
Abstract
Nonketotic hyperglycinemia is an autosomal recessive inborn error of glycine metabolism, characterized by deficient activity of the glycine cleavage enzyme system. Classic nonketotic hyperglycinemia is caused by mutations or genomic changes in genes that encode the protein components of the glycine cleavage enzyme system. We aimed to investigate clinical, biochemical, radiological findings and molecular genetic data in ten Turkish patients with classic nonketotic hyperglycinemia. Ten Turkish patients who were diagnosed with classic nonketotic hyperglycinemia in a single center from 2013 to 2019 were included in this study. Their clinical, radiological, electrophysiological and laboratory data were collected retrospectively. Sixty percent of the patients were in neonatal group, while 40 % of the patients were infantile. There were no late-onset patients. 90 % of the patients had the severe form. All patients had developmental delay and seizures. Mortality ratio was 30 % in all groups and 50 % in the neonatal group, while no mortality was seen in infantile group. Median (range) values of cerebrospinal fluid (CSF) glycine levels, plasma glycine levels and CSF/plasma glycine ratios were 148 (15–320) µmol/L, 896 (87-1910) µmol/L, 0.17 (0.09–0.21) respectively. Diffuse hypomyelination and corpus callosum anomaly were the most common cranial MRI findings and multifocal epileptic activity and burst supression pattern were the most common electroencephalographic findings. Six patients had variants in GLDC gene and four in AMT gene; five novel variants including AMT gene deletion were detected. Prognosis was poor and treatment was not effective, especially in the severe form. Classic nonketotic hyperglycinemia causes high morbidity and mortality. Neonatal-onset disease was more common and severe than infantile-onset disease. The ratio of AMT gene variants might be higher in Turkey than other countries. AMT gene deletion also plays a role in the etiology of classic nonketotic hyperglycinemia.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glycine encephalopathy (OMIM #605,899), more commonly known as nonketotic hyperglycinemia (NKH), is an autosomal recessive, debilitating disease of the glycine catabolic pathway. Decrease in the activity of the mitochondrial glycine cleavage system (GCS, EC 2.1.2.10) causes glycine accumulates in body fluids (Tada et al. 1969). On the glycine receptors in the brain stem and spinal cord, glycine serves as an inhibitory neurotransmitter, and contributes to the development of hiccups, apnea and hyptonia. Conversely, the glycinergic receptor is not inhibitory, but excitatory in neuronal stem cells. Deficiency of GCS may impair development of the embryonic brain, resulting in congenital brain malformations (Dinopoulos et al. 2005; Applegarth and Toone 2006). Glycine is an activator of the N-methyl-D-aspartate (NMDA) type glutamate receptor in the brain, cortex, cerebellum and basal ganglia, which is an excitatory receptor that can cause intractable seizures and excitotoxicity when overstimulated (Van Hove et al. 2002 [updated 2019]; Van Hove et al. 2016).
The estimated incidence of NKH among live births is highly variable around the globe, ranging from 1:187,500 in Malaysia to 1:9,684 in the Kairouan region of Tunisia, making up a worldwide incidence of 1:76,000 (von Wendt et al. 1979; Applegarth et al. 2000; Verissimo et al. 2013; Azize et al. 2014; Chiu et al. 2016; Coughlin et al. 2017; Nasrallah et al. 2020). The highest incidence reported so far is in an Arab village in northern Israel, with an estimated rate of 1:108 live births, possibly secondary to a founder effect in a large Palestinian kindred (Kure et al. 1999; Nasrallah et al. 2020).
Based on the molecular pathogenesis, NKH can be classified into classic NKH (caused by biallelic variants in GLDC, AMT, or GCSH genes, which code for the protein components of GCS), and variant NKH (associated with the synthesis and transport of the cofactor lipoate, related to LIAS, BOLA3, NFU1, GLRX5, ISCA2, IBA57, LIPT1 and LIPT2 genes). Classic NKH can be further classified into ‘severe’ and ‘attenuated’ forms, based on the clinical severity and developmental outcome. Patients with severe classic NKH have only minimal psychomotor development and universally develop refractory epilepsy; whereas those with the attenuated form make variable developmental progress and present with different symptoms, including attention deficits, hyperactivity, chorea and episodic lethargy (Van Hove et al. 2002 [updated 2019]; Van Hove et al. 2016). Based on the time of symptom onset, classic NKH can be divided into neonatal (< 2 weeks), infantile (2 weeks-3 months) and late-onset (> 3 months) disease (Van Hove et al. 2002 [updated 2019]; Van Hove et al. 2016). It has been reported for classic NKH that 85 % of neonatal onset cases are severe and 15 % are mild phenotypes, whereas 50 % of infantile cases are severe and 50 % are mild, and; all late onset cases are mild phenotype (Van Hove et al. 2002 [updated 2019]; Van Hove et al. 2016).
