
Abstract
Cholinergic, oxidative, nitrergic alterations, and neuroinflammation are some key neuropathological features common in schizophrenia disease. They involve complex biological processes that alter normal behavior. The present treatments used in the management of the disorder remain ineffective together with some serious side effects as one of their setbacks. Taurine is a naturally occurring essential β-amino acid reported to elicit antipsychotic property in first episode psychosis in clinical setting, thus require preclinical investigation. Hence, we set out to investigate the effects of taurine in the prevention and reversal of ketamine-induced psychotic-like behaviors and the associated putative neurobiological mechanisms underlying its effects. Adult male Swiss mice were sheared into three separate cohorts of experiments (n = 7): drug alone, preventive and reversal studies. Treatments consisted of saline (10 mL/kg/p.o./day), taurine (50 and 100 mg/kg/p.o./day) and risperidone (0.5 mg/kg/p.o./day) with concomitant ketamine (20 mg/kg/i.p./day) injections between days 8–14, or 14 days entirely. Behavioral hyperactivity, despair, cognitive impairment, and catalepsy were measured. Brain oxidative/nitrergic imbalance, immunoreactivity (COX-2 and iNOS), and cholinergic markers were determined in the striatum, prefrontal-cortex, and hippocampus. Taurine abates ketamine-mediated psychotic-like episodes without cataleptogenic potential. Taurine attenuated ketamine-induced decrease in glutathione, superoxide-dismutase and catalase levels in the striatum, prefrontal-cortex and hippocampus. Also, taurine prevented and reversed ketamine-mediated elevation of malondialdehyde, nitrite contents, acetylcholinesterase activity, and suppressed COX-2 and iNOS expressions in a brain-region dependent manner. Conclusively, taurine insulates against ketamine-mediated psychotic phenotype by normalizing brain central cholinergic neurotransmissions, oxidative, nitrergic and suppression of immunoreactive proteins in mice brains.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Schizophrenia is a very major neurological disease with wide range of genetic and epigenetic backgrounds (Wang et al. 2015). It is characterized by heterogeneous clinical manifestations such as hyperactivity, affective flattening, learning and memory impairments (Gobira et al. 2013). The disease has wide range of pathological features but not limited to alterations in central neurochemical functions (Chatterjee et al. 2012; Ben-Azu et al. 2018a, b). However, malnutrition, early life adverse events and negative peer group social stimulus have been identified as linkages for the onset of schizophrenia (Graham et al. 2015; Shivakumar et al. 2015; Yang et al. 2019), possibly by pushing vulnerable persons above their behavioral elastic limit into a schizophrenic state (Sánchez-Ramón et al. 2021). Although the brain appears as a small organ, it is the most metabolically active organ in the human body. Consequently, it generates a large amount of reactive oxygen and nitrergic species (ROS/RNS), of which both product moieties are believed to exacerbate neuronal excitotoxicity in different parts of the brain (de Araújo et al. 2021; Sánchez-Ramón et al. 2021). This is worsened by the oxidative potential of certain neurotransmitters like dopamine and glutamate, as well as their redox-sensitive receptors, including D2 dopamine and N-methyl-D-aspartate (NMDA) glutamate receptors (Nakao et al. 2021). The D2 and NMDA receptors have been recognized to play critical roles in the regulation of human behaviors and disease progression in certain neuropsychiatric conditions (Chatterjee et al. 2012; Monte et al. 2013; de Araújo et al. 2021; Nakao et al. 2021). This involves activation of microglia sensome, which is believed to exacerbate the release of pro-inflammatory cytokines including tumor necrosis factor-alpha (TNF-α) and interleukin (IL)-6, and expression of inflammatory proteins (e.g., cyclooxygenase-2, COX-2) (Yang et al. 2021; Zhang et al. 2021).
While polyunsaturated fatty acids (PUFAs) are known to play essential role in the functions and structural stability of neuronal membranes in the context of signal transduction mechanisms, synaptic plasticity, and behavioral developments (Yang et al. 2021), COX-2 on the other hand is largely reported to catabolize PUFAs and other metabolites that are involved in the regulation of the immune system (Zhang et al. 2021). Given the bilateral communication between COX-2 and PUFAs, growing evidence suggest a crossroad between the neuroinflammatory hypothesis and severity of schizophrenia disease (Frydecka et al. 2015), as well the mechanisms of action of some antipsychotic drugs (Cho et al. 2019; Mirabella et al. 2021; de Araújo et al. 2021). Notably, IL-6 is one of the pleiotropic cytokines released pathologically through the up-regulation of prostaglandin E2 (PGE2)-arachidonic acid pathway by COX-2 (Yang et al. 2021). It is known as one of the neuroinflammatory integrative factor of deterioration of schizophrenic disease (Frydecka et al. 2015; Ben-Azu et al. 2019). It also causes treatment resistance (Zhang et al. 2005; Potkin et al. 2020) via continuous disruption of neurotransmitter metabolism (Zalcman et al. 1994; Mirabella et al. 2021) including catabolism of tryptophan products (Nakao et al. 2021) and loss of synaptic machineries, including disassembling of cortical pyramidal neurons (Ben-Azu et al. 2019; Zhang et al. 2021). Deserves mentioning that many preclinical (Monte et al. 2013; Jeppesen et al. 2020) and clinical (Liu et al. 2014; Attari et al. 2017; Hong and Bang 2020) studies have hypothesized that anti-inflammatory drugs such as minocycline and non-steroidal anti-inflammatory drugs such as celecoxib as potential candidates in the treatment of schizophrenia patients (Müller et al. 2010). Notably, risperidone, a popular atypical antipsychotic drug and other counterparts have been largely reported to inhibit microglia release of pro-inflammatory cytokines such as TNF-α, IL-6, with suppression of COX-2, PGE2, and nuclear factor kappa-B (NF-kB) (MacDowell et al. 2013; Zhu et al. 2014; Ben-Azu et al. 2019; Racki et al. 2021). Additionally, previous reports showed that the anti-neuroinflammatory effect of risperidone alone (Ben-Azu et al. 2019), or in combination with minocycline (Zhu et al. 2014) resulted in protection of cortical pyramidal neurons and restoration of the negative and cognitive symptoms in mice exposed to single- or two-hit model of schizophrenia.
Furthermore, dysfunctional mitochondrial characterized by distorted oxidative phosphorylation, mitochondrial hypoplasia, and altered expression of mitochondria-related genes due to nutritional malnutrition, have also been identified as pathogenic factor for the progression of schizophrenia (Ben-Shachar and Laifenfeld 2004; Silva et al. 2019; Aucoin et al. 2020). There are evidences of irregular cellular metabolism and nitrergic-derived pro-oxidant in the prefrontal cortex of schizophrenia patients, including increased cellular hypoxia and glucose under-utilization (Ben-Shachar and Laifenfeld 2004; Silva et al. 2019). Notably, nitric oxide (NO) is an important pro-oxidant messenger molecule that is involve in different physiological and pathological activities involving oxidative damage, excitotoxicity, and peroxidation (Shahani and Sawa 2011; Sahebnasagh et al. 2022). NO is synthesized by three isoforms of nitric oxide synthases (NOS). In the brain, one of these isoforms called inducible NOS (iNOS) is highly expressed in the sensomes of inflammatory modulators such as microglial cells, infiltrating microphages, T-lymphocytes, as well as blood vessels of brain endothelium where it regulate cerebral blood flow (Szabadits et al. 2011; Sahebnasagh et al. 2022). However, iNOS-induced generation of NO is involved in the pathogenesis of schizophrenia (Nasyrova et al. 2015; Ishola et al. 2022). Because NO is involved as a second messenger regulator of NMDA, it has been reported that NO levels determine the extent of dopaminergic, glutamatergic and GABAergic activities in the central nervous system (CNS) (Szabadits et al. 2011; Chatterjee et al. 2012). Additionally, NO is involved in the synthesis, storage and release of neuro-modulators such as glutamate, acetylcholine, taurine and glycine (Chatterjee et al. 2012; Nasyrova et al. 2015). However, it has been identified that ROS and RNS are potent endogenous ligands that inhibit oxidative intermediates such as glutathione that are involved in neuroprotection and neuronal plasticity (Lidow 2003; Picón-Pagès et al. 2019). Studies have revealed marked disruption of NO in certain brain regions including cerebellum, hypothalamus, prefrontal cortex, hippocampus, and striatum of schizophrenia patients (Nasyrova et al. 2015) and mice brains (Ishola et al. 2021). Moreover, it has been suggested that the relative capacity of iNOS-induced pro-oxidants mediated depletion of neuroprotectants and disrupt neurochemical metabolism depend on the nutritional status of the brain (Wu and Prentice 2010; Dipasquale et al. 2013; Firth et al. 2018). Interestingly, the antipsychotic effect of risperidone is also linked to suppression of iNOS expression (Zhu et al. 2014; Ben-Azu et al. 2019).
Clinical (Krystal et al. 1994) and preclinical (Monte et al. 2013) studies have shown that ketamine-induced experimental schizophrenia is indeed characterize of induction of oxidative and nitrergic stresses via up-regulation of iNOS expression and NO release, depletion of endogenous antioxidants and derangement of glutamatergic neurotransmission (Krystal et al. 1994; Monte et al. 2013; Ben-Azu et al. 2018b). Also, ketamine-induced schizophrenia is linked some integrative aspects of neuroinflammation including release of inflammatory cytokines and proteins (Ben-Azu et al. 2019; de Araújo et al. 2021). On the other hand, antipsychotic drug actions are currently suggested to be largely dependent on the ability of neuroleptic drugs to reduce the activities of these pathways in experimental animals (Ben-Azu et al. 2018b; Ishola et al. 2021; de Araújo et al. 2021) and clinical (Müller et al. 2010; Attari et al. 2017; Hong and Bang 2020; Dietrich-Muszalska et al. 2021) settings. Prevailing evidence from “nutritional psychiatry indicates that dietary strategies seem to be a naturally feasible nutraceutical gift, and cost-effective approach to prevent and ameliorate schizophrenia pathology ( Graham et al. 2015; Shivakumar et al. 2015; Ben-Azu et al. 2016, 2018a, b, c, d, 2019; de Araújo et al. 2021; Ishola et al. 2021).
