
Abstract
The brain of diabetics revealed deterioration in many regions, especially the hippocampus. Hence, the present study aimed to evaluate the effects of gallic acid and p-coumaric acid against the hippocampal neurodegeneration in type 2 diabetic rats. Adult male albino rats were randomly allocated into four groups: Group 1 served as control ones and others were induced with diabetes. Group 2 considered as diabetic, and groups 3 and 4 were further orally treated with gallic acid (20 mg/kg b.wt./day) and p-coumaric acid (40 mg/kg b.wt./day) for six weeks. Diabetic rats revealed significant elevation in the levels of serum glucose, blood glycosylated hemoglobin and serum tumor necrosis factor-α, while the level of serum insulin was significantly declined. Furthermore, the brain of diabetic rats showed a marked increase in oxidative stress and a decrease of antioxidant parameters as well as upregulation the protein expression of Bax and downregulation the protein expression of Bcl-2 in the hippocampus. Treatment of diabetic rats with gallic acid and p-coumaric acid significantly ameliorated glucose tolerance, diminished the brain oxidative stress and improved antioxidant status, declined inflammation and inhibited apoptosis in the hippocampus. The overall results suggested that gallic acid and p-coumaric acid may inhibit hippocampal neurodegeneration via their potent antioxidant, anti-inflammatory and anti-apoptotic properties. Therefore, both compounds can be recommended as hopeful adjuvant agents against brain neurodegeneration in diabetics.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Diabetes mellitus (DM), among metabolic disorders, can be considered the most prevalent, growing, dangerous and costly worldwide medical problem. Uncontrolled or poorly controlled diabetes may cause serious deteriorations including neuropathy, nephropathy, retinopathy, etc., which are the main causes of morbidities and mortalities (Narayan et al. 2000; Zatalia and Sanusi 2013). Diabetic neuropathy affects nearly 50% of diabetic patients with either type 1 or type 2 diabetes and involves both the peripheral and central nervous systems (Abbott et al. 2011; Selvarajah et al. 2011). Furthermore, DM causes chronic damage of the brain that is displayed at neuropsychological, neurophysiological and structural level (Brands et al. 2004). The hippocampus is the center of alterations associated with diabetes in the brain (Foghi and Ahmadpour 2014). Many studies showed that both types of diabetes lead to impairment in the hippocampal-based memory and decrease in the brain volume of diabetics also, diabetes increases the risk of neurodegenerative diseases such as Alzheimer’s disease (Muriach et al. 2006; Gold et al. 2007; Choi et al. 2014; Patel et al. 2015).
Little is known about the mechanisms by which diabetes deteriorates the brain, however, the deficiency of insulin action in the brain besides damage induced by persistent hyperglycemia seemed to be connected with the pathogenesis of the brain dysfunction in diabetics (Brands et al. 2004). In the brain, insulin and insulin-like growth factor (IGF) signaling play essential roles in regulating and keeping cognitive function. The suppression of insulin/IGF signaling involved in brain neurodegeneration via increasing: 1) the formation of free radicals that damage lipids, proteins, DNA and RNA; 2) mitochondrial dysfunction; 3) levels of oxidative and endoplasmic reticulum (ER) stress; and 4) stimulation of pro-inflammatory and pro-death pathways (Brands et al. 2004; de la Monte 2012). Apoptosis can be suggested as a probable pathway for hyperglycemia-induced cell death in the neurons because of disturbance in the expression of apoptosis regulatory genes. The Bcl-2 family is one of the main regulators of apoptosis and includes a group of cytoplasmic proteins that control apoptosis such as Bcl-2 and Bax proteins. Bcl-2 stimulates cell survival, while Bax accelerates cell death. Furthermore, it has been reported that Bcl-2 and Bax expressed in the neurons of the nervous system (Kroemer 1997; Jafari et al. 2008).