The glycine cleavage system is located on the inner mitochondrial membranes of various tissues, including the liver, kidney, brain and placenta, and is comprised of a four-protein complex (P, T, H, L). The majority (80 %) of classic NKH cases are caused by pathogenic variants in GLDC, encoding the pyridoxal phosphate dependent P protein, and 20 % by those in AMT, encoding the tetrahydrofolate-requiring T protein. The most common disease-causing variants are missense, splicing and insertion deletion (indel) variants in both genes (Van Hove et al. 2002 [updated 2019]; Kure et al. 2006; Kanno et al. 2007; Van Hove et al. 2016; Bravo-Alonso et al. 2017). In GLDC, 20 % of disease-causing alleles are intragenic deletions, while intragenic deletions or duplications within AMT have not yet been reported (Van Hove et al. 2002 [updated 2019]; Kanno et al. 2007; Swanson et al. 2015; Van Hove et al. 2016; Bravo-Alonso et al. 2017; Coughlin et al. 2017). Nearly 5 % of patients with deficient GCS activity have no pathogenic variants in GLDC or AMT, which comprise variant NKH (Van Hove et al. 2002 [updated 2019]; Baker et al. 2014; Van Hove et al. 2016). Since most variants are private, reliable genotype-phenotype correlations have not been established.
In this present study we reviewed our classic NKH patients retrospectively to investigate genotype-phenotype features in ten Turkish patients.
Materials and methods
All patients diagnosed with classic NKH by clinical, biochemical and molecular analyses from 2013 to 2019 in our single center were included in this study. Their clinical, radiological, electrophysiological and laboratory data including plasma and cerebrospinal fluid (CSF) glycine levels, CSF/plasma glycine ratio and molecular genetic analyses were collected retrospectively. Plasma and CSF samples were taken simultaneously and if the CSF glycine level was elevated, patients with CSF/plasma glycine ratio (N ≤ 0.02) was calculated. Causes of false positive high CSF glycine levels, including bloody CSF or cases of impaired blood brain barrier like in case of neonatal ischemia or after prolonged seizures, were excluded. Ketotic hyperglycinemia such as organic acidemias were also excluded with normal urine organic acid analyses. Patients were classified into three clinical subtypes (neonatal, infantile and late onset) based on the onset of clinical symptoms and also classified as severe or attenuated phenotype based on severity of symptoms as mentioned in the introduction.
CSF and plasma glycine were assessed by ion-exchange chromatography using a single-column amino acid analyzer (Beckman 6300 analyzer, Beckman Insruments. Palo Alto, California, United States). Urine organic acid profile was analyzed by gas chromatography-mass spectrometry on a HP 5890 series II gas chromatograph coupled to a HP 5972 mass selective detector (Agilent, Atlanta, Georgia, United States).
Molecular analysis of the GLDC, AMT and GCSH genes were performed on genomic DNA by next generation sequencing (NGS) methods (for all patients except patient no 8), using DNA extracted from peripheral lymphocytes. Multiplex ligation probe amplification (MLPA) was performed on two patients (patient no: 5, 7) and planned for one patient (patient no: 9) if no mutations or only a single heterozygous mutation were detected by NGS. Patient 8 had been previously diagnosed with classic NKH with metabolic disorders gene panel. Confirmation with Sanger sequencing was not available in this patient (patient no: 8). NM_000170.2 was used as the reference sequence for GLDC gene and NM_000481.3 was used for AMT gene and NM_004483.4 was used for GCSH gene.
Next-generation sequencing (for all patients except patient no: 8)
DNA was isolated with the magnetic bead method (MagPurix- Zinexts, Taiwan). PCR amplification was performed, using in-house designed primers. Amplicons were checked by 2 % agarose gel electrophoresis. Sequencing was performed by the next-generation sequencing method by Miseq- Illumina equipment (Illumina, San Diego, CA, USA) by following manufacturer’s instructions. Data were evaluated by IGV 2.3 (Broad Institute) software. Variants were evaluated by ACMG criteria.
Multiplex ligation probe amplification (MLPA)
Exonic copy-number variants (CNVs) were evaluated by MLPA by using SALSA MLPA P209-C2 kit for patients 5, 7 who approved this second test and planned for patient 9.
Metabolic disorders gene panel sequencing (for patient no: 8)
Semiconductor sequencing technology (Ion Torrent, Thermo Fischer Scientific, USA) was used for the analysis of patient 8. Tests were either done with PGM by using 318 chip or by S5 using 540 chip. Human Gene Mutation Database was used as the primary database for the previously defined mutations. For the novel missense changes Polyphen system (for patient no: 8) was used for in silico analysis. Mean coverage range was around 150 with over 90 % on target and homogenous reads. Ion Reporter System (Thermo-Fischer) and Igenuity Variant Analysis (Qiagen) softwares were used for bioinformatic analysis.
Sanger sequencing (confirmation for patient no: 8)
Genomic DNA was isolated from peripheral blood with salting out excraction procedure. We performed Sanger sequencing to find out pathogenic variants on probands and his parents. Sequencing was carried out using BigDye 3.1 sequencing chemistry (Life Technologies), followed by capillary electrophoresis on the ABI 3500 capillary sequencer (Life Technologies). Primer sequences are available upon request.
