomics 16(1):598. doi:10.1186/s12864-015-1800-1Aug 13
Hao D, Cheng H, Yin Z, Cui S, Zhang D, Wang H, Yu D (2012a) Identification of single nucleotide polymorphisms and haplotypes associated with yield and yield components in soybean (Glycine max) landraces across multiple environments. Theor Appl Genet 124:447–458
Hao D, Chao M, Yin Z, Yu D (2012b) Genome-wide association analysis detecting significant single nucleotide polymorphisms for chlorophyll and chlorophyll fluorescence parameters in soybean (Glycine max) landraces. Euphytica 186(3):919–931
Hwang E-Y, Song Q, Jia G, Specht J, Hyten D, Costa J, Cregan PB (2014) A genome-wide association study of seed protein and oil content in soybean. BMC Genomics 15(1):1–12
Kim M, Hyten DL, Bent AF, Diers BW (2010) Fine mapping of the SCN resistance locus rhg1-b from PI 88788. The Plant genome 3:81–89
Kisha T, Sneller CH, Diers BW (1997) Relationship between genetic distance among parents and genetic variance in populations of soybean. Crop Sci 37:1317–1325
Li YH, Li W, Zhang C, Yang L, Chang RZ, Gaut BS, Qiu LJ (2010) Genetic diversity in domesticated soybean (Glycine max) and its wild progenitor (Glycine soja) for simple sequence repeat and single-nucleotide polymorphism loci. New Phytol 188:242–253
Lightfoot D, Meksem K (2002) US Patent Application Publication Number 2002144310
Liu S, Kandoth PK, Warren SD, Yeckel G, Heinz R, Alden J, Yang C, Jamai A, EI-Mellouki T, Juvale PS, Hill J, Baum TJ, Cianzio S, Whitham SA, Korkin D, Mitchum MG, Meksem K (2012) A soybean cyst nematode resistance gene points to a new mechanism of plant resistance to pathogens. Nature 492(7428):256–260
Lu W, Gai J, Li W (2006) Inheritance of resistance to race 1 and race 4 of cyst nematode (Heterodera glycines Ichinohe) in soybean. Acta Agron Sin 32(5):650–655
Matthews BF, Mac Donald MH, Genhardt JS, Devine TE (1998) Molecular markers residing close to the Rhg4 locus conferring resistance to soybean cyst nematode race 3 on linkage group A of soybean. Theor Appl Genet 97:1047–1052
Meksem K, Pantazopoulos P, Njiti VN, Hyten LD, Arelli PR, Lightfoot DA (2001) ‘Forrest’ resistance to the soybean cyst nematode is bigenic: saturation mapping of the Rhg1 and Rhg4 loci. Theor Appl Genet 103:710–717
Mitchum MG, Wrather JA, Heinz RD, Shannon JG, Danekas G (2007) Variability in distribution and virulence phenotypes of Heterodera glycines in Missouri during 2005. Plant Dis 91:1473–1476
Mudge J, Cregan PB, Kenworthy JP, Kenworthy WJ, Orf JH, Young ND (1997) Two microsatellite markers that flank the major soybean cyst nematode resistance locus. Crop Sci 37:1611–1615
Nyquist WE (1991) Estimation of heritability and prediction of selection response in plant populations. Crit Rev Plant Sci 10:235–322
Riggs RD, Schmitt DP (1988) Complete characterization of the race scheme for Heterodera glycines. J Nematol 20(3):392–395
Schmitt D, Shannon G (1992) Differentiating soybean responses to Heterodera glycines races. Crop Sci 32:275–277. doi:10.2135/cropsci19 92.0011183X003200010056x
Shi Z, Liu S, Noe J, Arelli P, Meksem K, Li Z (2015) SNP identification and marker assay development for high-throughput selection of soybean cyst nematode resistance. BMC Genomics 16:314
Smith CM, Boyko EV (2007) The molecular bases of plant resistance and defense responses to aphid feeding: current status. Entomol Exp Appl 122(1):1–16