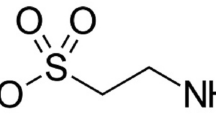
Taurine is an abundant non-proteogenic β-amino acid with sulphur compound. Endogenously, it is produced by cysteine in high quantity and serves as an important cytoprotective and nutritional agent for mental wellbeing particularly in excitable tissues (Wu and Prentice 2010). It is distributed in large amount in dietary products including animal sources (Wu and Prentice 2010). Intracellular concentration of taurine ranges between 5–50 mM with a plasma level of about 100 µM (Ghandforoush-Sattari et al. 2010). Prevailing evidence suggest robust connections between low taurine levels, cellular aging and neuropsychiatric diseases (Leon et al. 2009; Wu and Prentice 2010; Yang et al. 2019; Jakaria et al. 2019). In fact, epidemiological data revealed that individuals with early onset schizophrenia and first episode psychosis generally have low-quality diet and nutritional deficiency including reduced PUFAs and essential amino acids (Dipasquale et al. 2013; Firth et al. 2018). These malnutritions perhaps have been correlated with severity of the symptoms, reduced neural integrity and neurocognitive impairments in schizophrenia patients (Graham et al. 2015; Shivakumar et al. 2015; Rafiee et al. 2022). In this regards, some food-derived nutritional supplements in double-blind randomized controlled trial have been shown to improved schizophrenia symptoms (Berk et al. 2008).
Despite the cytoprotective roles of taurine in the central CNS (Wu and Prentice 2010; Jakaria et al. 2019), investigations of its therapeutic potential in the field of mental psychiatry only began in the last decades since its isolation in 1800s (Leon et al. 2009; Wu et al. 2009). Intriguingly, recent clinical studies revealed that taurine improved positive symptoms in first episode psychosis (O’Donnell et al. 2016). Preclinical investigation also showed that taurine prevents dizocilpine-evoked schizophrenia-like behavior by suppressing shoal dispersion and cortisol response in Zebra fishes (Franscescon et al. 2021). Previously, our research group also demonstrated the putative neuroprotective effects in antipsychotic-induced psychoneuroendocrine changes and cataleptic behavior (Oyovwi et al. 2021; Zebrowska-Lupina and Porowska 1987). Other relevant studies also showed that taurine possesses antidepressant (Wu et al. 2017), antioxidant (Jong et al. 2012), antiapoptotic (Takatani et al. 2004), anti-inflammatory (Marcinkiewicz and Kontny 2014) properties, and is plays key role in the regulation of neurochemical homeostasis (Bulley and Shen 2010). Notably, taurine is regarded as a natural inhibitory neuromodulatory amino acid in the CNS based on its ability to modulate NMDA receptors via activation of GABAergic and glycinergic pathways, and modulation of glutamatergic neurotransmission (Bulley and Shen 2010; del Olmo et al. 2000). These findings therefore provide the motivation for the preclinical study of taurine in ketamine-induced experimental psychosis. Here, we investigated the effects of taurine on ketamine-induced schizophrenia-like phenotypes, cholinergic deficits, oxidative/nitrergic damage and up-regulation of COX-2/iNOS pathways different brain regions relevant to the disease.
Materials and methods
Animal protocols
Six weeks old adult male Swiss albino mice of 24–29 g were obtained from the animal facility of the Faculty of Basic Medical Sciences, Delta State University (DELSU). Animals were acclimated to the study environment with standard night and day circle system. The study experimental protocol was approved by the DELSU Animal Care and Use Research Ethics Committee (REC/FBMS/DELSU/21/94). We estimated our sample size by an equation method (Charan and Kantharia 2013) which measures “E” as the degree of freedom of ANOVA. n = 7 were used for this study based on standard deviation, effect size and degree of significance of our studies on antipsychotic compounds (Ben-Azu et al. 2018b). Based on this, 7 animals/group were divided into 5 assemblages for the major experiments (preventive and reversal protocols), we obtained a value of 30. According to this method, 30 is an adequate value with a minimum benchmark of 10. Also, with the attrition formula, we confirmed that with 5% attrition level our sample size would be 6.3. Thus, we adopted n = 7.
Drugs, chemicals and treatments
Taurine and risperidone were purchased from Sigma-Aldrich, St. Louis, USA, and ketamine hydrochloride (KET) was bought from the German Rotex Medica company. Chemicals and reagents including Tris (hydroxymethyl)-amino-methane (Tris-buffer), and other regents used in this study were also purchased from Sigma Aldrich. The doses of taurine (50 and 100 mg/kg, p.o.) (Oyovwi et al. 2021), and risperidone (0.5 mg/kg, p.o.) and ketamine (20 mg/kg, i.p.) (Monte et al. 2013; Ben-Azu et al. 2018b) were selected based on findings from preliminary and previous preclinical and clinical reports. Risperidone was used as a standard control drug in this study because of its ability to prevent or reverse ketamine-induced oxidative, nitrergic alterations, neuroinflammation and schizophrenia-like behavior in mice (Monte et al. 2013; Zhu et al. 2014; Ben-Azu et al. 2019).
Experimental approach
The study was designed in three separate cohorts of experiments (drug alone, preventive and reversal) (Scheme 1). The preventive and reversal protocols of ketamine-induced schizophrenia behavior and pathology was designed based on earlier methods Monte et al. 2013(Ben-Azu et al. 2016, 2018b, c). Thus, the preventive approach was aimed at preventing or delaying onset while the reversal protocol targets already established schizophrenia conditions. In the experiment 1 (drug alone), animals were grouped into 4 cohort (n = 7). Cohort 1 received saline (10 mL/kg, p.o.), cohorts 2 and 3 were administered taurine (50 and 100 mg/kg) while cohort 4 was treated with risperidone (0.5 mg/kg) orally for 14 days. In the experiment 2 (preventive protocol), animals were arranged into 5 treatment cohorts (n = 7). Animals in cohorts 1 and 2 were pretreated with saline (10 mL/kg, p.o.), cohorts 3 and 4 received taurine (50 and 100 mg/kg, p.o) while cohort 5 received risperidone (0.5 mg/kg) for 14 days. From days 8–14, cohorts 2–5 were also engaged with ketamine (20 mg/kg,/day i.p.) after taurine treatment. In experiment 3 (reversal protocol), animals were also allotted into cohorts (n = 7) with opposite treatment approach to experiment 2. All administrations were done with 30 min interval between treatments.

Graphical description of experimental design
Behavioral assessments
On day 15, spontaneous motor activity (SMA), hyperlocomotion were accessed using the open-field test (OFT) box (28 × 28 × 25 cm) labelled with 16 (7 × 7 cm) squares, and the locomotive behavioral test was based on the number of line crossing for 5 min, as we have previously done by expert observers ( Monte et al. 2013; Ben-Azu et al. 2018b, 2019). The Y-maze test (YMT) was used to score the spatial memory of the mice based on previous studies ( Monte et al. 2013; Ben-Azu et al. 2018b, 2019). The YMT is a 3-symetrical armed (A, B, C) instrument demarcated at 120°, with behavioral adjudgment that is based on the patterns of arms visitation for 5 min. The cognitive score were presented as % correct alternation (Monte et al. 2013). Also, ketamine-induced behavioral despair was assessed based on previous report using forced swim test (FST) (Ben-Azu et al. 2018b). In the pre-test session, we acclimatized the animals to the experimental set-up containing water at 25 °C to a depth of 30 cm for a period of 5 min, dried and taken back to their home cages as previously described (Ben-Azu et al. 2018a, d). After 24 h mice were returned to the transparent Plexiglas cylinder with the same swimming condition and made to swim for 6 min. However, 1 min was ignored while immobility period (passive floats) was measured for 5 min (Ben-Azu et al. 2018a, d).
The study also accessed the effect of taurine on extrapyramidal potential which is a common feature of antipsychotic drugs especially the first typical agents. Cataleptogenic potential was done with wood block (Height = 6 cm; Width = 4 cm; Length = 16 cm) catalepsy test as described based on decent latency (catalepsy time) (Ben-Azu et al. 2018a, d).
Estimation of brain biochemicals (oxidative, nitrergic stress and cholinergic biomarkers) in the striatum, prefrontal cortex and hippocampus of mice brains
All oxidative and biochemical data were assayed as described in our previous study ( Monte et al. 2013; Ben-Azu et al. 2018a). After euthanasia of mice, relevant brain areas (striatum, prefrontal cortex and hippocampus) were quickly isolated, centrifuged with buffer phosphate solution (PBS) (10% w/v, 0.1 M, pH 7.4). For GSH, supernatant was reacted with trichloric and nitrobenzoic acid reagents and the reacting mixture was read spectrophotometrically (INESA 752 N Model) at 412 nm based on previous method (Monte et al. 2013). The method combining Tris-KCL (0.15 M), TCA (0.5 mL) and TBA (0.75%) was adopted to assay for malondialdehyde (MDA) levels and thereafter the product was expressed as nanomole/mg protein of MDA (Monte et al. 2013). The superoxide dismutase (SOD) and catalase (CAT) activities were estimated based on superoxide inhibition of adrenaline and hydrogen period by an enzyme source, respectively (Ben-Azu et al., 2018a). Nitrite content (Monte et al. 2013) and acetylcholinesterase activity (Ben-Azu et al. 2018a) were assayed using Griess solution and acetyl-thiocholine iodide as active reagent together with nitrobenzene mixture, respectively.
Immunohistochemical studies
Following anesthesia, perfusion of mice was done by trans-cardiac method with PBS together with 10% buffered formaldehyde. Brain samples were replaced after 24 h with 25% sucrose solution. Brain areas of interest [striatum, prefrontal cortex and hippocampus, (cornus ammonis 1 CA1)] were paraffin processed and embedded in blocks. The expressions of cyclooxygenase-2 (COX-2) and inducible nitric oxide synthase (iNOS) was processed as previously described using their immunohistochemistry antibodies respectively (Ben-Azu et al. 2019). Immunopositive cell expressions were examined with image J software (NIH, Bethesda, MD, USA) after obtainment of images with a computer-linked Olympus microscope to a German Leica IC50 Digital Camera (Edelstein et al. 2014).
Statistical analysis
Following test for normality distribution with Shapiro–Wilk test, we expressed data as Mean ± S.E.M. (standard error of mean) of n = 7 per group. Graphs of behavioral data were presented as scattered plot for clear demonstration of behavioral distribution following one-way ANOVA and Bonferroni post-hoc test. Two-way ANOVA was used to analyze all biochemical densitometry of immunoreaction data for the different brain regions and graphs are presented in bar charts with GraphPad version 5 Software (Inc. La Jolla, CA 92,037 USA) using p < 0.05 as statistically significant level.
Results
Effects of taurine on motor function, cognition, and behavioral despair-like behavior in naïve mice
The effects of taurine on OFT, YMT and FST are shown in Fig. 1a–c. Treatment with taurine (50 and 100 mg/kg) did not produce any significant change in SMA when compared to saline. But administration of risperidone (0.5 mg/kg) significantly (p < 0.05) reduced SMA when compared with the saline group (Fig. 1a). Taurine (50 and 100 mg/kg) did not alter cognitive function in the YMT (Fig. 1b) nor cause any obvious changes in the immobility time (Fig. 1c) when compared to saline groups.