The sequences which were alligned to the hq19 reference genome, have been analysed via Integrative Genomic Viewer (IGV) software. The variants were interpreted with the recommendations of American College of Medical Genetics and Genomics and the Association for Molecular Pathology (Richards et al. 2015).
Variants’ pathogenicity was evaluated using SIFT (https://sift.bii.a-star.edu.sg/), PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/), PROVEAN (http://provean.jcvi.org/index.php), DANN (https://cbcl.ics.uci.edu/public_data/DANN/), FATHMM (http://fathmm.biocompute.org.uk/fathmm-xf/) and Mutation Taster (http://www.mutationtaster.org/) prediction algorithms. We assessed frequency in control populations and datasets including the ExAC database, the 1000 Genomes Project (https://gnomad.broadinstitute.org/), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and HGMD (http://www.hgmd.cf.ac.uk/ac/index.php).
Electroencephalography (EEG) recordings were obtained by the NIHON-Cohden machine of 18 channels in the dark, during wakefulness and sleep. Sagittal and axial T1W, T2W and FLAIR brain images were acquired via the magnetic resonance (MR) device (Siemens, Germany) performing at 1.5 Tesla. For cranial magnetic resonance spectroscopy (MRS), a measurement was performed at 3.6 ppm (TE = 135 ms) for glycine signal imaging.
Statistical evaluation was done using Statistical Package for Social Sciences (SPSS) for Windows 20 (IBM SPSS Inc., Chicago, IL). The normal distribution of the data was evaluated with the Kolmogorov-Smirnov test. Numerical variables without normal distribution were presented as median (min-max). Categorical variables were specified as numbers and percentages. Student T test was used to compare numerical variables that showed normal distribution between the groups with disease-onset before or after 2 weeks of life, and Mann-Whitney U test was used to compare numerical variables that did not show normal distribution. In statistical analysis, p < 0.05 was considered significant.
Results
All the patients had developmental delay and seizures. Clinical signs and symptoms were poor sucking/feeding and increased deep tendon reflexes (DTR) in 80 %, microcephaly and hypotonia in 70 %, pathological reflexes in 60 %, hiccups in 40 %, micrognathia in 30 %, vomiting and low set ears in 20 % and spasticity and nystagmus in 10 % of patients. Abnormal echocardiographic findings were detected in 56 % (n: 5/9) of patients including mitral insufficiency, patent ductus arteriosus, patent foramen ovale and dilated cardiomyopathy. Pyloric stenosis and pes equinovarus deformity were detected in 10 % of patients.
Thirty-three percent of patients with neonatal classic NKH (n = 6) had a history of premature birth. All patients with neonatal classic NKH had the severe form of the disease, while patients with infantile NKH (n = 4) presented as a severe form in 75 % of cases and an attenuated form in 25 % of cases.
The CSF/plasma glycine ratio was > 0.08 in nine patients and was not available in one patient (patient no: 8) who was diagnosed by metabolic disorders gene panel screening. The median CSF glycine level was 148 (15–320) µmol/L, the median plasma glycine level was 896 (87-1910) µmol/L and the median CSF/plasma glycine ratio was 0.17 (0.09–0.21). In neonatal classic NKH, the median of glycine level was 228 (26–320) µmol/L, the median plasma glycine level was 1468 (159–1910) µmol/L and the median CSF/plasma glycine ratio was 0.19 (0.09–0.21). In infantile classic NKH, the median CSF glycine level was 74 (15–178) µmol/L, the median plasma glycine level was 522 (87–923) µmol/L and the median CSF/plasma glycine ratio was 0.17 (0.14–0.19). Plasma, CSF and CSF/plasma glycine ratio were higher in the neonatal group than the infantile group, but the difference was not statistically significiant (p > 0.05).
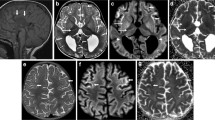
Cranial MRI findings were diffuse hypomyelination in 80 % (n: 8/10), corpus callosum dysgenesis in 60 % (n: 6/10), ventricular dilatation in10 % (n: 1/10), hydrocephalus in 10 % (n: 1/10), and mega cisterna magna in 10 % (n: 1/10). Two of the four patients who underwent cranial MRS had a glycine peak (patient no: 2 and 4) but MRS was normal in two patients (patient no: 3 and 9), one of whom had normal plasma glycine level under treatment at the time of MRS acquisition (patient no: 3). EEG findings were multifocal epileptic activity in 100 % (n: 10/10), burst suppression pattern in 70 % (n: 7/10), hypsarrhythmia in 50 % (n: 5/10) and cerebral dysfunction in 20 % (n: 2/10).