Sonah H, O’Donoughue L, Cober E, Rajcan I, Belzile F (2015) Identification of loci governing eight agronomic traits using a GBS-GWAS approach and validation by QTL mapping in soya bean. Plant Biotechnol J 13(2):211–221
Vuong TD, Sonah H, Meinhardt CG, Deshmukh R, Kadam S, Nelson RL, Shannon JG, Nguyen HT (2015) Genetic architecture of cyst nematode resistance revealed by genome-wide association study in soybean. BMC Genomics 16:593. doi:10.1186/s12864-015-1811-y
Wen Z, Tan R, Yuan J, Bales C, Du W, Zhang S, Chilvers MI, Schmidt C, Song Q, Cregan PB, Wang D (2014) Genome-wide association mapping of quantitative resistance to sudden death syndrome in soybean. BMC Genomics 15(1):809
Wen Z, Boyse JF, Song Q, Cregan PB, Wang D (2015) Genomic consequences of selection and genome-wide association mapping in soybean. BMC Genomics 16:671. doi:10.1186/s12864-015-1872-ySep 3
Winter SMJ, Shelp BJ, Anderson TR, Welacky TW, Rajcan I (2007) QTL associated with horizontal resistance to soybean cyst nematode in Glycine soja PI464925B. Theor Appl Genet 114(3):461–472
Wrather JA, Koenning SR (2006) Estimates of disease effects on soybean yields in the United States 2003 to 2005. J Nematol 38(2):173–180
Yan J, Yang X, Shah T, Sánchez-Villeda H, Li J, Warburton M, Zhou Y, Crouch JH, Xu Y (2010) High-throughput SNP genotyping with the Golden Gate assay in maize. Mol Breed 25:441–451
Yu J, Pressoir G, Briggs WH, Vroh Bi I, Yamasaki M, Doebley JF, McMullen MD, Gaut BS, Nielsen DM, Holland JB, Kresovich S, Buckler ES (2006) A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet 38:203–208
Yue P, Sleper DA, Arelli PR (2001) Mapping resistance to multiple races of Heterodera glycines in soybean PI 89772.2001. Crop Sci 41(5):1589–1595
Zhang Z, Ersoz E, Lai CQ, Todhunter RJ, Tiwari HK, Gore MA, Bradbury PJ, Yu J, Arnett DK, Ordovas JM, Buckler ES (2010) Mixed linear model approach adapted for genome-wide association studies. Nat Genet 42:355–360
Acknowledgments
This work was supported by the State Key Basic Research and Development Plan of China (no. 2006A10A111); National Natural Science Foundation of China (no. 30971815); MOE 111 Project (no. B08025); China Agriculture Research System (CARS-04-CES18); China Postdoctoral Science Foundation Funded Project (no. 2011M500717); Shandong Provincial Natural Science Foundation, China (ZR2012CM034); and Postdoctoral Innovation Project Special Foundation of Shandong Province (no. 201103022). We thank Dr. Paul Collins for language editing.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Supplementary Table S1
(XLSX 16 kb)
Supplementary Table S2
(DOCX 15 kb)
Supplementary Figure S1
Comparison of number of cysts evaluated in two rounds (GIF 62 kb)
Supplementary Figure S2
Genome-wide average LD decay estimated in association panel. Decay of LD (measured as r2) as a function of distance between SNPs (GIF 273 kb)
Supplementary Figure S3
A composite map of earlier reported GWAS loci and loci identified in present study. (GIF 47 kb)
Rights and permissions
About this article

Cite this article
Zhang, J., Wen, Z., Li, W. et al. Genome-wide association study for soybean cyst nematode resistance in Chinese elite soybean cultivars. Mol Breeding 37, 60 (2017). https://doi.org/10.1007/s11032-017-0665-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-017-0665-1