According to several in vitro and in vivo studies, in animals and human, dietary phenolic compounds modulate hyperglycemia, dyslipidemia and insulin resistance, and ameliorate oxidative stress and inflammation markers. In addition, they can inhibit the progression of many complications of diabetes involving retinopathy, nephropathy, neuropathy and cardiovascular disease (Bahadoran et al. 2013). Gallic acid (GA, 3,4,5-trihydroxybenzoic acid), a plant polyphenol, found with high content in tea, grapes and wine (Ma et al. 2003). GA received much attention due to its potent antioxidant, antihyperglycemic, antihyperlipidemic, anti-inflammatory and neuroprotective effects (Latha and Daisy 2011; Mansouri et al. 2013). p-Coumaric acid (PCA, 4-hydroxyphenyl-2-propenoic acid), a natural phenolic acid, presents in a plenty of foods such as grapes, carrot, spinach, coffee, tomato and garlic (Alamed et al. 2009). PCA has several biological actions such as antioxidant and radical scavenging, anti-inflammatory and neuroprotective activities (Yoon et al. 2014; Guven et al. 2015). Although these findings, no major research has been performed until now concerning the anti-neurodegenerative effects of gallic acid and p-coumaric acid in the diabetic brain. Thus, the present investigation aimed to assess the beneficial effects of these phenolic compounds against hippocampal neurodegeneration in type 2 diabetic rats.
Materials and methods
Chemicals
Nicotinamide, streptozotocin and gallic acid were purchased from Sigma Chemical Co., St Louis, MO, USA. p-Coumaric acid was purchased from Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA. All other chemicals were of analytical grade and were obtained from standard commercial supplies.
Experimental animals
Adult male albino rats (Rattus norvegicus) of age four months and body weight about 130 ± 10 g were used in the present study. Rats were obtained from National Research Center (NRC), Doki, Giza, Egypt. The chosen animals were housed in well-aerated cages at the normal atmospheric temperature (25 ± 5 °C), humidity (55 ± 5%) and normal 12 h light/dark cycle. During the entire period of study, animals were provided with water and normal basal diet with known composition ad libitum and allowed to adapt for one week. Animal experiments were approved by the Animal Care and Use Committee at Beni-Sueif University.
Induction of diabetes mellitus
Experimental type 2 diabetes was induced in rats starved for 16 h by single intraperitoneal (i.p.) injection of streptozotocin (STZ) (65 mg/kg b.wt.), freshly dissolved in cold citrate buffer (pH 4.5) after 15 min of i.p. injection of nicotinamide (120 mg/kg b.wt.) prepared in 0.9% saline (Masiello et al. 1998). Seven days after STZ injection, the blood glucose levels were measured in the rats to detect the hyperglycemia state.
Animals grouping and treatment schedule
The animals were randomly allocated into four groups as discussed follows:
-
Group 1: considered as control rats.
-
Group 2: considered as diabetic rats.
-
Group 3: considered as diabetic rats treated with gallic acid (20 mg/kg b.wt.; Latha and Daisy 2011) orally for six weeks.
-
Group 4: considered as diabetic rats treated with p-coumaric acid (40 mg/kg b.wt.; Ambika et al. 2013) orally for six weeks.
After one week of diabetes induction, all treatments were dissolved in 0.5% carboxymethyl cellulose and given daily by gastric intubation. Every week, the dosage was adjusted according to any change in the weight of the body to keep similar dose over the whole period of investigation for all treated groups.
Sample collection and tissue preparation
By the end of the sixth week, animals were starved for 12 h and were sacrificed by cervical dislocation under anesthesia with diethyl ether. Blood samples were collected then sera were isolated for analysis of some biochemical parameters. The brain was excised quickly after dissection of the sacrificed animals. Half of brain was fixed in neutral buffered formalin (10%) for paraffin section preparation, staining and histological examination. Hippocampus was isolated from the second half and kept in RNA later at −70 °C for protein expression assay. Part of the brain was homogenized at 4 °C with ten times (w/v) in 0.9% saline. The homogenate was centrifuged (3000 r.p.m. for five minutes) for removing cellular debris, the supernatant was stored at −20 °C, and used for brain biochemical assay.
Biochemical examinations
Blood glucose level was estimated spectrophotometrically according to Trinder (1969) using reagent kit purchased from Spinreact Co. (Spain). Blood glycosylated hemoglobin (HbA1c) was measured according to the method of Bisse and Abraham (1985) using reagent kits purchased from Biosystems S.A. (Spain). Insulin and tumor necrosis factor alpha (TNF-α) levels in serum were estimated using specific ELISA kits (R&D Systems Inc., USA) according to the manufacturer’s instructions. Lipid peroxidation (LPO) level was assayed in the brain homogenate by measuring the formation of malondialdehyde (MDA) according to the method of Preuss et al. (1998). The level of nitric oxide (NO) in the brain homogenate was estimated as nitrite using reagent kit purchased from Biodiagnostic Co. (Egypt). Reduced glutathione (GSH) content (Beutler et al. 1963), and the activities of antioxidant enzymes such as catalase (CAT) (Cohen et al. 1970), superoxide dismutase (SOD) (Marklund and Marklund 1974), glutathione peroxidase (GPx) (Matkovics et al. 1998) and glutathione-S-transferase (GST) (Mannervik and Guthenberg 1981) were determined in the homogenate of brain. The insulin resistance was calculated by homeostasis model assessment of insulin resistance (HOMA-IR) according to the formula of Haffner et al. (2002).