Mutations or genomic changes ratio was 60 % (n: 6/10) in GLDC and 40 % (n: 4/10) in AMT while no mutations in GCSH gene were detected. 80 % (n: 8/10) of patients had homozygous and 20 % (n: 2/10) had heterozygous mutations or genomic alterations in GLDC or AMT gene. Novel homozygous c.1777 C > T;p.G593*, c.2237 A > G;p.D746G, c.2237 A > T;p.D746V and previously identified homozygous c.1867T > C;p.Y623H, c.2113G > A;p.V705M and heterozygous c.2640T > G;p.D880E mutations in the GLDC gene were detected. Novel homozygous total AMT gene deletion, heterozygous c.163T > C;p.W55R mutation and previously known homozygous c.434 A > T;p.N145I and homozygous c.631G > A;p.E211K with heterozygous c.959G > A;p.R320H mutations were detected in AMT gene. MLPA was performed for the GLDC gene for one patient (patient no: 5) to detect the second mutation, but it was normal. MLPA was planned for the AMT and GLDC genes for another patient (patient no: 9) to detect the second mutation. The in silico analysis of the novel mutations in our patients with classic NKH are shown in Table 2. The results of bioinformatic analysis strongly suggest that the documented mutations are disease causing.
Low protein diet (glycine-restricted diet), sodium benzoate (200–500 mg/kg/day) and dextromethorphan (5–10 mg/kg/day) and multiple antiepileptic drugs (≥ 3 drugs) were given with a little clinical response. Only one patient was seizure free and made developmental progress as she was classified in infantile group with attenuated form of classic NKH (patient no: 4). The remaining had severe develepmantal delay and refractory seizures or expired.
Discussion
The differential diagnosis for classic NKH are sepsis, ischemia, brain hemorrhage, brain injury, hypoxic ischemic encephalopathy, congenital stroke, central nervous system infection, infantile epileptic encephalopathies, ketotic hyperglycinemia including propionic, methylmalonic and isovaleric acidemias and beta-ketothiolase deficiency, pyridoxine responsive seizures, vanishing white matter disease, variant NKH and transient NKH. In addition, valproate theraphy (inhibits the GCS), contamination of CSF with blood or with serum (as evidence by increased CSF protein levels) and starvation make pitfalls in measuring CSF glycine levels (Korman and Gutman 2002; Aburahma et al. 2011; Baker et al. 2014; Van Hove et al. 2016). We mostly excluded these disorders and false elevations by taking only mutation positive classic NKH patients in this study.
Incidence of classic NKH is not known in Turkey. Because of higher rate of consanguinity (23 %), it might be more frequent than that estimated in European and other countries. Phenylketonuria (1:6000) and biotinidase deficiency (1:6000), which were included in the national neonatal screening program in our country and determined to have a high frequency, can be given as examples. In this study, consanguinity ratio was higher resulting higher homozygous mutation ratio than other reported studies (Bravo-Alonso et al. 2017; Shbarou et al. 2019; Nasrallah et al. 2020).
Neonatal and severe classic NKH was found to be the most common form of classic NKH as reported previously (Van Hove et al. 2002 [updated 2019]; Hoover-Fong et al. 2004; Shbarou et al. 2019; Nasrallah et al. 2020). High morbidity and mortality in this subgroup of patients in our study supports the dire severity of neonatal classic NKH.
Increased levels of CSF glycine are highly indicative of NKH. The lowest CSF glycine found in a patient with classic NKH was 26 µmol/L (normal < 20 µmol/L) in the literature (Van Hove et al. 2002 [updated 2019]; Swanson et al. 2015; Van Hove et al. 2016; Bravo-Alonso et al. 2017). An elevated CSF/plasma glycine level is a further indication of NKH, but is only valid if the CSF glycine level is elevated. Exceptional patients with attenuated NKH have had normal CSF glycine levels, but this is very rare and the sensitivity of elevated CSF glycine is > 99 %, making it the preferred diagnostic test (Van Hove et al. 2016). Similar to the literature, the lowest CSF glycine level (before treatment) was found 26 µmol/L in our patients with homozygous mutations in this study. According to our experience, CSF glycine levels (after excluding pittfalls) was more reliable than CSF/plasma glycine ratio since it can give erroneous results. Therefore, we do not suggest looking at this ratio for a diagnosis, monitoring and/or prognosis especially in patients without elevated CSF glycine levels. In the presence of similar clinical, radiological and electrophysiological findings of NKH, patients with CSF glycine ≥ 25–50 are possible, 50–100 probable and ≥ 100 definitely make you to think diagnosis of NKH, after excluding secondary elevation of CSF glycine. Thereafter GCS activity and/or genetic testing should be made for the exact diagnosis. Prognostic indicators of severe outcome include high CSF glycine level (> 230 µM) and presence of malformations on brain MRI, whereas prognostic indicators of attenuated NKH are low CSF/plasma glycine ratio (< 0.08), late onset (≥ 4 months) and absence of epilepsy were reported (Swanson et al. 2015; Van Hove et al. 2016; Shbarou et al. 2019). However, intrafamilial phenotypic variability was also previously discussed (Shbarou et al. 2019). In this study, although plasma, CSF and CSF/plasma glycine ratio were higher in neonatal group than infantile group, it was not statistically significiant. Mortality ratio was higher in neonatal group than infantile group and being neonatal group was associated severe outcome.