Effects of taurine on behavioral activities: locomotion in the OFT (a) spatial working memory in the YMT (b) and behavioral despair in the FST (c). Plots represent mean ± SEM (n = 7). #P < 0.05, ##P < 0.01 versus saline group. RIS = Risperidone
Taurine prevents and reverses on ketamine-induced hyperlocomotion, spatial memory impairment and behavioral despair
Figure 2a–e showed that intraperitoneal injection of ketamine (20 mg/kg) significantly (p < 0.05) increased the number of line crossings in the OFT, reduced memory function in the YMT and caused behavioral despair in the preventive and reversal protocols when compared with saline-treated groups. In the OFT, 50 mg/kg (p < 0.05) and 100 mg/kg (p < 0.01) of taurine and risperidone (0.5 mg/kg, p.o.) (p < 0.001) significantly prevented ketamine-induced hyperlocomotion (Fig. 2a). But, in the reversal protocol, only taurine (100 mg/kg, p.o.) and risperidone significantly reversed ketamine-mediated hyperlocomotion (Fig. 2b). For the YMT, taurine (50 and 100 mg/kg) and risperidone significantly prevented (Fig. 2c) and reversed (Fig. 2d) the spatial memory impairment induced by ketamine when compared with ketamine treated groups (Fig. 2d). Furthermore, taurine (50 and 100 mg/kg) significantly prevented (Fig. 2e) and reversed (Fig. 2e) ketamine-induced behavioral despair in the FST when compared with KET-treated group.

Effects of taurine on ketamine-induced hyperlocomotion (a, b), spatial memory impairment (c, d) and behavioral despair (e, f) in the preventive and reversal treatments. Plots represent mean ± SEM (n = 7). #P < 0.05, ##P < 0.01 versus saline group; *P < 0.05, **P < 0.01, ***P < 0.001 versus ketamine (KET) group. RIS = Risperidone
Effects of taurine on cataleptogenic behavior in naïve and ketamine treated mice
The effects of taurine on cataleptogenic potential as estimated by period of inactivity is shown in Fig. 3a–c. Treatments with both doses of taurine, and risperidone did not prolong the decent latency on the wood block when compared with saline (Fig. 3a), or preventive (Fig. 3b) and reversal (Fig. 3c) of ketamine groups, suggesting absence of cataleptogenic potential.

Effect of taurine on cataleptogenic potential in naïve (a) and ketamine treated mice in the preventive (b) and reversal (c) treatments. Plots represent mean ± SEM (n = 7). RIS = Risperidone
Taurine increases superoxide dismutase and catalase activities in naïve and ketamine-treated mice brains
The effects of taurine on ketamine-induced deactivation of SOD and CAT activities in the striatum, prefrontal cortex and hippocampus of naïve and ketamine treated mice in the preventive and reversal treatments are represented Fig. 4a–f. In the drug treatment alone, taurine (50 and 100 mg/kg, p.o.) significantly (p < 0.001) increased SOD activities in the striatum and prefrontal cortex when compared with saline group. Risperidone failed to alter SOD level in the cortex, but it increased it the striatum (Fig. 4a). Taurine (50 and 100 mg/kg, p.o.) but not risperidone significantly (p < 0.001) enhanced CAT activity in the striatum when compared with saline group. Also, in the prefrontal cortex taurine (100 mg/kg) significantly up-regulated CAT levels in the prefrontal cortex (p < 0.001) and hippocampus (p < 0.01) when compared with saline groups (Fig. 4b).

Effects of taurine on superoxide dismutase (SOD) and catalase (CAT) activities in naïve (a, b) and ketamine treatments in the preventive (c, d) and reversal (e, f) protocols. Bars represent mean ± SEM (n = 7). #P < 0.05, ##P < 0.01, ###P < 0.001 versus saline group; *P < 0.05, **P < 0.01, **P < 0.001 versus ketamine (KET) group. RIS = Risperidone
Bonferroni analysis revealed that ketamine reduced SOD level in the preventive and reversal treatments in the striatum and prefrontal cortex when compared with saline controls (Fig. 4c, e). Ketamine reduced CAT activity only in the reversal treatment. Also, ketamine reduced CAT activity in the striatum, prefrontal cortex but not in the hippocampus in the preventive. Ketamine reduced CAT activity in the three brain regions in the reversal study when compared with saline controls (Fig. 4d, f).
In the preventive treatment, taurine (50 and 100 mg/kg) significantly increased the SOD (Fig. 4c) and CAT (Fig. 4d) activities in the striatum and prefrontal cortex when compared with ketamine treatment. Risperidone significantly increased SOD and CAT activities prefrontal cortex, but not in the striatum (Fig. 4c, d). In the hippocampus, taurine (50 and 100 mg/kg) and risperidone significantly increased SOD activity when compared with ketamine treated mice in the preventive treatment (Fig. 4c). Taurine (100 mg/kg) increased CAT activity in the hippocampus when compared with ketamine group (Fig. 4d).
In the reversal treatment, only risperidone increased SOD activity in the striatum when compared with ketamine-treated mice. In the prefrontal cortex, taurine (100 mg/kg) and risperidone also significantly reversed the effect of ketamine on SOD activity. Both taurine (50 and 100 mg/kg) and risperidone enhanced SOD activity when compared with ketamine-treated group (Fig. 4e). As regards CAT activity in the reversal experiment, taurine (50 and 100 mg/kg) and risperidone significantly increased CAT activities in the striatum, prefrontal cortex and hippocampus when compared with ketamine groups (Fig. 4f).
Effects of taurine on glutathione and malondialdehyde concentrations in naïve and ketamine-treated mice brains
The effects of taurine on ketamine-induced changes on GSH and MDA concentrations in the striatum, prefrontal cortex and hippocampus of naïve and ketamine treated mice in the preventive and reversal treatments are shown Fig. 5a–f. In the drug treatment alone, two-way ANOVA revealed that taurine (50 and 100 mg/kg, p.o.) profoundly enhanced GSH levels in the striatal-cortical regions when compared with saline group. In the hippocampus, only taurine (50 mg/kg, p.o.) increased GSH level (Fig. 5a). As indicated by two-way ANOVA, there were concomitant reduction of MDA in the striatum and prefrontal cortex by 100 and 50 mg/kg of taurine respectively (Fig. 5b). No changes were found with risperidone in the three brain regions (Fig. 5a, b).

Effects of taurine on glutathione (GSH) and malondialdehyde (MDA) concentrations in naïve (a, b) and treatments in the preventive (c, d) and reversal (e, f) protocols. Bars represent mean ± SEM (n = 7). #P < 0.05, ##P < 0.01, ###P < 0.001 versus saline group; *P < 0.05, **P < 0.01, **P < 0.001 versus ketamine (KET) group. RIS = Risperidone
In the pre- and reversal treatments ketamine (20 mg/kg, i.p.) notably decreased GSH concentration in the three brain areas in both treatment protocols (Fig. 5c, e). In comparison with ketamine in the preventive treatment, taurine (100 mg/kg, p.o.) and risperidone profoundly increased the GSH concentrations in the striatum and prefrontal cortex (Fig. 5c). However, treatment with the lower dose of taurine (50 mg/kg, p.o.) differentially increased GSH level in the striatum (p < 0.05) and prefrontal cortex (p < 0.01) respectively (Fig. 5c). In the hippocampus, only 100 mg/kg (p < 0.05) of taurine increased GSH levels (Fig. 5c). For the reversal effects in comparison with ketamine alone (Fig. 5d), taurine (50 and 100 mg/kg, p.o.) and risperidone significantly increased the GSH concentrations in the striatum. In the prefrontal cortex, only 100 mg/kg of taurine and risperidone considerably increased the GSH concentrations. In the hippocampus, there was a dose-dependent elevation of GSH by 50 mg/kg (p < 0.01) and 100 mg/kg (p < 0.001) of taurine as well as risperidone relative to KET control (Fig. 5e).
Furthermore, ketamine also increased MDA levels in both protocols when compared with saline groups, in the striatum, prefrontal cortex and hippocampus (Fig. 5d, f). However, taurine (50 and 100 mg/kg) significantly reduced MDA levels in the striatum and prefrontal cortex when compared with ketamine-treated in the preventive treatment, akin to the effect of risperidone (Fig. 5d). In the hippocampus, only 50 mg/kg of taurine (p < 0.05) as well as risperidone (p < 0.001) reduced MDA levels when compared with ketamine control group (Fig. 5e). For the reversal study (Fig. 5f), however, taurine (50 and 100 mg/kg) and risperidone significantly (p < 0.001) both reduced MDA levels in the striatum and prefrontal cortex, although no effect was seen with the lower dose of taurine (50 mg/kg) in the striatum. In the hippocampus, taurine (50 and 100 mg/kg, p.o.) and risperidone, both significantly depleted MDA concentrations (Fig. 5f).
Effects of taurine on nitrite and acetylcholinesterase levels in naïve and ketamine-treated mice brains
As presented in Fig. 6a–b, no marked changes found between saline groups and treatments with both taurine (50 and 100 mg/kg) and risperidone in the striatum and prefrontal cortex (Fig. 6a). But 50 mg/kg of taurine increased nitrite levels in the hippocampus relative to saline (Fig. 6a). Nevertheless, both doses of taurine significantly reduced AChE activity only in the prefrontal cortex when compared with saline group (Fig. 6b).

Effects of taurine on nitrite and acetylcholinesterase (AChE) levels in naïve (a, b) and ketamine treatments in the preventive (c, d) and reversal (e, f) protocols. Bars represent mean ± SEM (n = 7). #P < 0.05, ##P < 0.01, ###P < 0.001 versus saline group; *P < 0.05, **P < 0.01, **P < 0.001 versus ketamine (KET) group. RIS = Risperidone
In the preventive treatment (Fig. 6c, d), ketamine increased nitrite levels in the striatum, prefrontal cortex (Fig. 6c). Taurine (50 and 100 mg/kg) and risperidone attenuated the increase in nitrite levels in the striatal-cortical regions compared with ketamine-treated mice (Fig. 6c). But only 100 mg/kg of taurine was found to reduce nitrite levels in the hippocampus (Fig. 6c). In the reversal study, post hoc examination showed there were discrete effects in the striatum, prefrontal cortex and hippocampus by ketamine, as evidenced by significant increase in nitrite levels in the three brain compartments by ketamine (Fig. 6e). Taurine (50 and 100 mg/kg) profoundly reduced the nitrite concentrations in the striatum and prefrontal cortex in a similar manner to risperidone when compared with ketamine group. In the hippocampus, both taurine (100 mg/kg) (p < 0.05) and risperidone (p < 0.01) also decreased nitrite level (Fig. 6e).
