
Abstract
Elevated concentrations of ammonia in the brain as a result of hyperammonemia leads to cerebral dysfunction involving a spectrum of neuropsychiatric and neurological symptoms (impaired memory, shortened attention span, sleep-wake inversions, brain edema, intracranial hypertension, seizures, ataxia and coma). Many studies have demonstrated ammonia as a major player involved in the neuropathophysiology associated with liver failure and inherited urea cycle enzyme disorders. Ammonia in solution is composed of a gas (NH3) and an ionic (NH4 +) component which are both capable of crossing plasma membranes through diffusion, channels and transport mechanisms and as a result have a direct effect on pH. Furthermore, NH4 + has similar properties as K+ and, therefore, competes with K+ on K+ transporters and channels resulting in a direct effect on membrane potential. Ammonia is also a product as well as a substrate for many different biochemical reactions and consequently, an increase in brain ammonia accompanies disturbances in cerebral metabolism. These direct effects of elevated ammonia concentrations on the brain will lead to a cascade of secondary effects and encephalopathy.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Elevated concentrations of ammoniaFootnote 1 in the brain consequently leads to central nervous system dysfunction and encephalopathy. Ammonia accumulates to toxic levels in the brain arising from the blood during hyperammonemic conditions as seen in a large number of diseases including liver failure and inborn errors of the urea cycle (Cooper and Plum 1987). Ammonia is a metabolite which is mostly produced within the gut during protein digestion and deamination. The urea cycle within the liver regulates the concentration of ammonia in the systemic circulation maintaining blood ammonia levels in the low 50–100 uM range. Therefore, reduced hepatic capacity for ammonia removal leads to hyperammonemia, increased levels of brain ammonia and consequently a spectrum of neuropsychiatric and neurological symptoms including impaired memory, shortened attention span, sleep-wake inversions, brain edema, intracranial hypertension, seizures, ataxia and coma. During liver failure, brain ammonia concentrations can attain 1–5 mM. Many deaths of patients with liver failure occur due to neurological complications such as brain edema, increased intracranial pressure and brain stem herniation, which have been demonstrated to be related to arterial ammonia concentrations in both clinical (Bernal et al. 2007; Bhatia et al. 2006; Jalan et al. 2004; Clemmesen et al. 1999) and animal studies (Sen et al. 2006; Jover et al. 2006; Rose et al. 1999).
Ammonia
Ammonia is a unique molecule which can act both as a weak base (NH3) or weak acid (NH4 +), has electrolytic conductance, and is a product as well as a substrate for many biochemical reactions. Ammonia is composed of a gaseous (NH3) and ionic (NH4 +) component, both of which are transportable across phospholipid bilayers (cell membranes). This in turn alters intracellular as well as extracellular pH. In aqueous solutions, NH3 is in equilibrium with NH4 + (NH3 + H+ ↔ NH4 +) where the ratio of NH3/ NH4 + is a function of pH defined by the Henderson-Hasselbach equation:
At 37°C the pKa of ammonia is 9.15 (Bromberg et al. 1960), therefore under normal physiological conditions (pH 7.4), more than 98% of ammonia is present as NH4 +.
Distribution
Since both the gas (NH3) and the ion (NH4 +) are capable of entering into the cell, this defines ammonia as a complex molecule in comparison to other weak acids and bases.
Ammonia as a gas (NH3) is lipid soluble and therefore enters the brain through diffusion. Interestingly, NH4 + has very similar ionic properties to those of K+ with a comparable ionic radius and a similar diffusion coefficient. These parallel properties allow NH4 + to compete with K+ on membrane ion channels such as inward rectifying and voltage-gated K+ channels. Furthermore, NH4 + has been demonstrated to cross cell membranes by substituting K+ in the ATPase transporters Na+/K+ and H+/K+ (Moser 1987). In addition, the NH4 +/Cl− and Na+/K+/Cl− cotransporters have also demonstrated to transport NH4 + (Aickin et al. 1982; Kelly et al. 2008).