Abnormal myelination, gyral malformation, progressive atrophy, parenchymal volume loss, corpus callosum dysgenesis and hydrocephalus on brain MRI have been reported in classic NKH (Van Hove et al. 2000; Roy et al. 2004; Mohammad and Abdelkhalek 2017; Shbarou et al. 2019; Stence et al. 2019). Lack of growth of the corpus callosum (shorter length and decreased thickness) was common, and this growth failure was found directly related to the severity of the clinical phenotype (Stence et al. 2019). Diffuse hypomyelination, corpus callosum dysgenesis and rarely hydrocephalus were observed in our patients, similar to the literature. Increased glycine peak in cranial MRS is typical and useful in the diagnosis and monitoring of treatment in NKH have been reported (Shin et al. 2012; Stence et al. 2019). Severe patients were found to have the greatest glycine/creatine ratios (Stence et al. 2019). In our study two patients had a glycine peak but normal in two patients whom one of under therapy. Treatment might decrease the glycine peak in the brain MRS parallel to the decrease in plasma and CSF glycine levels in this patient. Therefore, since lumbar puncture (L/P) is an invasive procedure, brain MRS can be used instead of lumbar puncture in diagnosis and monitoring of treatment in classic NKH.
Abnormal EEG findings are observed in at least 90 % of cases in the neonatal period. Burst suppression pattern is a common EEG finding in NKH, like early myoclonic encephalopathy (Hoover-Fong et al. 2004). We observed mostly multifocal epileptic activity and burst suppression pattern, similar to the literature (Genç-Sel et al. 2018; Shbarou et al. 2019; Nasrallah et al. 2020).
Sequencing and MLPA analysis of the genes involved in the GCS is an excellent confirmatory test in classic NKH (Kure et al. 2006). Nearly 500 unique pathogenic variants were deposited in the Leiden Open Variant Database (LOVD) (Coughlin et al. 2017). Both GLDC and AMT gene alterations were highly heterogeneous, including many private mutations (Nanao et al. 1994; Applegarth and Toone 2004; Conter et al. 2006; Kure et al. 2006; Bravo-Alonso et al. 2017; Coughlin et al. 2017). However recurring pathogenic variants and the founder variants in certain populations were also reported (Kure et al. 1992; Azize et al. 2014; Swanson et al. 2015; Bravo-Alonso et al. 2017; Coughlin et al. 2017). The S564I mutation was found to responsible for high incidence of NKH in Finland and R515S was found in 5 % of Caucasian mutant alleles in GLDC gene (Kure et al. 1992; Toone et al. 2000). In classic NKH, missense mutations associated with residual enzyme activity have been associated with an attenuated phenotype (Kure et al. 2006; Coughlin et al. 2017). In our study, 80 % of patients were found to have homozygous and 20 % had heterozygous mutations. Because of typical clinical, biochemical and radiological findings, two patients with heterozygous mutations (patient no: 5 and 9) were also thought strongly likely to have classic NKH and not to be heterozygous carriers. There might be an allele dropout or the presence of intronic variations affecting mRNA processing or deletion-duplication in the related genes in these two patients. In spite of GLDC/AMT gene alterations nearly 80 %/20 % in literatures; we found 60 %/40 % in this study (Bravo-Alonso et al. 2017; Coughlin et al. 2017). We speculated that AMT gene alterations might be higher in our country than others. Novel mutations were homozygous total AMT gene deletion and heterozygous c.163T > C;p.W55R mutation in AMT gene and homozygous c.1777 C > T;p.G593*, c.2237 A > G;p.D746G and c.2237 A > T;p.D746V mutations in GLDC gene were identified (Table 2). Homozygous c1867T > C;p.Y623H mutation was detected in GLDC gene related to a mild clinical phenotype, while the remaining was related to severe phenotype. Large deletions or duplications of AMT gene have not been reported, but would be expected to be pathogenic due to loss of function disease mechanism (Van Hove et al. 2002 [updated 2019]; Bravo-Alonso et al. 2017). In this study total AMT gene deletion was encountered for the first time.