Western blot analysis
Hippocampi were homogenized with ice-cold homogenization buffer by polytron homogenizer then homogenates were centrifuged (10,000 r.p.m. for 15 min) and supernatants were separated. Proteins were measured by Bio-Rad Protein Assay Kit. Hippocampal lysates containing 50 μg of proteins were onto to SDS-PAGE gel then transported to polyphenyl diene difluoride (PVDF) membrane by electrophoresis. Then, blots were blockaded with 5% non-fat dry milk in tris-buffered saline with tween (TBST) for 3 h at room temperature. Consequently, blots were probed with specific primary antibodies including mouse monoclonal antiserum against Bcl-2 (Thermo-scientific, USA), rabbit polyclonal antiserum against Bax (Thermo-scientific, USA), and mouse and rabbit monoclonal anti-serums against β-actin (Cell Signaling, USA), at 1:1000 dilutions for 2 h in room temperature. TBST (0.1%) was used for washing membranes (3 times). Then, blots were incubated with anti-mouse and rabbit horseradish peroxidase labeled IgG (Cell Signaling, USA) that acted as secondary antibodies (1:3000 dilutions for 1 h in room temperature). Lastly, bands of protein were visualized by an enhanced chemiluminescence reagent (Pierce ECL western blotting substrate) and Alliance Gel-doc (Alliance 4.7 Gel doc, UVtec UK). Semi-quantitative analysis of protein bands was performed by UV Tec software (UK). All blots were standardized against intensities of matching β-actin protein bands.
Histological investigation
Paraffin sections were routinely prepared from fixed brain specimens. The prepared sections were stained with hematoxylin and eosin stain method according to Bancroft and Stevens (2008).
Statistical analysis
Data were expressed as mean ± standard error (SE) and subjected to One-Way Analysis of Variance (ANOVA) by a computer software package, SPSS version 20, (IBM Corp 2011) and followed by Duncan’s Multiple Range Test (DMRT) to detect the significant differences between groups. The differences were considered significant at P < 0.05.
Results
As illustrated in Table 1, there was a significant increase in the levels of serum glucose and blood HbA1c in diabetic rats as compared to control rats. Treatment of diabetic rats with gallic acid and p-coumaric markedly ameliorated these altered parameters. On the contrary, serum insulin level was obviously declined in diabetic rats and was significantly increased as a result of administration of the tested agents. Diabetic rats revealed a significant elevation of HOMA-IR that was dropped significantly after supplementation with gallic acid and p-coumaric acid. These results indicate the potential antidiabetic effects of the tested agents. However, gallic acid seemed to be more potent than p-coumaric acid.
The levels of LPO and NO, markers of oxidative stress, were significantly elevated in the brain of diabetic rats and were significantly diminished after treatment with gallic acid and p-coumaric acid. The brain of diabetic rats exhibited a significant depletion in GSH content as compared to control ones. On the other hand, both treatments produced the marked increase in GSH concentration in treated diabetic rats. The activities of antioxidant enzymes including CAT, SOD, GPx and GST showed a significant decrease in the brain of diabetic rats as compared to control rats. Oral administration of gallic acid and p-coumaric acid potentially improved these altered activities. Based on the above-mentioned data, GA and PCA have powerful free radical scavenging and antioxidant activities in the diabetic brain (Table 2).
Consistent with the previous results, the serum level of pro-inflammatory cytokine, TNF-α, was significantly elevated in diabetic rats and was decreased upon treatment with gallic acid and p-coumaric acid, reflecting their anti-inflammatory effects as showed in Table 1.