Furthermore, as regards AChE activity in the preventive study, ketamine increased AChE activity in the prefrontal cortex, hippocampus but not striatum, which were however eventually mitigated by taurine (50 and 100 mg/kg) in a brain region-dependent manner (Fig. 6d). In the reversal study, ketamine also increased AChE activity in the striatum, prefrontal cortex and hippocampus in comparison with saline (Fig. 6f). Notably, these changes were reversed by taurine (50 and 100 mg/kg) in the prefrontal cortex (p < 0.001) and hippocampus (p < 0.01) (Fig. 6f). In the striatum, taurine (50 and 100 mg/kg) did not elicit any significant changes. Only risperidone was able to reverse ketamine-induced decrease in cholinergic transmission (Fig. 6f).
Taurine reduces ketamine-induced expression of COX-2 in the cortical regions of mice brains
As shown in Fig. 7a–d, ketamine caused a significant increase in COX-2 expression in the prefrontal cortex in the preventive study, but no changes were seen in the hippocampus and striatum in relation to saline cohorts (Fig. 7a). In the reversal study, ketamine increased COX-2 expression in the prefrontal cortex and hippocampus, suggesting inflammation of cortical brain areas (Fig. 7b). Taurine (100 mg/kg) and risperidone prevented ketamine-mediated elevated expression of COX-2 in the prefrontal cortex (Fig. 7c). Likewise, taurine (100 mg/kg) and risperidone suppressed COX-2 activities in the reversal study (Fig. 7d).


Illustrative photomicrographs of the effect of taurine on ketamine-induced immunohistochemical changes and expressions of COX-2 immunopositive cells in the striatum, prefrontal cortex and hippocampus of mice brains in the preventive (a) and reversal (b) treatments. Saline = SAL (10 mL/kg), Ketamine = KET (20 mg/kg), Taurine = TAU (100 mg/kg), Risperidone = RIS (0.05 mg/kg). Higher intensity indicates high COX-2 immunopositive cell expression while lower intensity indicates low CPX-2 immunopositive cell expression. Taurine reduces ketamine-induced expression of COX-2 in mice brains in the preventive (c) and reversal (d) protocols. Bars represent mean ± SEM (n = 7). #P < 0.05 versus saline group; *P < 0.05, **P < 0.01, **P < 0.001 versus ketamine (KET) group. RIS = Risperidone
Taurine decreases ketamine-induced expression of iNOS in mice brains
Two-way ANOVA showed that ketamine increased iNOS expressions in the three brain sub-fields examined in both protocols (Fig. 8a–b) in relation to saline cohorts. In the preventive study, taurine (100 mg/kg) and risperidone (0.5 mg/kg) inhibited ketamine-induced immunopositive cell expressions of iNOS in the striatum and prefrontal cortex, but only risperidone reduced the iNOS level of expression in the hippocampus (Fig. 8c). Unlike taurine (100 mg/kg), risperidone suppressed the iNOS in the striatum and prefrontal cortex. In the hippocampus, taurine (100 mg/kg) but not risperidone reduced iNOS expressions comparatively to ketamine groups respectively in the reversal treatment (Fig. 8d).


Illustrative photomicrographs of the effect of taurine on ketamine-induced immunohistochemical changes and expressions of iNOS immunopositive cells in the striatum, prefrontal cortex and hippocampus of mice brains in the preventive (a) and reversal (b) treatments. Saline = SAL (10 mL/kg), Ketamine = KET (20 mg/kg), Taurine = TAU (100 mg/kg), Risperidone = RIS (0.05 mg/kg). Higher intensity indicates high COX-2 immunopositive cell expression while lower intensity indicates low CPX-2 immunopositive cell expression. Taurine reduces ketamine-induced expression of iNOS in mice brains in the preventive (c) and reversal (d) protocols. Bars represent mean ± SEM (n = 7). #P < 0.05, ##P < 0.01, ###P < 0.001 versus saline group; *P < 0.05, **P < 0.01, **P < 0.001 versus ketamine (KET) group. RIS = Risperidone
Discussion
Here, we show that taurine, an essential β-amino acid prevents, and reverses ketamine induced experimental psychosis based suppression of schizophrenia-like behavior and inhibition of brain metabolic changes including reduction of oxidative/nitrergic stress levels, increased cholinergic neurotransmission and suppression of COX-2 and iNOS expression.
During development, taurine plays important roles as one of the most important amino acids in all aspects of behavioral development and neuronal physiology including control and refinement of neuronal wiring, glial cell development, neurogenesis, membrane stability, osmoregulation and neuroprotection (Hussy et al. 2000; Bulley and Shen 2010; Schaffer et al. 2014; Li et al. 2017). Taurine is transported from the periphery into neurons of CNS including the blood brain barrier (BBB) by taurine transporter (TAUT) and it is abundantly expressed in the cerebellum, cortex and hippocampus (Ohtsuki 2004). Of note, taurine is involve in enhancement of emotion memory and learning in rodents (Li et al. 2017) and contribute to the protection of large population of excitable tissues cells from excitatory amino acid-induced excitotoxicity, especially the brain (Rahmeier et al. 2016). Given the relevant of taurine, it is not surprising that conditions resulting from taurine depletion during development or adulthood mental stress create a possibility for a second hit which in turn leads to spectrum of psychiatric disorders such as schizophrenia (O’Donnell et al. 2016). Moreover, compiling preclinical evidence suggests that low brain levels of taurine in the striatum, temporal cortex and parietal cortex could be associated with behavioral changes relevant to schizophrenia behavior, which suggests a strong connection between taurine and molecular psychiatry (Yang et al. 2019). Interestingly, a clinical study by O'Donnell et al. (2016) show that taurine improved symptomology and cognition in schizophrenia patients based on DMS-IV first-episode psychotic disorder. Thus, hypofunctionality of taurine transmitter could be de facto new target for development of antipsychotic therapy.
In this study, biochemical and behavioral strategies were employed to study the ameliorative effect of taurine on ketamine-mediated experimental psychosis in adult mice. Psychotic mice showed excessive behavioral hyperactivity as evidenced by enhanced locomotion, which is regularly linked to the positive symptoms (Ishola et al. 2021). Also, the mice exhibit increased behavior in the FST based on increased duration of immobility in the apparatus, as well as cognitive impairment in the YMT as represented by disorganized spontaneous alternation behavior. These observations showed that schizophrenia-like behaviors were successfully produced in the mice following repeated intraperitoneal injections of ketamine, which is consistent with previous investigations (Monte et al. 2013; Ben-Azu et al. 2018a, b, c; de Araújo et al. 2021; Ishola et al. 2021; Oshodi et al. 2021).
Ketamine induced experimental psychosis has been consistently linked to hypofunctionality of the downstream NMDA receptors located on GABAergic inhibitory interneurons, a primary mechanism that is responsible for the sub-cortical neuronal excitation particularly in the striatal-limbic areas because of increased glutamate and dopamine release (Ben-Azu et al. 2018b). On the other hand, NMDA receptor blockade in the tegmental areas plays an important role in the negative and cognitive symptoms due to reduced dopamine release in the prefrontal cortex (Neill et al. 2010). As in mice, brain NMDA receptor blockade in human subjects by ketamine show symptoms of endogenous psychosis evident by behavioral hyperactivity, altered perception, disrupted verbal fluency and cognitive disabilities (Krystal et al. 1994). Most interestingly, postmortem biochemical and structural brain analyses of schizophrenia patients show altered glutamate, dopamine and GABA homeostasis as well as, reduced frontal cortex NMDA receptor subunits, disarray and loss of hippocampal or cortical neurons orientation, depleted synaptic markers and entorhinal cortex dysplasia (Harrison 2000). These findings have been proven in ketamine model of schizophrenia and thus confirms the face, etiological and constructive validity of ketamine in experimental schizophrenia pathology (Ben-Azu et al. 2018a, b, c; de Araújo et al. 2021; Oshodi et al. 2021).
In this study, we found that preventive and reversal treatments with taurine prevented and reversed ketamine-mediated psychotic phenotype including reduction in the hyperlocomotor activity, reduced behavioral despair and cognitive plasticity, indicating an obvious antipsychotic potential of taurine against psychotic behavior. These findings are synonymous to the effects of taurine in other related investigations where it was found to demonstrate conditional anxiolytic (Francisco and Guedes 2015), cognitive improvement (Zhang et al. 2016) and antidepressant (Wu et al. 2017) effects in rodents. It is of interest to report that consecutive administration of taurine for 14 days did not produce any significant effects as regards behavioral despair and cognitive enhancement but elicits significant tendency of reduced hyperactivity in the OFT. Previous reports have shown that taurine supplementation produces no significant behavioral changes in naïve animals especially on measures of depression and anxiety (Whirley and Einat 2008; Wu et al. 2017) but Whirley and Einat (2008) reported that taurine produces transient decrease in activity, which are indeed in agreement with our findings. Additionally, the test for catalepsy using wood block, which is a behavioral paradigm used for evaluation of extrapyramidal side effects (EPSs) (Gobira et al. 2013), revealed that repeated administration of taurine alone is devoid of catalepsy effect. One major problem associated with long-term use of antipsychotic drugs is the induction of EPS due to D2 dopaminergic receptor blockade (Kapur et al. 2000). In this study, we also observed that the therapeutic effect of taurine in both the preventive and reversal treatments was not associated with cataleptogenic behavior unlike risperidone that showed heightened tendency of catalepsy in the mice. This effect could be obvious when higher doses and prolonged duration are applied (Moe et al. 2017). Contrastingly, it is interesting to state that taurine has been reported to rather attenuate cataleptic effect of first-generation antipsychotic-induced catalepsy in rodents. Together, this findings suggest the anti-catalepsy potential of taurine (Zebrowska-Lupina and Porowska 1987).
Oxidative stress has been identified to cause dysregulation of neuronal lipids and mitochondrial metabolic shifts, thereby causing alterations in neurochemical homeostasis through altered receptor activities (Upthegrove and Khandaker 2020; Nakao et al. 2021). Of note, NMDA receptor dysfunction is cross-linked with alterations in oxidative pathway via multiple mechanisms of Ca2+-regulated signaling including Ca2+/calmodulin-dependent protein kinase II (Ca2+/CaMKII) (Mouri et al. 2007). Notably, CaMKII is important in the regulation of glutamate, GABA and glycine neurotransmissions (Bulley and Shen 2010). Interestingly, taurine regulates glutamate mediated Ca2+ permeability or excitation by GABA-mediated hyperpolarization via intracellular second messenger system (Bulley and Shen 2010). Previous studies have been reported that ketamine induces changes in glutamate-GABA-CaMKII pathways (Lazarevic et al. 2021). It is hypothesized that ketamine-induced down-regulation of Ca2+/CaMKII pathway could be mediated via inhibition of intracellular uptake of taurine in the CNS (Satsu et al. 2004; Yao and Wu 2001), thereby causing delayed neuromodulatory responses to neuronal changes (Li et al. 2002; Satsu et al. 2004). Furthermore, it is sensible to state that taurine depletion might also play an important role in the up-regulation of the superoxide precursor enzyme, NDPH oxidase-2 (Nox-2), and superoxide formation via calcium-dependent depolarization and NMDA receptor hypofunction-derived oxidative stress (Bulley and Shen 2010). Moreover, since Ca2+ is the main regulator of taurine-sensitive ensembles, elevated levels of Nox-2-mediated ROS generation and mitochondrial membrane collapse (El Idrissi 2008) could cause enduring loss of cortical GABAergic and glutamate phenotypes including parvalbumin interneurons, inactivation of excitatory amino acid and cysteine transporters, thereby leading to low levels of glutathione in the brain and altered cognitive executive behavioral functions (Behrens et al. 2007; Valtcheva and Venance 2019).