Although more than a half century has passed since the disease was first described, most of patients still die before the age of five. A curative treatment method to change the prognosis of classic NKH is not available yet. Low protein (glycine and serine restricted diet or a specific glycine-free formula to achieve substrate reduction), sodium benzoate (200–750 mg/kg/day), dextrometorphane (5–35 mg/kg/day) and ketamine (for noncompetitive NMDA receptor antagonist), strychnine and diazepam (1.5-3 mg/kg/day) (to compete with glycine for postsynaptic receptor sites), choline (1–4 g/day), folic acid (2 mg/day) (for single-carbon unit transfer reactions), pyridoxine and tetrahydrofolate (to stimulate residual glycine cleavage activity), tryptophan (50–100 mg/kg) (which is metabolized to kynurenic acid to antagonize glycine at the NMDA receptor), imipramine (20 mg/day) (to antagonize the NMDA receptor) along with other antiepileptic medications have been used with a little success (Matalon et al. 1983; Wiltshire et al. 2000; Van Hove et al. 2005). In our study, glycine restricted diet, sodium benzoate (200–500 mg/kg/day) and dextrometorphane (5–10 mg/kg/day) treatments with multiple antiepileptic drugs were given in all of the patients and a decrease in blood glycine levels was observed. However, although there was a partial benefit in seizure control, prognosis was poor especially in severe cases. Severe developmental delay and mortality could not be prevented. We agree that benzoate treatment results in a reduction of seizure frequency and an improvement of alertness but in severe NKH, it does not ameliorate developmental delay (Wolff et al. 1986; Van Hove et al. 1995, 2016; Hennermann et al. 2012). Because the brain injury caused NKH begins in utero rendering postnatal treatment unsuccessful. Folat therapy to pregnant mother was suggested for deficiency of methylene tetrahydrofolate as a result of defective GCS (Applegarth and Toone 2006). In recent years ketogenic diet with standard pharmacological therapy has been found useful in providing seizure control and improved quality of life in patients of NKH with retractable seizures (Cusmai et al. 2012; Kava et al. 2019; Shbarou et al. 2019). We could not have any experience with ketogenic diet in our NKH patients yet.
Limitation of this study is a lack of second mutations in two patients (patient no: 5 and 9), lack of confirmation with Sanger sequencing in one patient (patient no: 8), lack of cranial MRS in six patients and in one patient before treatment (patient no: 3).
In conclusion, classic NKH causes high morbidity and mortality. New curative treatment modalities shoud be investigated for this devastating disorder, especially for the severe form. The ratio of AMT gene mutations or genomic changes might be higher in Turkey than other countries. AMT gene deletions also play a role in the etiology of classic NKH.
Data availability
All data generated or analysed during this study are included in this published article.
References
Aburahma S, Khassawneh M, Griebel M, Sharp G, Gibson J (2011) Pitfalls in measuring cerebrospinal fluid glycine levels in infants with encephalopathy. J Child Neurol 26:703–706. https://doi.org/10.1177/0883073810389041
Applegarth DA, Toone JR (2004) Glycine encephalopathy (nonketotic hyperglycinaemia): review and update. J Inherit Metab Dis 27:417–422. https://doi.org/10.1023/b:boli.0000031222.38328.59
Applegarth DA, Toone JR (2006) Glycine encephalopathy (nonketotic hyperglycinemia): comments and speculations. Am J Med Genet A 140:186–188. https://doi.org/10.1002/ajmg.a.31030
Applegarth DA, Toone JR, Lowry RB (2000) Incidence of inborn errors of metabolism in British Columbia, 1969–1996. Pediatrics 105:e10. https://doi.org/10.1542/peds.105.1.e10
Azize NA, Ngah WZ, Othman Z, Ngah WZ, Othman Z, Desa N, Chin BC et al (2014) Mutation analysis of glycine decarboxylase, aminomethyltransferase and glycine cleavage system protein-H genes in 13 unrelated families with glycine encephalopathy. J Hum Genet 59:593–597. https://doi.org/10.1038/jhg.2014.69
Baker PR II, Friederich MW, Swanson MA, Shaikh T, Bhattacharya K, Scharer GH, Aicher J et al (2014) Variant non ketotic hyperglycinemia is caused by mutations in LIAS, BOLA3 and the novel gene GLRX5. Brain 137:366–379. https://doi.org/10.1093/brain/awt328
Bravo-Alonso I, Navarrete R, Arribas-Carreira L, Perona A, Abia D, Couce ML, Garcia-Cazorla A et al (2017) Nonketotic hyperglycinemia: Functional assessment of missense variants in GLDC to understand phenotypes of the disease. Hum Mutat 38:678–691. https://doi.org/10.1002/humu.23208