Figures 1 and 2 revealed the effect of treatments on Bax and Bcl-2 protein expression levels in the hippocampus. In diabetic rats, Bax protein expression level significantly increased as compared to control ones. In contrast, Bcl-2 protein expression level significantly declined. On the other hand, supplementation with gallic acid and p-coumaric acid significantly downregulated the protein expression of Bax and upregulated the protein expression of Bcl-2 in the hippocampus of diabetic rats.

Western blot analysis of Bax and β-actin proteins expression level of control, diabetic and diabetic rats treated with gallic acid and p-coumaric acid. Top: Gel photograph of Western blot analysis of Bax and β-actin proteins. Bottom: Corresponding densitometric analysis of Western blot bands of Bax and β-actin proteins expression level, represented as the Bax percentage of β-actin protein. * P < 0.05 vs. Control, # P < 0.05 vs. Diabetic

Western blot analysis of Bcl-2 and β-actin proteins expression level of control, diabetic and diabetic rats treated with gallic acid and p-coumaric acid. Top: Gel photograph of Western blot analysis of Bcl-2 and β-actin proteins. Bottom: Corresponding densitometric analysis of Western blot bands of Bcl-2 and β-actin proteins expression levels, represented as the Bcl-2 percentage of β-actin protein. * P < 0.05 vs. Control, # P < 0.05 vs. Diabetic
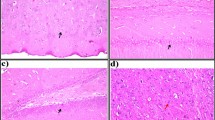
The hippocampus of control rats showed normal histological architecture (Fig. 3a & b). Diabetic rats revealed marked histopathological changes in hippocampus including focal necrosis and decrease in the number of pyramidal cells. There was also an obvious many shrunken irregular pyramidal cells with pyknotic nuclei and loss their processes. Other shrunken cells appeared with pyknotic nuclei and surrounded by halo shape (Fig. 3c). Oral treatment with gallic acid and p-coumaric acid preserves the pyramidal cells with nearly normal cell size, vesicular nuclei and neural process except few shrunken pyramidal cells. (Fig. 3d & e).

a–e Photomicrographs of hippocampal sections of rats in different experimental groups. a & b The hippocampus of control rats is formed of cornu ammonis (CA) which includes CA1, CA2, CA3 and CA4. The pyramidal cells, the principle cells, have a triangular body with large vesicular nuclei (arrow). CA4 projects into the concavity of dentate gyrus (c) Diabetic rats show marked histopathological changes in hippocampus including focal necrosis (curved arrow), many shrunken irregular pyramidal cells with pyknotic nuclei and surrounded by halo shape (arrow head). Gallic acid (d) or p-coumaric acid (e) administration to diabetic rats preserves the pyramidal cells with nearly normal cell size, vesicular nuclei and neural process except few shrunken pyramidal cells
Discussion
Because of difficulties in human research, animal models of diabetes are suitable and essential research tools for understanding the molecular pathogenesis and therapeutic approaches of diabetes and its associated complications (Chatzigeorgiou et al. 2009). For induction of experimental diabetes in animals, nicotinamide-streptozotocin (NA-STZ) model is frequently utilized which causes a moderate degree of hyperglycemia with clinical symptoms as type 2 diabetes. This model is based on the protective effects of NA against STZ cytotoxic effects in β-cells. NA-STZ model has been reported to be a good model to study the complications of diabetes such as diabetic neuropathy (Srinivasan and Ramarao 2007; Sharma et al. 2012).
In our study, fasted diabetic rats revealed a significant elevation in serum glucose level that was parallel with high HOMA-IR value. However, serum insulin level was markedly diminished in diabetic rats as compared to control ones. Oral supplementation with gallic acid (GA) and p-coumaric acid (PCA) to diabetic rats significantly ameliorated the altered parameters. The present results are concomitant with many previous studies that reported the antihyperglycemic and insulinotropic effects of GA and PCA in STZ-induced diabetic rats (Latha and Daisy 2011; Ambika et al. 2013). Blood HbA1c level remains the standard biochemical marker to evaluate long-term glycemic control in diabetic patients, and it helps to assess the risk of the progression or development of diabetic complications (Calisti and Tognetti 2004). The HbA1c level was markedly elevated in the diabetic rats as compared to control rats. On the other hand, treatment with GA and PCA caused an obvious decrease of the elevated HbA1c level. This finding could be due to potential glycemic control by the tested agents.