In addition to ammonia being capable of crossing cell membranes through diffusion as well as through K+ ion channels and transporters, there is increasing evidence that a specific ammonia transporter exists in mammals. The human nonerythroid Rhesus (Rh) glycoprotein B (RhBG) and C (RhCG) have been identified as mammalian ammonia transporters (for review see Bakouh et al. 2006). It has also been demonstrated that ammonia can be transported through aquaporin channels, specifically aquaporin-8 (Saparov et al. 2007). However, the regulation as well as the specificity (NH3 vs NH4 +) of these transporters still needs to be confirmed.
Consequential effects of ammonia toxicity
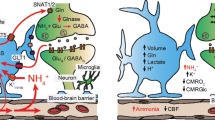
Animal models of liver failure develop hyperammonemia and hepatic encephalopathy which are associated with an array of biochemical, physiological and molecular changes (Fig. 1). Similar effects are observed when either cultured neurons or glia cells are treated with pathophysiological concentrations of ammonia (Sánchez-Pérez and Felipo 2006; Rama Rao et al. 2003; Chan et al. 2003). For further readings on the effects of ammonia toxicity refer to reviews by Felipo and Butterworth (2002) and Cooper and Lai (1987).

Cerebral effects due to ammonia toxicity
Direct effects of ammonia
Ammonia has direct effects on cell function which can consequently initiate a cascade of pathophysiological pathways as mechanisms as demonstrated in Fig. 1.
pH
When ammonia (pH 7.4) is applied to cultured cells (ex. astrocytes), NH3 (<2%) rapidly diffuses across the plasma membrane and establishes an equilibrium inside the cell (NH3 + H+ ↔ NH4 +) by combining with cytosolic H+ to form NH4 +. Consequently intracellular pH [pH]i increases leading to intracellular alkalinization (Fig. 2). Similar results are observed when other weak bases are applied to cultured cell preparations such as trimethylamine (Rose et al. 2005). However, the ammonia-induced transient increase in [pH]i recovers towards baseline since NH4 + (>98%) is capable of crossing cell membranes (at a slower rate than NH3 diffusion) through different channels and transporters (see above). The entry of NH4 + re-establishes a new equilibrium inside the cell (NH3 + H+ ↔ NH4 +), releasing H+ and subsequently lowering [pH]i. The amplitude of increased [pH]i (degree of alkalinization) upon ammonia application is dependent upon a) the rate of entry of NH3 vs NH4 + into the cell b) the concentration and ratio of NH3 vs NH4 + (pH and temperature dependent) and c) the quantitative expression of ammonia “transporters and channels” permitting NH4 + to enter the cell. These will also reflect time to recovery of [pH]i and degree of acidification. For example, ammonia applied to cultured cells with an over-expression of K+-channels/transporters (or ammonia transporters) will result in a smaller amplitude of increased [pH]i and a faster recovery of [pH]i, leading to intracellular acidification. Since the ionic component of other weak acids is not capable of crossing cell membranes, the initial alkalinization upon application of trimethylamine, does not fully recover. This is supported with experiments by co-applying barium chloride (a blocker of inwardly-rectifying K+ channels) along with ammonia and observing an initial alkalinization followed by a slow recovery to baseline. In conclusion, ammonia due to its similarities with K+, alters both intracellular and extracellular pH distinctively from other weak acids and bases.

The effect of ammonia on intracellular pH. Transmembrane fluxes of NH3 exceed fluxes of NH4 + therefore resulting in intracellular alkalinization (A). The amplitude of increased [pH]i (degree of alkalinization) as well as degree of secondary acidification (B) upon ammonia application is dependent upon the rate of entry and concentration of NH3 and NH4 + as well as the quantitative expression of ammonia “transporters and channels” which allow NH4 + to enter the cell
Membrane potential
Since K+ and NH4 + ions are comparable in many aspects, increased concentrations of NH4 + may affect K+ homeostasis and therefore compete with K+ on Na+/K+ and H+/K+ ATPase exchangers as well as Na+/K+/Cl− and K+/Cl− co-transporters. This in turn may have an impact on the Nernst potential for potassium and resting membrane potential.