Chiu CF, Lin JL, Lin JJ, Tseng MH, Lo FS, Chiang MC (2016) Nonketotic Hyperglycinemia of Infants in Taiwan. Pediatr Neonatol 57:420–426. https://doi.org/10.1016/j.pedneo.2015.10.008
Conter C, Rolland MO, Cheillan D, Bonnet V, Maire I, Froissart R (2006) Genetic heterogeneity of the GLDC gene in 28 unrelated patients with glycine encephalopathy. J Inherit Metab Dis 29:135–142. https://doi.org/10.1007/s10545-006-0202-6
Coughlin CR II, Swanson MA, Kronquist K, Acquaviva C, Hutchin T, Rodríguez-Pombo P, Väisänen ML et al (2017) The genetic basis of classic nonketotic hyperglycinemia due to mutations in GLDC and AMT [published correction appears in Genet Med. 2018 Jan 04;:]. Genet Med 19:104-111. https://doi.org/10.1038/gim.2016.74
Cusmai R, Martinelli D, Moavero R, Carlo Dionisi Vici CD, Vigevano F, Castana C, Elia M, Bernabei S, Bevivino E (2012) Ketogenic diet in early myoclonic encephalopathy due to non ketotic hyperglycinemia. Eur J Paediatr Neurol 16:509–513. https://doi.org/10.1016/j.ejpn.2011.12.015
Dinopoulos A, Matsubara Y, Kure S (2005) Atypical variants of nonketotic hyperglycinemia. Mol Genet Metab 86:61–69. https://doi.org/10.1016/j.ymgme.2005.07.016
Genç Sel Ç, Kılıç M, Yüksel D, Aksoy A, Kasapkara CS, Ceylaner S, Karli-Oguz K (2018) Nonketotic hyperglycinemia: Clinical range and outcome of a rare neurometabolic disease in a single-center. Brain Dev 40:865–875. https://doi.org/10.1016/j.braindev.2018.06.007
Hennermann JB, Berger JM, Grieben U, Scharer G, Van Hove JL (2012) Prediction of long-term outcome in glycine encephalopathy: a clinical survey. J Inherit Metab Dis 35:253–261. https://doi.org/10.1007/s10545-011-9398-1
Hoover-Fong JE, Shah S, Van Hove JL, Applegarth D, Toone J, Hamosh A (2004) Natural history of nonketotic hyperglycinemia in 65 patients. Neurology 63:1847–1853. https://doi.org/10.1212/01.wnl.0000144270.83080.29
Kanno J, Hutchin T, Kamada F, Narisawa A, Aoki Y, Matsubara Y, Kure S (2007) Genomic deletion within GLDC is a major cause of non-ketotic hyperglycinaemia. J Med Genet 44:e69. https://doi.org/10.1136/jmg.2006.043448
Kava MP, Robertson A, Greed L, Balasubramaniam S (2019) Ketogenic diet, a potentially valuable therapeutic option for the management of refractory epilepsy in classical neonatal nonketotic hyperglycinemia: a case report. Eur J Clin Nutr 73:961–965. https://doi.org/10.1038/s41430-018-0286-8
Korman SH, Gutman A (2002) Pitfalls in the diagnosis of glycine encephalopathy (non-ketotic hyperglycinemia). Dev Med Child Neurol 44:712–720. https://doi.org/10.1017/s0012162201002808
Kure S, Takayanagi M, Narisawa K, Tada K, Leisti J (1992) Identification of a common mutation in Finnish patients with nonketotic hyperglycinemia. J Clin Invest 90:160–164. https://doi.org/10.1172/JCI115831
Kure S, Rolland MO, Leisti J, Mandel H, Sakata Y, Tada K, Matsubara Y, et al (1999) Prenatal diagnosis of non-ketotic hyperglycinaemia: enzymatic diagnosis in 28 families and DNA diagnosis detecting prevalent Finnish and Israeli-Arab mutations. Prenat Diagn 19:717–720. doi: https://doi.org/10.1002/(sici)1097-0223(199908)19:8<717::aid-pd625>3.0.co;2-l.
Kure S, Kato K, Dinopoulos A, Gail C, deGrauw TJ, Christodoulou J, Bzduch V et al (2006) Comprehensive mutation analysis of GLDC, AMT, and GCSH in nonketotic hyperglycinemia. Hum Mutat 27:343–352. https://doi.org/10.1002/humu.20293
Matalon R, Naidu S, Hughes JR, Michals K (1983) Nonketotic hyperglycinemia: treatment with diazepam–a competitor for glycine receptors. Pediatrics 71:581–584
Mohammad SA, Abdelkhalek HS (2017) Nonketotic hyperglycinemia: spectrum of imaging findings with emphasis on diffusion-weighted imaging. Neuroradiology 59:1155–1163. https://doi.org/10.1007/s00234-017-1913-0
Nanao K, Okamura-Ikeda K, Motokawa Y, Danks DM, Baumgartner ER, Takada G, Hayasaka K (1994) Identification of the mutations in the T-protein gene causing typical and atypical nonketotic hyperglycinemia. Hum Genet 93:655–658. https://doi.org/10.1007/BF00201565
Nasrallah F, Hadj-Taieb S, Chehida AB, Jelassi A, Massoued SB, Charfi M, Zidi W et al (2020) Nonketotic Hyperglycinemia in Tunisia. Report upon a Series of 69 Patients. Neuropediatrics. https://doi.org/10.1055/s-0040-1712489
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW et al (2015) ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17:405–424. https://doi.org/10.1038/gim.2015.30
Roy D, Al-Asmari A, Ghazal YK, Al-Oqiel S (2004) Nonketotic hyperglycinemia in Suleimaniah Children’s Hospital, Riyadh, Saudi Arabia. Ann Saudi Med 24:378–381. https://doi.org/10.5144/0256-4947.2004.378
Shbarou RM, Boustany RM, Daher RT, Pakdel P, Noureddine A, Karam PE (2019) Outcome of nonketotic hyperglycinemia in Lebanon: 14-year retrospective review. Neuropediatrics 50:235–243. https://doi.org/10.1055/s-0039-1692207