The brain is specifically susceptible to oxidative damage due to its elevated metabolic rate, high lipid content and relative lack of antioxidant enzymes system when compared to other organs. Thus, oxidative stress is considered the main player of many neurodegenerative diseases (Dugan et al. 1995). Many studies on both diabetic humans and experimentally induced diabetic rats have revealed that hyperglycemia induces oxidative stress that may disturb brain function (Biessels et al. 2002). Raza et al. (2015) reported the increase of oxidative and nitrosative stress in tissues of Zucker diabetic fatty rats, especially in the brain homogenate. More specifically, it has been shown that hippocampus of STZ-induced diabetic rats has increased oxidative stress and impaired antioxidant defense systems (Samarghandian et al. 2014).
In line with many authors, the current results showed that MDA and NO levels in brain of diabetic rats significantly increased while the activities of SOD, CAT, GPx and GST as well as GSH content markedly reduced as compared to those of control ones (Prince et al. 2011; Oyagbemi et al. 2016). These findings indicated the contribution of oxidative stress in the neuronal damage of the diabetic brain. The decreased activity of SOD and CAT could be attributed to an elevated production of reactive oxygen species (ROS) that can inhibit the activity of these enzymes (Wohaieb and Godin 1987) while the lowered activities of GSH-dependent enzymes, GPx and GST, may be due to elevated lipid peroxidation or decreased GSH (Prince et al. 2011). The alteration of GSH levels may be linked to increased polyol pathway activity resulting in NADPH deficiency which is essential for the enzymatic depletion of oxidized glutathione (Preet et al. 2005).
Treatment of diabetic rats with GA and PCA markedly decreased oxidative stress and improved antioxidant parameters. These results revealed that both tested agents have potent free radical scavenging and antioxidant properties in the diabetic brain. Oyagbemi et al. (2016) revealed that GA ameliorated oxidative stress in the brain of rats (cerebrum and cerebellum) via quenching ROS and RNS production and improving antioxidant status. GA also increased the activities of SOD, CAT, and GSH-dependent enzymes and blocks the formation of free radicals generated in the STZ-induced diabetic brain (Prince et al. 2011). In addition, PCA revealed neuroprotective effects in a rat model of embolic cerebral ischemia via its antioxidant activity (Guven et al. 2015). The antioxidant and free radical scavenging properties of phenolic acids may be attributed to direct free radical scavenging activity, inactivation the enzymes responsible for the production of ROS and/or upregulation of the antioxidant enzymes (Thyagaraju and Muralidhara 2008; Saibabu et al. 2015). Depending on these data, it could be suggested that both agents have neuroprotective effects against oxidative stress-induced neurodegeneration in the diabetic brain.
High levels of ROS and persistent activation of nuclear factor kappa B (NF-휅B) have been detected in the hippocampus of STZ-administered rats (Alvarez-Nölting et al. 2012). The activated NF-κB regulates numerous pro-inflammatory mediators including pro-inflammatory cytokines including interleukins (ILs) and tumor necrosis factor-α (TNF-α), cyclooxygenase-2 (COX-2) and inducible nitric oxide synthase (iNOS), also its activation exacerbates inflammation as well as oxidative stress and promotes apoptosis (Yun et al. 2008). The level of serum TNF-α was significantly increased in diabetic rats and was significantly reduced upon treatment with GA and PCA, reflecting the anti-inflammatory activities of both agents. Consequently, the neuroprotective effects of GA and PCA could be attributed to their anti-inflammatory effects. This finding is supported by the results of Mansouri et al. (2013) who found that GA treatment considerably ameliorated memory and long-term potentiation in the traumatic brain rats via decreasing the levels of TNF-α, IL-1β and IL-6 in the brain. Moreover, Yoon et al. (2014) indicated that PCA derived from Corni fructus suppressed the Aβ25-35-stimulated expression of pro-inflammatory mediators such as COX-2 and iNOS in PC12 cells.