Given this linkages, it is not surprising that conditions or agents that resulted in taurine and glutathione depletion would lead to spectrum of neuropsychiatric disorders associated with oxidative damage (Yang et al. 2019). In line with previous studies (Monte et al. 2013; Ben-Azu et al. 2018b), ketamine was found to reduce glutathione levels together with superoxide dismutase and catalase activities with increased malondialdehyde and nitrite concentrations in the striatum, prefrontal cortex and hippocampus respectively, suggesting a state of neuronal oxidative stress. As with mice, postmortem reports show that low levels of glutathione are commonly found in the cerebrospinal fluid (Do et al. 2000), caudate region (Yao and Wu 2001) and prefrontal cortex (Gawryluk et al. 2011) of schizophrenia patients, which are believed to contribute to the severity of the disease (Nakao et al. 2021). Interestingly, enhanced glutathione functionality has been reported in the brains of first-episode psychotic patients specifically in the temporal lobes, suggesting compensational antioxidant activity possibly due to oxidative stress (Nakao et al. 2021). Most notably, vast majority of clinical findings have shown that schizophrenia patients are enriched with increased neuronal nitric oxide synthase (nNOS) immunoreactivity accompanied with nitric oxide overload in the dentate nucleus and purkinje cells (Bernstein et al. 2001; Zhang et al. 2012). In fact, altered striatal glutamatergic system could be a resultant effect of nitrergic alterations (Chatterjee et al. 2012; Ben-Azu et al. 2018b). Although we found that seven days injections of ketamine did not cause any significant change in the nitrite hippocampal region, in line with the finding of de Araújo et al., (de Araújo et al. 2021) it is explainable that prolonged pathological state could be responsible for the nitrergic imbalance in the hippocampus compared to control as observed in the reversal protocol consisting of fourteen days ketamine injections.
It is known that chronic NMDA receptor activation triggers Ca2+ influx thereby leading elevated of levels of NO and excessive presynaptic glutamate release via a Ca2+-regulated nNOS pathway (Chatterjee et al. 2012; O’Dell et al. 1991). Though, iNOS-mediated production of NO is Ca2+-independent, microglia (the resident immune cells of the brain) release of NO possesses neuro-toxic effects which indirectly relies on the concentration of cyclic adenosine monophosphate (cAMP) and directly on iNOS expression (Nasyrova et al. 2015). Postmortem brain investigations have revealed increased levels of NO in the brains of schizophrenia patients, thus supporting the association between increased activity of iNOS and schizophrenia (Nasyrova et al. 2015). Moreover, exacerbated endogenous nitrergic tone in the striatum has been hypothesized to be partly responsible for the increase dopamine release in the midbrain (West and Grace 2000). This mechanism is thus responsible for ketamine-induced increased dopamine release, glutamate-mediated excitotoxicity and psychotic-like behavior (Chatterjee et al. 2012; Monte et al. 2013; Ben-Azu et al. 2018b; Oshodi et al. 2021). Most notably, neuronal blockade with NO antagonist is known to causes a prolongation of cortical inhibition of dopamine release and modulation of glutamatergic afferent drive (West and Grace 2000), further suggesting that inhibition of NO release is an antipsychotic-related mechanism (Monte et al. 2013; Ben-Azu et al. 2018b). The findings that the preventive and reversal treatments with taurine attenuated ketamine-induced oxidative (increased antioxidant markers/low pro-oxidant) and nitrergic (decreased iNOS expressions/NO levels) stress suggest a neuroprotective property possibly through nutritional amino acid and cysteine-dependent replenishment of glutathione (Wu et al. 2009), normalization of glutamate neurotransmission, as well as inhibition of iNOS-induced neuroinflammation through “brain amino acid sensing mechanism” (Bulley and Shen 2010; Tsurugizawa et al. 2014).
Indeed, glutathione is a tripeptide antioxidant molecule comprising of cysteine, glycine and glutamate (Herrmann et al. 2012; Wei et al. 2020; Aoyama 2021). Endogenously, taurine is produced by cysteine in high quantity especially excitable tissue like the brain and serves an important cytoprotective role as a nutritional agent in maintaining mental homeostasis (Wu and Prentice 2010). To date, enhancement of glutathione-regulated activity and normalization of glutamate neurotransmission are related to some of the key mechanisms associated with second generation antipsychotic drugs including risperidone, a standard drug used in the treatment of schizophrenia (Herrmann et al. 2012; Monte et al. 2013). In the light of this, investigations show that taurine efficiently reduces intracellular Ca2+ overload and glutamate-induced depolarization, apoptosis and neuroinflammation by modulating cAMP pathways, Ca2+ channels, NMDA receptors and enhancement of antiapoptotic factors through chaplain-dependent machineries. Thus, these findings suggest that taurine elicits its neuroprotective functions through a multipronged mechanisms (El Idrissi 2008; Wu et al. 2009; Jakaria et al. 2019; Rafiee et al. 2022). Moreover, other studies also showed that that taurine protects against oxidative stress in different organs in rodent and human disease conditions (Zhang et al. 2014; Niu et al. 2018; Maleki et al. 2020).
It is also worthy of note that ability of taurine to suppress ketamine-induced up-regulation of COX-2 expression denotes anti-neuroinflammatory mechanism. COX-2, which is an important aspect of membrane hypothesis of schizophrenia plays a prominent role in neuroinflammation-induced disruption of membrane phospholipids and receptor transduction mechanism. Also, COX-2-induced increased IL-6 and phospholipase A2 activities particularly contributes to the hypodopaminergic and hypofunctionality of the prefrontal cortex, breakdown of PUFAs, which play critical roles in the initiation of schizophrenia behavior. However, selective inhibition of COX-2 is linked to improvement in schizophrenia symptoms via reduction of PGE synthesis, microglia-mediated release of pro-inflammatory cytokines, NO and iNOS expression (Zhang and Zhao 2014). Therefore, the suppression of ketamine-induced COX-2 immunoreactivity in certain regions of mice brains by taurine might be partially mediated through inhibition of COX-2 expression and this mechanism might be relevant in the anti-psychotic effect in first episode psychotic patients (O’Donnell et al. 2016).
Furthermore, ketamine-induced experimental psychosis is long established and linked to increased activity of AChE, a metabolizing enzyme that regulates synaptic pool of acetylcholine (Chatterjee et al. 2012). Acetylcholine is a well-known neurotransmitter responsible for the control of cognitive and social functions (Contestabile 2011). Earlier studies have shown that ketamine-induced schizophrenia-related memory dysfunction is commonly associated with increased brain AChE activity (Ben-Azu et al. 2018a, b, 2019; Chatterjee et al. 2012). Moreover, ketamine-induced schizophrenia-like behavior is also believed to be mediated via blockade of 7-alpha nicotinic acetylcholine receptors (α-7nAChR), a regulator of cholinergic-neuroimmune pathway (Borovikova et al. 2000; Jiang et al. 2021). In this study, we also showed that ketamine-induced behavioral alterations were accompanied with reduced cholinergic neurotransmission as evidenced by elevated levels of AChE activity in the three brain regions respectively, although with more severe increase in the striatum in the reversal treatment and its consistent with previous findings (Ben-Azu et al. 2018b). Since acetylcholine is involve in the regulation of glutamatergic activity, this disease model could possibly be the resultant effect of a more prolonged blockade of α-7nAChR by ketamine (Coates and Flood 2001) thereby leading deregulation cholinergic activity (Chatterjee et al. 2012) and loss of the anti-neuroinflammatory neuroprotective effect of acetylcholine in this brain (Jiang et al. 2021). In this study, the preventive and reversal treatments with taurine was found to also attenuate ketamine-induced increased cortical and subcortical AChE activities. This finding also indicates one of the mechanisms responsible for taurine’s ability to mitigate ketamine-induced cognitive impairment in mice. Interestingly, other report has also shown that taurine prevents ethanol-induced increment in AChE activity and oxidative damage zebrafish brain as part of its mechanism of neuronal protection (Rosemberg et al. 2010).
To the best of our knowledge after a rigorous literature search, this is first pre-clinical study with mice that provides scientific evidence for the potential benefit of taurine as a safe naturally occurring treatment for psychosis. While this paper represents a first important effort to prove the clinical effects of taurine in first episode psychosis (O’Donnell et al. 2016) without determining endogenous taurine levels, our goal is to continue this research. So, further pre-clinical investigation will continue as a stable project to provide molecular evidence including effects on neurochemical transmissions, other neuroimmune activities and neurotrophic factors in experimental psychosis. At this stage, we thus achieved that taurine prevents ketamine-induced schizophrenia-like behavior, an effect that is devoid of cataleptogenic potential. Notably, some possible underlying mechanisms might involve the enhancement of cholinergic neurotransmission, inhibition of oxidative and nitrergic imbalances, and suppression of COX-2/iNOS immunopositive cells in mice brains.
Data availability
Data will be made available upon reasonable request.
Code availability
Not applicable.