Shin JH, Ahn SY, Shin JH, Sung SI, Jung JM, Kim JK, Kim ES et al (2012) Sequential magnetic resonance spectroscopic changes in a patient with nonketotic hyperglycinemia. Korean J Pediatr 55:301–305. https://doi.org/10.3345/kjp.2012.55.8.301
Stence NV, Fenton LZ, Levek C, Tong S, Coughlin CR II, Hennermann JB, Wortmann SB et al (2019) Brain imaging in classic nonketotic hyperglycinemia: Quantitative analysis and relation to phenotype. J Inherit Metab Dis 42:438–450. https://doi.org/10.1002/jimd.12072
Swanson MA, Coughlin CR Jr, Scharer GH, Szerlong HJ, Bjoraker KJ, Spector EB, Creadon-Swindell G et al (2015) Biochemical and molecular predictors for prognosis in nonketotic hyperglycinemia [published correction appears in Ann Neurol 2016;79:505]. Ann Neurol 78:606–618. https://doi.org/10.1002/ana.24485
Tada K, Narisawa K, Yoshida T, Yokoyama K, Nakagawa H, Tanno K, Mochizukİ K et al (1969) Hyperglycinemia: a defect in glycine cleavage reaction. Tohuku J Exp Med 98:289–296
Toone JR, Applegarth DA, Coulter-Mackie MB, James ER (2000) Biochemical and molecular investigations of patients with nonketotic hyperglycinemia. Mol Genet Metab 70:116–121. https://doi.org/10.1006/mgme.2000.3000
Van Hove JL, Kishnani P, Muenzer J, Wenstrup RJ, Summar ML, Brummond MR, Lachiewicz AM et al (1995) Benzoate therapy and carnitine deficiency in non-ketotic hyperglycinemia. Am J Med Genet 59:444–453. https://doi.org/10.1002/ajmg.1320590410
Van Hove JL, Kishnani PS, Demaerel P, Kahler SG, Miller C, Jaeken J, Rutledge SL (2000) Acute hydrocephalus in nonketotic hyperglycinemia. Neurology 54:754–756. https://doi.org/10.1212/wnl.54.3.754
Van Hove JLK, Coughlin C II, Swanson M, Hennermann JB (2002) [updated 2019] Nonketotic Hyperglycinemia In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. GeneReviews® [Internet]. University of Washington, Seattle, 1993–2020
Van Hove JL, Vande Kerckhove K, Hennermann JB, Mahieu V, Declercq P, Mertens S, De Becker M et al (2005) Benzoate treatment and the glycine index in nonketotic hyperglycinaemia. J Inherit Metab Dis 28:651–663. https://doi.org/10.1007/s10545-005-0033-x
Van Hove JLK, Hennermann JB, Coughlin IICR (2016) Nonketotic Hyperglycinemia (Glycine Encephalopathy) and Lipoate Deficiency Disorders. In: Saudubray JM, Baumgartner MR, Walter J (eds) Inborn Metabolic Diseases, Diagnosis and Treatment, 6th edn. Springer, Berlin, pp 349–357
Veríssimo C, Garcia P, Simões M, Robalo C, Henriques R, Diogo L, Grazina M (2013) Nonketotic hyperglycinemia: a cause of encephalopathy in children. J Child Neurol 28:251–254. https://doi.org/10.1177/0883073812441063
von Wendt L, Hirvasniemi A, Similä S. Nonketotic hyperglycinemia (1979) A genetic study of 13 Finnish families. Clin Genet 15:411–417. https://doi.org/10.1111/j.1399-0004.1979.tb01773.x
Wiltshire EJ, Poplawski NK, Harrison JR, Fletcher JM (2000) Treatment of late-onset nonketotic hyperglycinaemia: effectiveness of imipramine and benzoate. J Inherit Metab Dis 23:15–21. https://doi.org/10.1023/a:1005690611675
Wolff JA, Kulovich S, Yu AL, Qiao CN, Nyhan WL (1986) The effectiveness of benzoate in the management of seizures in nonketotic hyperglycinemia. Am J Dis Child 140:596–602. https://doi.org/10.1001/archpedi.1986.02140200106038
Acknowledgements
We sincerely thank the family of the patient for their participation, after informed consent.
Author information
Authors and Affiliations
Contributions
H.B., Y.Y., A.O., C.S.K., A.K., A.Z., D.Y. and M.K. has been involved general evaluation of patient, analyses of biochemical data and drafting the article. S.C. has been involved molecular analyses of patient and analyses of data.
Corresponding author
Ethics declarations
This case has not been and will not be published elsewhere in substantially the same form. All co-authors reviewed the final approval of the version from prior to submission. All authors have been involved in (a) conception and design, or analysis and interpretation of data and (b) drafting the case or revising it critically for important intellectual content and agree to submission. All authors have provided the information on ‘Declarations’.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Conflict of interest
The authors declare that they have no conflict of interest.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bayrak, H., Yıldız, Y., Olgaç, A. et al. Genotypic and phenotypic features in Turkish patients with classic nonketotic hyperglycinemia. Metab Brain Dis 36, 1213–1222 (2021). https://doi.org/10.1007/s11011-021-00718-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11011-021-00718-3