Apoptosis could be a probable mechanism for hyperglycemia-induced cell death in the hippocampus. In the current study, the level of Bax protein expression in the hippocampus of diabetic rats significantly elevated, while Bcl-2 protein expression level significantly declined as compared to control rats. These data run in parallel with the study of Jafari et al. (2008) who showed that after 8 weeks of experimentally induced diabetes using STZ, Bax expression in hippocampus of diabetic rats was significantly elevated at both mRNA and protein levels, while the expression of Bcl-2 and Bcl-xL was considerably declined at the levels of both mRNA and protein. Oral administration of GA and PCA significantly downregulated the protein expression level of Bax and upregulated the protein expression level of Bcl-2. These findings are in line with the study of Hong et al. (2012) who found that GA and PCA isolated from Corni fructus showed neuroprotective activity against Aβ (25–35)-induced neurotoxicity in PC12 Cells through antioxidant and anti-apoptotic activities. Thus, GA and PCA may have anti-apoptotic effects against hyperglycemia-induced hippocampal neurodegeneration via downregulation the protein expression of Bax and upregulation the protein expression of Bcl-2.
Concerning the present histological examination of hippocampal sections, diabetic rats exhibited marked histopathological alterations in hippocampus including disorganization, cell loss and a decrease in the number of pyramidal cells indicating cell death. This could be attributed to apoptosis as supported by the present results of Bax and Bcl-2 protein expression levels. In agreement with our findings, electron microscopic studies showed neuronal cell death, after 21 days, in the hippocampus of STZ-induced diabetes in rats (Golembewski et al. 2007). This study supports the fact that diabetes induced by STZ is capable of inducing apoptosis in the hippocampus of diabetic rats (Jafari et al. 2008). Otherwise, the supplementation of diabetic rats with GA and PCA reversed the histopathological changes in the hippocampus. Our results agreed with those of Sarkaki et al. (2014) who stated that GA could improve cell viability of hippocampus and cerebral cortex by attenuating oxidative stress in the hypoperfusion brain tissues. Furthermore, Guven et al. (2015) reported that PCA is a neuroprotective agent because of its powerful antioxidant and anti-apoptotic properties. Thus, treatment with both agents can inhibit the progress of hippocampal neuronal damage in diabetic rats.
Taken together, it can be concluded that gallic acid and p-coumaric acid may attenuate the progress of hippocampal neurodegeneration in the brain of diabetic rats through their potential antioxidant, anti-inflammatory and anti-apoptotic activities. Hence, gallic acid and p-coumaric acid can be considered as promising adjuvant agents against the progression of neurodegeneration in the brain by diabetes.
References
Abbott CA, Malik RA, van Ross ER et al (2011) Prevalence and characteristics of painful diabetic neuropathy in a large community-based diabetic population in the UK. Diabetes Care 34:2220–2224
Alamed J, Chaiyasit W, McClements DJ et al (2009) Relationships between free radical scavenging and antioxidant activity in foods. J Agric Food Chem 57:2969–2976
Alvarez-Nölting R, Arnal E, Barcia JM et al (2012) Protection by DHA of early hippocampal changes in diabetes: possible role of CREB and NF-κB. Neurochem Res 37(1):105–115
Ambika S, Saravanan R, Thirumavalavan K (2013) Antidiabetic and antihyperlipidemic effect of p-hydroxycinnamic acid on streptozotocin-induced diabetic Wistar rats. Biomed Aging Pathol 3(4):253–257
Bahadoran Z, Mirmiran P, Azizi F (2013) Dietary polyphenols as potential nutraceuticals in management of diabetes: a review. J Diabetes Metab Disord 12:43. doi:10.1186/2251-6581-12-43
Bancroft JD, Stevens A (2008) Theory and practice of histological techniques, 6th edn. Chruchill Liveingstone, Edinburgh, pp 126–127
Beutler E, Duron O, Kelly BM (1963) Improved method for determination of blood glutathione. J Lab Clin Med 61:882–888