References
Aoyama K (2021) Glutathione in the Brain. Int J Mol Sci 22:5010
Attari A, Mojdeh A, Khalifeh, Soltani FAS, Najarzadegan MR (2017) Aspirin inclusion in antipsychotic treatment on severity of symptoms in schizophrenia: a randimized clinical trial. Iran J Psychiatry Behav Sci 2017(11):e5848
Aucoin M, LaChance L, Cooley K, Kidd S (2020) Diet and Psychosis: A Scoping Review. Neuropsychobiology 79:20–42
Behrens MM, Ali SS, Dao DN, Lucero J, Shekhtman G, Quick KL, Dugan LL (2007) Ketamine-induced loss of phenotype of fast-spiking interneurons is mediated by NADPH-oxidase. Science 318:1645–1647. https://doi.org/10.1126/science.1148045
Ben-Azu B, Aderibigbe AO, Ajayi AM, Iwalewa EO (2016) Neuroprotective effects of the ethanol stem bark extracts of Terminalia ivorensis in ketamine-induced schizophrenia-like behaviors and oxidative damage in mice. Pharm Biol 54:2871–2879. https://doi.org/10.1080/13880209.2016.1190382
Ben-Azu B, Aderibigbe AO, Omogbiya IA, Ajayi AM, Owoeye O, Olonode ET, Iwalewa EO (2018a) Probable mechanisms involved in the antipsychotic-like activity of morin in mice. Biomed Pharmacother 105:1079–1090. https://doi.org/10.1016/j.biopha.2018.06.057
Ben-Azu B, Aderibigbe AO, Eneni A-EO, Ajayi AM, Umukoro S, Iwalewa EO (2018b) Morin Attenuates Neurochemical Changes and Increased Oxidative/Nitrergic Stress in Brains of Mice Exposed to Ketamine: Prevention and Reversal of Schizophrenia-Like Symptoms. Neurochem Res 43:1745–1755. https://doi.org/10.1007/s11064-018-2590-z
Ben-Azu B, Aderibigbe AO, Ajayi AM, Eneni A-EO, Umukoro S, Iwalewa EO (2018c) Involvement of GABAergic, BDNF and Nox-2 mechanisms in the prevention and reversal of ketamine-induced schizophrenia-like behavior by morin in mice. Brain Res Bull 139:292–306. https://doi.org/10.1016/j.brainresbull.2018.03.006
Ben-Azu B, Aderibigbe AO, Omogbiya IA, Ajayi AM, Iwalewa EO (2018d) Morin Pretreatment Attenuates Schizophrenia-Like Behaviors in Experimental Animal Models. Drug Res (stuttg) 68:159–167. https://doi.org/10.1055/s-0043-119127
Ben-Azu B, Aderibigbe AO, Ajayi AM, Eneni A-EO, Omogbiya IA, Owoeye O, Umukoro S, Iwalewa EO (2019) Morin decreases cortical pyramidal neuron degeneration via inhibition of neuroinflammation in mouse model of schizophrenia. Int Immunopharmacol 70:338–353. https://doi.org/10.1016/j.intimp.2019.02.052
Ben-Shachar D, Laifenfeld D (2004) Mitochondria, synaptic plasticity, and schizophrenia. Int Rev Neurobiol 59:273–296. https://doi.org/10.1016/S0074-7742(04)59011-6
Berk M, Copolov D, Dean O, Lu K, Jeavons S, Schapkaitz I, Anderson-Hunt M, Judd F, Katz F, Katz P, Ording-Jespersen S, Little J, Conus P, Cuenod M, Do KQ, Bush AI (2008) N-Acetyl Cysteine as a Glutathione Precursor for Schizophrenia—A Double-Blind, Randomized, Placebo-Controlled Trial. Biol Psychiat 64:361–368. https://doi.org/10.1016/j.biopsych.2008.03.004
Bernstein HG, Krell D, Braunewell KH, Baumann B, Gundelfinger ED, Diekmann S, Danos P, Bogerts B (2001) Increased number of nitric oxide synthase immunoreactive Purkinje cells and dentate nucleus neurons in schizophrenia. J Neurocytol 30:661–670. https://doi.org/10.1023/a:1016520932139
Borovikova LV, Ivanova S, Nardi D, Zhang M, Yang H, Ombrellino M, Tracey KJ (2000) Role of vagus nerve signaling in CNI-1493-mediated suppression of acute inflammation. Auton Neurosci 85:141–147. https://doi.org/10.1016/S1566-0702(00)00233-2
Bulley S, Shen W (2010) Reciprocal regulation between taurine and glutamate response via Ca2+-dependent pathways in retinal third-order neurons. J Biomed Sci 17(Suppl 1):S5. https://doi.org/10.1186/1423-0127-17-S1-S5
Charan J, Kantharia ND (2013) How to calculate sample size in animal studies? J Pharmacol Pharmacother 4:303–306. https://doi.org/10.4103/0976-500X.119726
Chatterjee M, Verma R, Ganguly S, Palit G (2012) Neurochemical and molecular characterization of ketamine-induced experimental psychosis model in mice. Neuropharmacology 63:1161–1171. https://doi.org/10.1016/j.neuropharm.2012.05.041
Cho M, Lee TY, Kwak YB, Yoon YB, Kim M, Kwon JS (2019) Adjunctive use of anti-inflammatory drugs for schizophrenia: A meta-analytic investigation of randomized controlled trials. Aust N Z J Psychiatry 53:742–759. https://doi.org/10.1177/0004867419835028
Coates KM, Flood P (2001) Ketamine and its preservative, benzethonium chloride, both inhibit human recombinant alpha7 and alpha4beta2 neuronal nicotinic acetylcholine receptors in Xenopus oocytes. Br J Pharmacol 134:871–879. https://doi.org/10.1038/sj.bjp.0704315
Contestabile A (2011) The history of the cholinergic hypothesis. Behav Brain Res 221:334–340. https://doi.org/10.1016/j.bbr.2009.12.044
de Araújo FYR, Chaves Filho AJM, Nunes AM, de Oliveira GV, Gomes PXL, Vasconcelos GS, Carletti J, de Moraes MO, de Moraes ME, Vasconcelos SMM, de Sousa FCF, de Lucena DF, Macedo DS (2021) Involvement of anti-inflammatory, antioxidant, and BDNF up-regulating properties in the antipsychotic-like effect of the essential oil of Alpinia zerumbet in mice: a comparative study with olanzapine. Metab Brain Dis 36:2283–2297. https://doi.org/10.1007/s11011-021-00821-5
del Olmo N, Bustamante J, del Río RM, Solís JM (2000) Taurine activates GABA(A) but not GABA(B) receptors in rat hippocampal CA1 area. Brain Res 864:298–307. https://doi.org/10.1016/s0006-8993(00)02211-3
Dietrich-Muszalska A, Kolodziejczyk-Czepas J, Nowak P (2021) Comparative study of the effects of atypical antipsychotic drugs on plasma and urine biomarkers of oxidative stress in schizophrenic patients. Neuropsychiatr Dis Treat 17:555–565. https://doi.org/10.2147/NDT.S283395
Dipasquale S, Pariante CM, Dazzan P, Aguglia E, McGuire P, Mondelli V (2013) The dietary pattern of patients with schizophrenia: a systematic review. J Psychiatr Res 47:197–207. https://doi.org/10.1016/j.jpsychires.2012.10.005
Do KQ, Trabesinger AH, Kirsten-Krüger M, Lauer CJ, Dydak U, Hell D, Holsboer F, Boesiger P, Cuénod M (2000) Schizophrenia: glutathione deficit in cerebrospinal fluid and prefrontal cortex in vivo. Eur J Neurosci 12:3721–3728. https://doi.org/10.1046/j.1460-9568.2000.00229.x
Edelstein AD, Tsuchida MA, Amodaj N, Pinkard H, Vale RD, Stuurman N (2014) Advanced methods of microscope control using μManager software. J Biol Methods 1:e10–e10. https://doi.org/10.14440/jbm.2014.36
El Idrissi A (2008) Taurine increases mitochondrial buffering of calcium: role in neuroprotection. Amino Acids 34:321–328. https://doi.org/10.1007/s00726-006-0396-9
Firth J, Rosenbaum S, Ward PB, Curtis J, Teasdale SB, Yung AR, Sarris J (2018) Adjunctive nutrients in first-episode psychosis: A systematic review of efficacy, tolerability and neurobiological mechanisms. Early Interv Psychiatry 12:774–783. https://doi.org/10.1111/eip.12544
Francisco EDS, Guedes RCA (2015) Neonatal taurine and alanine modulate anxiety-like behavior and decelerate cortical spreading depression in rats previously suckled under different litter sizes. Amino Acids 47:2437–2445. https://doi.org/10.1007/s00726-015-2036-8
Franscescon F, Souza TP, Müller TE, Michelotti P, Canzian J, Stefanello FV, Rosemberg DB (2021) Taurine prevents MK-801-induced shoal dispersion and altered cortisol responses in zebrafish. Prog Neuropsychopharmacol Biol Psychiatry 111:110399. https://doi.org/10.1016/j.pnpbp.2021.110399
Frydecka D, Misiak B, Pawlak-Adamska E, Karabon L, Tomkiewicz A, Sedlaczek P, Kiejna A, Beszłej JA (2015) Interleukin-6: the missing element of the neurocognitive deterioration in schizophrenia? The focus on genetic underpinnings, cognitive impairment and clinical manifestation. Eur Arch Psychiatry Clin Neurosci 265:449–459. https://doi.org/10.1007/s00406-014-0533-5
Gawryluk JW, Wang J-F, Andreazza AC, Shao L, Young LT (2011) Decreased levels of glutathione, the major brain antioxidant, in post-mortem prefrontal cortex from patients with psychiatric disorders. Int J Neuropsychopharmacol 14:123–130. https://doi.org/10.1017/S1461145710000805
Ghandforoush-Sattari M, Mashayekhi S, Krishna CV, Thompson JP, Routledge PA (2010) Pharmacokinetics of oral taurine in healthy volunteers. J Amino Acids 2010:346237. https://doi.org/10.4061/2010/346237
Gobira PH, Ropke J, Aguiar DC, Crippa JAS, Moreira FA (2013) Animal models for predicting the efficacy and side effects of antipsychotic drugs. Braz J Psychiatry 35(Suppl 2):S132-139. https://doi.org/10.1590/1516-4446-2013-1164
Graham KA, Keefe RS, Lieberman JA, Calikoglu AS, Lansing KM, Perkins DO (2015) Relationship of low vitamin D status with positive, negative and cognitive symptom domains in people with first-episode schizophrenia. Early Interv Psychiatry 9:397–405. https://doi.org/10.1111/eip.12122
Harrison PJ (2000) Postmortem studies in schizophrenia. Dialogues Clin Neurosci 2:349–357
Herrmann AP, Lunardi P, Pilz LK, Tramontina AC, Linck VM, Okunji CO, Gonçalves CA, Elisabetsky E (2012) Effects of the putative antipsychotic alstonine on glutamate uptake in acute hippocampal slices. Neurochem Int 61:1144–1150. https://doi.org/10.1016/j.neuint.2012.08.006
Hong J, Bang M (2020) Anti-inflammatory strategies for schizophrenia: A review of evidence for therapeutic applications and drug repurposing. Clin Psychopharmacol Neurosci 18:10–24