Biessels GJ, van der Heide LP, Kamal A et al (2002) Ageing and diabetes: implications for brain function. Eur J Pharmacol 441:1–14
Bisse E, Abraham EC (1985) New less temperature-sensitive micro-chromatographic method for the separation and quantitation of glycosylated hemoglobins using a non-cyanide buffer system. J Chromatogr B Biomed Sci Appl 344:81–91
Brands MW, Bell TD, Gibson B (2004) Nitric oxide may prevent hypertension early in diabetes by counteracting renal actions of superoxide. Hypertension 43(1):57–63
Calisti L, Tognetti S (2004) Measure of glycosylated hemoglobin. Acta Biomed Ateneo Parmense 76:59–62
Chatzigeorgiou A, Halapas A, Kalafatakis K et al (2009) The use of animal models in the study of diabetes mellitus. In Vivo 23:245–258
Choi J, Chandrasekaran K, Demarest TG et al (2014) Brain diabetic neurodegeneration segregates with low intrinsic aerobic capacity. Ann Clin Transl Neurol 1(8):589–604
Cohen G, Dembiec D, Marcus J (1970) Measurement of catalase activity in tissue extracts. Anal Biochem 34:30–38
de la Monte SM (2012) Brain insulin resistance and deficiency as therapeutic targets in Alzheimer’s disease. Curr Alzheimer Res 9(1):35–66
Dugan LL, Sensi SL, Canzoniero LM et al (1995) Mitochondrial production of reactive oxygen species in cortical neurons following exposure to N-methyl-D-aspartate. J Neurosci 15(10):6377–6388
Foghi K, Ahmadpour S (2014) Diabetes mellitus type 1 and neuronal degeneration in ventral and dorsal hippocampus. Iran J Pathol 9(1):33–37
Gold SM, Dziobek I, Sweat V et al (2007) Hippocampal damage and memory impairments as possible early brain complications of type 2 diabetes. Diabetologia 50(4):711–719
Golembewski EK, Wales SQ, Aurelian L et al (2007) The HSV-2 protein ICP10PK prevents neuronal apoptosis and loss of function in an in vivo model of neurodegeneration associated with glutamate excitotoxicity. Exp Neurol 203(2):381–393
Guven M, Bozkurt AA, Akman T et al (2015) Neuroprotective effect of p-coumaric acid in rat model of embolic cerebral ischemia. Iran J Basic Med Sci 18:356–363
Haffner SM, Greenberg AS, Weston WM et al (2002) Effect of rosiglitazone treatment on nontraditional markers of cardiovascular disease in patients with type 2 diabetes mellitus. Circulation 106:679–684
Hong SY, Jeong WS, Jun M (2012) Protective effects of the key compounds isolated from Corni fructus against β-amyloid-induced neurotoxicity in PC12 cells. Molecules 17:10831–10845
IBM Corp (2011) IBM SPSS statistics for windows, version 20.0. IBM Corp, Armonk
Jafari AI, Sankian M, Ahmadpour S et al (2008) Evaluation of Bcl-2 family gene expression and caspase-3 activity in hippocampus STZ-induced diabetic rats. Exp Diabetes Res 638467:1–6
Kroemer G (1997) The proto-oncogene Bcl-2 and its role in regulating apoptosis. Nat Med 3(6):614–620
Latha RC, Daisy P (2011) Insulin-secretagogue, antihyperlipidemic and other protective effects of gallic acid isolated from Terminalia bellerica Roxb. in streptozotocin-induced diabetic rats. Chem Biol Interact 189(1):112–118
Ma J, Luo XD, Protiva P et al (2003) Bioactive novel polyphenols from the fruit of Manilkara zapota (sapodilla). J Nat Prod 66:983–986
Mannervik B, Guthenberg C (1981) Glutathione transferase (human placenta). Methods Enzymol 77:231–235
Mansouri MT, Naghizadeh B, Ghorbanzadeh B et al (2013) Gallic acid prevents memory deficits and oxidative stress induced by intracerebroventricular injection of streptozotocin in rats. Pharmacol Biochem Behav 111:90–96
Marklund S, Marklund G (1974) Involvement of superoxide anion radical in the autooxidation of pyrogallol and convenient assay for superoxide dismutase. Eur J Biochem 47:469–474
Masiello P, Broca C, Gross R et al (1998) Experimental NIDDM: development of a new model in adult rats administered streptozotocin and nicotinamide. Diabetes 47:224–229
Matkovics B, Sasvari M, Kotorman M et al (1998) Further prove on oxidative stress in alloxan diabetic rat tissues. Acta Physiol Hung 85:183–192
Muriach M, Bosch-Morell F, Alexander G et al (2006) Lutein effect on retina and hippocampus of diabetic mice. Free Radic Biol Med 41(6):979–984