Hussy N, Deleuze C, Desarménien MG, Moos FC (2000) Osmotic regulation of neuronal activity: a new role for taurine and glial cells in a hypothalamic neuroendocrine structure. Prog Neurobiol 62:113–134. https://doi.org/10.1016/s0301-0082(99)00071-4
Ishola IO, Ben-Azu B, Adebayo OA, Ajayi AM, Omorodion IL, Edje KE, Adeyemi OO (2021) Prevention and reversal of ketamine-induced experimental psychosis in mice by the neuroactive flavonoid, hesperidin: The role of oxidative and cholinergic mechanisms. Brain Res Bull 177:239–251
Jakaria M, Azam S, Haque ME, Jo SH, Uddin MS, Kim IS, Choi DK (2019) Taurine and its analogs in neurological disorders: Focus on therapeutic potential and molecular mechanisms. Redox Biol 24:101223
Jeppesen R, Christensen RHB, Pedersen EMJ, Nordentoft M, Hjorthøj C, Köhler-Forsberg O, Benros ME (2020) Efficacy and safety of anti-inflammatory agents in treatment of psychotic disorders - A comprehensive systematic review and meta-analysis. Brain Behav Immun 90:364–380
Jiang Y, Ma H, Wang X, Wang Z, Yang Y, Li L, Feng T (2021) Protective Effect of the α7 Nicotinic Receptor Agonist PNU-282987 on Dopaminergic Neurons Against 6-Hydroxydopamine, Regulating Anti-neuroinflammatory and the Immune Balance Pathways in Rat. Front Aging Neurosci 12:606927
Jong CJ, Azuma J, Schaffer S (2012) Mechanism underlying the antioxidant activity of taurine: prevention of mitochondrial oxidant production. Amino Acids 42:2223–2232. https://doi.org/10.1007/s00726-011-0962-7
Kapur S, Zipursky R, Jones C, Remington G, Houle S (2000) Relationship between dopamine D(2) occupancy, clinical response, and side effects: a double-blind PET study of first-episode schizophrenia. Am J Psychiatry 157:514–520. https://doi.org/10.1176/appi.ajp.157.4.514
Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, Heninger GR, Bowers MB, Charney DS (1994) Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry 51:199–214. https://doi.org/10.1001/archpsyc.1994.03950030035004
Lazarevic V, Yang Y, Flais I, Svenningsson P (2021) Ketamine decreases neuronally released glutamate via retrograde stimulation of presynaptic adenosine A1 receptors. Mol Psychiatry 26:7425–7435. https://doi.org/10.1038/s41380-021-01246-3
Leon R, Wu H, Jin Y, Wei J, Buddhala C, Prentice H, Wu J-Y (2009) Protective function of taurine in glutamate-induced apoptosis in cultured neurons. J Neurosci Res 87:1185–1194. https://doi.org/10.1002/jnr.21926
Li G, Liu Y, Olson JE (2002) Calcium/calmodulin-modulated chloride and taurine conductances in cultured rat astrocytes. Brain Res 925:1–8. https://doi.org/10.1016/s0006-8993(01)03235-8
Li X-W, Gao H-Y, Liu J (2017) The role of taurine in improving neural stem cells proliferation and differentiation. Nutr Neurosci 20:409–415. https://doi.org/10.1080/1028415X.2016.1152004
Lidow MS (2003) Calcium signaling dysfunction in schizophrenia: a unifying approach. Brain Res Brain Res Rev 43:70–84. https://doi.org/10.1016/s0165-0173(03)00203-0
Liu F, Guo X, Wu R, Ou J, Zheng Y, Zhang B, Xie L, Zhang L, Yang L, Yang S, Yang J, Ruan Y, Zeng Y, Xu X, Zhao J (2014) Minocycline supplementation for treatment of negative symptoms in early-phase schizophrenia: a double blind, randomized, controlled trial. Schizophr Res 153:169–176. https://doi.org/10.1016/j.schres.2014.01.011
MacDowell KS, García-Bueno B, Madrigal JL, Parellada M, Arango C, Micó JA, Leza JC (2013) Risperidone normalizes increased inflammatory parameters and restores anti-inflammatory pathways in a model of neuroinflammation. Int J Neuropsychopharmacol 16:121–135
Maleki V, Mahdavi R, Hajizadeh-Sharafabad F, Alizadeh M (2020) The effects of taurine supplementation on oxidative stress indices and inflammation biomarkers in patients with type 2 diabetes: a randomized, double-blind, placebo-controlled trial. Diabetol Metab Syndr 12:9. https://doi.org/10.1186/s13098-020-0518-7
Marcinkiewicz J, Kontny E (2014) Taurine and inflammatory diseases. Amino Acids 46:7–20. https://doi.org/10.1007/s00726-012-1361-4
Mirabella F, Desiato G, Mancinelli S, Fossati G, Rasile M, Morini R, Markicevic M, Grimm C, Amegandjin C, Termanini A, Peano C, Kunderfranco P, di Cristo G, Zerbi V, Menna E, Lodato S, Matteoli M, Pozzi D (2021) Prenatal interleukin 6 elevation increases glutamatergic synapse density and disrupts hippocampal connectivity in offspring. Immunity 54:2611-2631.e8. https://doi.org/10.1016/j.immuni.2021.10.006
Moe AAK, Medely GA, Reeks T, Burne THJ, Eyles DW (2017) Short- and long-term effects of risperidone on catalepsy sensitisation and acquisition of conditioned avoidance response: Adolescent vs adult rats. Pharmacol Res 121:1–13. https://doi.org/10.1016/j.phrs.2017.04.013
Monte AS, de Souza GC, McIntyre RS, Soczynska JK, dos Santos JV, Cordeiro RC, Ribeiro BMM, de Lucena DF, Vasconcelos SMM, de Sousa FCF, Carvalho AF, Macêdo DS (2013) Prevention and reversal of ketamine-induced schizophrenia related behavior by minocycline in mice: Possible involvement of antioxidant and nitrergic pathways. J Psychopharmacol 27:1032–1043. https://doi.org/10.1177/0269881113503506
Mouri A, Noda Y, Noda A, Nakamura T, Tokura T, Yura Y, Nitta A, Furukawa H, Nabeshima T (2007) Involvement of a dysfunctional dopamine-D1/N-methyl-d-aspartate-NR1 and Ca2+/calmodulin-dependent protein kinase II pathway in the impairment of latent learning in a model of schizophrenia induced by phencyclidine. Mol Pharmacol 71:1598–1609. https://doi.org/10.1124/mol.106.032961
Müller N, Krause D, Dehning S, Musil R, Schennach-Wolff R, Obermeier M, Möller H-J, Klauss V, Schwarz MJ, Riedel M (2010) Celecoxib treatment in an early stage of schizophrenia: results of a randomized, double-blind, placebo-controlled trial of celecoxib augmentation of amisulpride treatment. Schizophr Res 121:118–124. https://doi.org/10.1016/j.schres.2010.04.015
Nakao A, Matsunaga Y, Hayashida K, Takahashi N (2021) Role of Oxidative Stress and Ca2+ Signaling in Psychiatric Disorders. Front Cell Dev Biol 9:615569. https://doi.org/10.3389/fcell.2021.615569
Nasyrova RF, Ivashchenko DV, Ivanov MV, Neznanov NG (2015) Role of nitric oxide and related molecules in schizophrenia pathogenesis: biochemical, genetic and clinical aspects. Front Physiol 6:139. https://doi.org/10.3389/fphys.2015.00139
Neill JC, Barnes S, Cook S, Grayson B, Idris NF, McLean SL, Snigdha S, Rajagopal L, Harte MK (2010) Animal models of cognitive dysfunction and negative symptoms of schizophrenia: focus on NMDA receptor antagonism. Pharmacol Ther 128:419–432. https://doi.org/10.1016/j.pharmthera.2010.07.004
Niu X, Zheng S, Liu H, Li S (2018) Protective effects of taurine against inflammation, apoptosis, and oxidative stress in brain injury. Mol Med Rep 18:4516–4522. https://doi.org/10.3892/mmr.2018.9465
O’Dell TJ, Hawkins RD, Kandel ER, Arancio O (1991) Tests of the roles of two diffusible substances in long-term potentiation: evidence for nitric oxide as a possible early retrograde messenger. Proc Natl Acad Sci USA 88:11285–11289
O’Donnell CP, Allott KA, Murphy BP, Yuen HP, Proffitt T-M, Papas A, Moral J, Pham T, O’Regan MK, Phassouliotis C, Simpson R, McGorry PD (2016) Adjunctive taurine in first-episode psychosis: A phase 2, double-blind, randomized, placebo-controlled study. J Clin Psychiatry 77:e1610–e1617. https://doi.org/10.4088/JCP.15m10185
Ohtsuki S (2004) Physiological function of blood-brain barrier transporters as the CNS supporting and protecting system. Yakugaku Zasshi 124:791–802. https://doi.org/10.1248/yakushi.124.791
Oshodi TO, Ben-Azu B, Ishola IO, Ajayi AM, Emokpae O, Umukoro S (2021) Molecular mechanisms involved in the prevention and reversal of ketamine-induced schizophrenia-like behavior by rutin: the role of glutamic acid decarboxylase isoform-67, cholinergic, Nox-2-oxidative stress pathways in mice. Mol Biol Rep 48:2335–2350
Oyovwi MO, Nwangwa EK, Ben-Azu B, Edesiri TP, Emojevwe V, Igweh JC (2021) Taurine and coenzyme Q10 synergistically prevent and reverse chlorpromazine-induced psycho-neuroendocrine changes and cataleptic behavior in rats. Naunyn Schmiedebergs Arch Pharmacol 394:717–734. https://doi.org/10.1007/s00210-020-02003-z
Picón-Pagès P, Garcia-Buendia J, Muñoz FJ (2019) Functions and dysfunctions of nitric oxide in brain. Biochim Biophys Acta Mol Basis Dis 1865:1949–1967
Potkin SG, Kane JM, Correll CU, Lindenmayer J-P, Agid O, Marder SR, Olfson M, Howes OD (2020) The neurobiology of treatment-resistant schizophrenia: paths to antipsychotic resistance and a roadmap for future research. NPJ Schizophr 6:1–10. https://doi.org/10.1038/s41537-019-0090-z
Racki V, Marcelic M, Stimac I, Petric D, Kucic N (2021) Effects of haloperidol, risperidone, and aripiprazole on the immunometabolic properties of BV-2 microglial cells. Int J Mol Sci 22(9):4399. https://doi.org/10.3390/ijms22094399
Rafiee Z, García-Serrano AM, Duarte JMN (2022) Taurine supplementation as a neuroprotective strategy upon brain dysfunction in metabolic syndrome and diabetes. Nutrients 14:1292. https://doi.org/10.3390/nu14061292
Rahmeier FL, Zavalhia LS, Tortorelli LS, Huf F, Géa LP, Meurer RT, Machado AC, Gomez R, Fernandes MDC (2016) The effect of taurine and enriched environment on behaviour, memory and hippocampus of diabetic rats. Neurosci Lett 630:84–92. https://doi.org/10.1016/j.neulet.2016.07.032
Rosemberg DB, da Rocha RF, Rico EP, Zanotto-Filho A, Dias RD, Bogo MR, Bonan CD, Moreira JCF, Klamt F, Souza DO (2010) Taurine prevents enhancement of acetylcholinesterase activity induced by acute ethanol exposure and decreases the level of markers of oxidative stress in zebrafish brain. Neuroscience 171:683–692. https://doi.org/10.1016/j.neuroscience.2010.09.030
Sahebnasagh A, Saghafi F, Negintaji S, Hu T, Shabani-Borujeni M, Safdari M, Ghaleno HR, Miao L, Qi Y, Wang M, Liao P, Sureda A, Simal-Gándara J, Nabavi SM, Xiao J (2022) Nitric oxide and immune responses in cancer: Searching for new therapeutic strategies. Curr Med Chem 29:1561–1595
Sánchez-Ramón S, Faure F, Jolles S, Leboyer M, Tremblay M-È (2021) Editorial: The crossroads between immunological disorders and neuropsychiatric diseases. A case for schizophrenia. Front Cell Neurosci 15:733997. https://doi.org/10.3389/fncel.2021.733997
Satsu H, Manabe M, Shimizu M (2004) Activation of Ca2+/calmodulin-dependent protein kinase II is involved in hyperosmotic induction of the human taurine transporter. FEBS Lett 569:123–128. https://doi.org/10.1016/j.febslet.2004.05.062
Schaffer SW, Ito T, Azuma J (2014) Clinical significance of taurine. Amino Acids 46:1–5. https://doi.org/10.1007/s00726-013-1632-8
Shahani N, Sawa A (2011) Nitric oxide signaling and nitrosative stress in neurons: role for S-nitrosylation. Antioxid Redox Signal 14:1493–1504. https://doi.org/10.1089/ars.2010.3580
Shivakumar V, Kalmady SV, Amaresha AC, Jose D, Narayanaswamy JC, Agarwal SM, Joseph B, Venkatasubramanian G, Ravi V, Keshavan MS, Gangadhar BN (2015) Serum vitamin D and hippocampal gray matter volume in schizophrenia. Psychiatry Res 233:175–179. https://doi.org/10.1016/j.pscychresns.2015.06.006
Silva ELFS, Brito MD, Yuzawa JMC, Rosenstock TR (2019) Mitochondrial Dysfunction and Changes in High-Energy Compounds in Different Cellular Models Associated to Hypoxia: Implication to Schizophrenia. Sci Rep 9:18049
Szabadits E, Cserép C, Szőnyi A, Fukazawa Y, Shigemoto R, Watanabe M, Itohara S, Freund TF, Nyiri G (2011) NMDA Receptors in hippocampal GABAergic synapses and their role in nitric oxide signaling. J Neurosci 31:5893–5904. https://doi.org/10.1523/JNEUROSCI.5938-10.2011
Takatani T, Takahashi K, Uozumi Y, Shikata E, Yamamoto Y, Ito T, Matsuda T, Schaffer SW, Fujio Y, Azuma J (2004) Taurine inhibits apoptosis by preventing formation of the Apaf-1/caspase-9 apoptosome. Am J Physiol Cell Physiol 287:C949-953. https://doi.org/10.1152/ajpcell.00042.2004
Tsurugizawa T, Uneyama H, Torii K (2014) Brain amino acid sensing. Diabetes Obes Metab 16(Suppl 1):41–48. https://doi.org/10.1111/dom.12336
Upthegrove R, Khandaker GM (2020) Cytokines, oxidative stress and cellular markers of inflammation in schizophrenia. Curr Top Behav Neurosci 44:49–66. https://doi.org/10.1007/7854_2018_88
Valtcheva S, Venance L (2019) Control of long-term plasticity by glutamate transporters. Front Synaptic Neurosci 9(11):10. https://doi.org/10.3389/fnsyn.2019.00010
Wang L, Alachkar A, Sanathara N, Belluzzi JD, Wang Z, Civelli O (2015) A Methionine-Induced Animal Model of Schizophrenia: Face and Predictive Validity. Int J Neuropsychopharmacol 18:pyv054. https://doi.org/10.1093/ijnp/pyv054
Wei Y, Lu M, Mei M, Wang H, Han Z, Chen M, Yao H, Song N, Ding X, Ding J, Xiao M, Hu G (2020) Pyridoxine induces glutathione synthesis via PKM2-mediated Nrf2 transactivation and confers neuroprotection. Nat Commun 11:941
West AR, Grace AA (2000) Striatal nitric oxide signaling regulates the neuronal activity of midbrain dopamine neurons in vivo. J Neurophysiol 83:1796–1808. https://doi.org/10.1152/jn.2000.83.4.1796
Whirley BK, Einat H (2008) Taurine trials in animal models offer no support for anxiolytic, antidepressant or stimulant effects. Isr J Psychiatry Relat Sci 45:11–18
Wu J-Y, Prentice H (2010) Role of taurine in the central nervous system. J Biomed Sci 17(Suppl 1):S1. https://doi.org/10.1186/1423-0127-17-S1-S1
Wu J-Y, Wu H, Jin Y, Wei J, Sha D, Prentice H, Lee H-H, Lin C-H, Lee Y-H, Yang L-L (2009) Mechanism of neuroprotective function of taurine. Adv Exp Med Biol 643:169–179. https://doi.org/10.1007/978-0-387-75681-3_17
Wu G-F, Ren S, Tang R-Y, Xu C, Zhou J-Q, Lin S-M, Feng Y, Yang Q-H, Hu J-M, Yang J-C (2017) Antidepressant effect of taurine in chronic unpredictable mild stress-induced depressive rats. Sci Rep 7:4989. https://doi.org/10.1038/s41598-017-05051-3
Yang J, Guo H, Sun D, Duan J, Rao X, Xu F, Manyande A, Tang Y, Wang J, Wang F (2019) Elevated glutamate, glutamine and GABA levels and reduced taurine level in a schizophrenia model using an in vitro proton nuclear magnetic resonance method. Am J Transl Res 11:5919–5931
Yang X, Li M, Jiang J, Hu X, Qing Y, Sun L, Yang T, Wang D, Cui G, Gao Y, Zhang J, Li X, Shen Y, Qin S, Wan C (2021) Dysregulation of phospholipase and cyclooxygenase expression is involved in Schizophrenia. EBioMedicine 64:103239. https://doi.org/10.1016/j.ebiom.2021.103239
Yao WD, Wu CF (2001) Distinct roles of CaMKII and PKA in regulation of firing patterns and K(+) currents in Drosophila neurons. J Neurophysiol 85:1384–1394. https://doi.org/10.1152/jn.2001.85.4.1384
Zalcman S, Green-Johnson JM, Murray L, Nance DM, Dyck D, Anisman H, Greenberg AH (1994) Cytokine-specific central monoamine alterations induced by interleukin-1, -2 and -6. Brain Res 643:40–49. https://doi.org/10.1016/0006-8993(94)90006-x
Zebrowska-Lupina I, Porowska A (1987) Anticataleptic effect of taurine: interaction with antiparkinsonian agents. Pol J Pharmacol Pharm 39:329–335
Zhang L, Zhao J (2014) Profile of minocycline and its potential in the treatment of schizophrenia. Neuropsychiatr Dis Treat 10:1103–1111. https://doi.org/10.2147/NDT.S64236
Zhang XY, Zhou DF, Cao LY, Wu GY, Shen YC (2005) Cortisol and cytokines in chronic and treatment-resistant patients with schizophrenia: Association with psychopathology and response to antipsychotics. Neuropsychopharmacol 30:1532–1538. https://doi.org/10.1038/sj.npp.1300756
Zhang XY, Zhou DF, Shen YC, Zhang PY, Zhang WF, Liang J, Chen DC, Xiu MH, Kosten TA, Kosten TR (2012) Effects of risperidone and haloperidol on superoxide dismutase and nitric oxide in schizophrenia. Neuropharmacology 62:1928–1934. https://doi.org/10.1016/j.neuropharm.2011.12.014
Zhang Z, Liu D, Yi B, Liao Z, Tang L, Yin D, He M (2014) Taurine supplementation reduces oxidative stress and protects the liver in an iron-overload murine model. Mol Med Rep 10:2255–2262. https://doi.org/10.3892/mmr.2014.2544
Zhang Y, Li D, Li H, Hou D, Hou J (2016) Taurine pretreatment prevents isoflurane-induced cognitive impairment by inhibiting ER stress-mediated activation of apoptosis pathways in the hippocampus in aged rats. Neurochem Res 41:2517–2525. https://doi.org/10.1007/s11064-016-1963-4
Zhang Y, Shi H, Yang G, Yang Y, Li W, Song M, Shao M, Su X, Lv L (2021) Associations between expression of indoleamine 2, 3-dioxygenase enzyme and inflammatory cytokines in patients with first-episode drug-naive Schizophrenia. Transl Psychiatry 11:1–8. https://doi.org/10.1038/s41398-021-01688-x
Zhu F, Zheng Y, Ding YQ, Liu Y, Zhang X, Wu R, Guo X, Zhao J (2014) Minocycline and risperidone prevent microglia activation and rescue behavioral deficits induced by neonatal intrahippocampal injection of lipopolysaccharide in rats. PLoS One 9(4):e93966. https://doi.org/10.1371/journal.pone.0093966
Funding
Benneth Ben-Azu was supported by an International Brain Research Organization African Regional Committee (IBRO-ARC) 2019 Grant for a Postdoctoral Fellowship on Schizophrenia Research at the University of Victoria, BC Canada.
Author information
Authors and Affiliations
Contributions
All authors contributed to this research work and the development of the final manuscript.
MOO, BBA, BSC, OSO, OPO, AO, DTE and EOO designed the study and wrote the protocols; MOO, BBA, BSC, OSO, OPO, AO, DTE, EOO, KJ, TAJ and EOO performed the experiments; MOO, BSC, OSO, OPO, AO, DTE, EOO, KJ, EGM and EOO did the behavioral study; BBA, ATE, IAO, KEE and MOO did the biochemical study; BBA and TAJ performed the immunohistochemistry. MOO, BBA, BSC, OSO, OPO, AO, DTE, EOO, EGM and MOO analyzed the data and literature searches; BBA, MOO, BSC, OSO, OPO, AO, DTE, EOO, KJ and EOO contributed reagents/materials/analysis tools; BBA, OSO, OPO, AO, DTE, EOO, KJ wrote the first draft of the manuscript; BBA, EGM, MOO, IOA completed the final draft of the paper.
Corresponding author
Ethics declarations
Ethical approval
The study protocol was approved by the Delta State University Animal Care and Use Research Ethics Committee (REC/FBMS/DELSU/21/94) in accordance with the directions from National institutes of Health Guide for Care and Use of Laboratory Animals (Publication No. 85–23, revised 1985).
Conflicts of interest
Authors declare that they have no conflict of interest.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ben-Azu, B., Adebayo, O.G., Jarikre, T.A. et al. Taurine, an essential β-amino acid insulates against ketamine-induced experimental psychosis by enhancement of cholinergic neurotransmission, inhibition of oxidative/nitrergic imbalances, and suppression of COX-2/iNOS immunoreactions in mice. Metab Brain Dis 37, 2807–2826 (2022). https://doi.org/10.1007/s11011-022-01075-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11011-022-01075-5