Narayan KM, Gregg EW, Fagot-Campagna A et al (2000) Diabetes—a common, growing, serious, costly, and potentially preventable public health problem. Diabetes Res Clin Pract 50:S77–S84
Oyagbemi AA, Omobowale TO, Saba AB et al (2016) Gallic acid ameliorates cyclophosphamide-induced neurotoxicity in Wistar rats through free radical scavenging activity and improvement in antioxidant defense system. J Diet Suppl 13(4):402–419
Patel SS, Parashar A, Udayabanu M (2015) Urtica dioica leaves modulates muscarinic cholinergic system in the hippocampus of streptozotocin-induced diabetic mice. Metab Brain Dis 30(3):803–811
Preet A, Gupta BL, Siddiqui MR et al (2005) Restoration of ultrastructural and biochemical changes in alloxan-induced diabetic rat sciatic nerve on treatment with Na3VO4 and Trigonella: a promising antidiabetic agent. Mol Cell Biochem 278:21–31
Preuss HG, Jarrel ST, Scheckenbach R et al (1998) Comparative effects of chromium, vanadium and Gymnema sylvestre on sugar-induced blood pressure elevations in SHR. J Am Coll Nutr 17(2):116–123
Prince M, Stanely P, Kumar MR et al (2011) Effects of gallic acid on brain lipid peroxide and lipid metabolism in streptozotocin-induced diabetic Wistar rats. J Biochem Mol Toxicol 25(2):101–107
Raza H, John A, Howarth FC (2015) Increased oxidative stress and mitochondrial dysfunction in zucker diabetic rat liver and brain. Cell Physiol Biochem 35(3):1241–1251
Saibabu V, Fatima Z, Khan LA et al (2015) Therapeutic potential of dietary phenolic acids. Adv Pharmacol Sci 823539:1–10
Samarghandian S, Azimi-Nezhad M, Samini F (2014) Ameliorative effect of saffron aqueous extract on hyperglycemia, hyperlipidemia, and oxidative stress on diabetic encephalopathy in streptozotocin induced experimental diabetes mellitus. Biomed Res Int 920857:1–12
Sarkaki A, Fathimoghaddam H, Mansouri SM et al (2014) Gallic acid improves cognitive, hippocampal long-term potentiation deficits and brain damage induced by chronic cerebral hypoperfusion in rats. Pak J Biol Sci 17(8):978–990
Selvarajah D, Wilkinson ID, Davies J et al (2011) Central nervous system involvement in diabetic neuropathy. Curr Diab Rep 11(4):310–322
Sharma AK, Sharma A, Kumari R et al (2012) Sitagliptin, sitagliptin and metformin, or sitagliptin and amitriptyline attenuate streptozotocin-nicotinamide induced diabetic neuropathy in rats. J Biomed Res 26(3):200–210
Srinivasan K, Ramarao P (2007) Animal models in type 2 diabetes research: an overview. Indian J Med Res 125(3):451–472
Thyagaraju BM, Muralidhara (2008) Ferulic acid supplements abrogate oxidative impairments in liver and testis in the streptozotocin-diabetic rat. Zool Sci 25:854–860
Trinder P (1969) Determination of glucose in blood using glucose oxidase with an alternative oxygen acceptor. Ann Clin Biochem 6:24–27
Wohaieb SA, Godin DV (1987) Alterations in free radical tissue defense mechanisms in streptozocin-induced diabetes in rat. Effects of insulin treatment. Diabetes 36:1014–1018
Yoon JH, Youn K, Ho CT et al (2014) p-Coumaric acid and ursolic acid from Corni fructus attenuated β-amyloid 25-35-induced toxicity through regulation of the NF-κB signaling pathway in PC12 cells. J Agric Food Chem 62(21):4911–4916
Yun KJ, Koh DJ, Kim SH et al (2008) Anti-inflammatory effects of sinapic acid through the suppression of inducible nitric oxide synthase, cyclooxygase-2, and proinflammatory cytokines expressions via nuclear factor-κB inactivation. J Agric Food Chem 56:10265–10272
Zatalia SR, Sanusi H (2013) The role of antioxidants in the pathophysiology, complications, and management of diabetes mellitus. Acta Med Indones 45:141–147
Acknowledgements
The authors are very grateful to Dr. Manal Abdul-Hamid, Professor of Histology and Cytology, Zoology Department, Faculty of Science, Beni-Suef University for her help in reading and criticizing the histopathology part of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Abdel-Moneim, A., Yousef, A.I., Abd El-Twab, S.M. et al. Gallic acid and p-coumaric acid attenuate type 2 diabetes-induced neurodegeneration in rats. Metab Brain Dis 32, 1279–1286 (2017). https://doi.org/10.1007/s11011-017-0039-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11011-017-0039-8