High concentrations of ammonia can raise the membrane potential, depolarizing both neurons and astrocytes. It has been demonstrated that 2 mM NH4Cl can depolarize hippocampal neurons by approximately 10 mV. An increase in positive ions (NH4 +)/positive charge inside the cell depolarizes the membrane potential (Fig. 3a) but not sufficient enough to generate an action potential (Fan and Szerb 1993). Astrocytes, who have a lower resting membrane potential than neurons, exhibit a concentration-dependent increase in membrane potential following application of 5, 10 and 20 mM of NH4Cl (Fig. 3b) (Allert et al. 1998). Ammonia (2 mM) added to the superfusate caused a very small depolarization (1.6 mV) of the glial cells in a slice of the retina of the honey-bee drone (Coles et al. 1996). Activation of the glutamate ionotropic receptor N-methyl-D-aspartate (NMDA) has been demonstrated to play an important role in the pathophysiology of hepatic encephalopathy (Llansola et al. 2007). The opening of the channel is controlled by a powerful voltage-dependent block by external magnesium ions (Mayer et al. 1984; Nowak et al. 1984). It is believed ammonia, by raising the membrane potential, removes the magnesium block rendering NMDA receptors susceptible to activation. To which degree the magnesium block is removed due to ammonia needs to be clarified as it has been demonstrated half the magnesium block is removed upon raising the membrane potential to −20 mV (Mayer et al. 1984). In conclusion, pathophysiological concentrations of ammonia directly raise the membrane potential in both astrocytes and neurons, however, not sufficiently to activate voltage-gated channels or generate an action potential in neurons.


Direct effects of ammonia consequently lead to secondary effects
Metabolism
Ammonia is an important substrate as well as a product for at least 16 different enzymatic reactions in the brain (Cooper and Plum 1987). Increased concentrations of ammonia in the brain results in alterations in metabolism affecting regulatory activities of important enzymes such as glutaminase, glutamine synthetase and glutamate dehydrogenase.
Summary
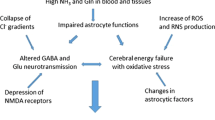
Ammonia plays a major role in the pathogenesis of hepatic encephalopathy as well as other hyperammonemia-induced encephalopathies. Ammonia-lowering strategies remain the key therapeutic approach. Ammonia neurotoxicity has been demonstrated to be associated with a number of physiological, biochemical and molecular changes in the brain. It is the initiating/direct effects of ammonia on cell function which consequentially leads to additional secondary effects and stimulates a cascade of pathophysiological pathways leading to cerebral dysfunction (Fig. 4).
Notes
Throughout the text, “ammonia” is written referring to total ammonia; gas (NH3) + ion (NH4 +).
References
Aickin CC, Deisz RA, Lux HD (1982) Ammonium action on post-synaptic inhibition in crayfish neurones: implications for the mechanism of chloride extrusion. J Physiol 329:319–339
Allert N, Köller H, Siebler M (1998) Ammonia-induced depolarization of cultured rat cortical astrocytes. Brain Res 782:261–270
Bakouh N, Benjelloun F, Cherif-Zahar B, Planelles G (2006) The challenge of understanding ammonium homeostasis and the role of the Rh glycoproteins. Transfus Clin Biol 13:139–146
Bernal W, Hall C, Karvellas CJ, Auzinger G, Sizer E, Wendon J (2007) Arterial ammonia and clinical risk factors for encephalopathy and intracranial hypertension in acute liver failure. Hepatology 46:1844–1852
Bhatia V, Singh R, Acharya SK (2006) Predictive value of arterial ammonia for complications and outcome in acute liver failure. Gut 55:98–104
Bromberg PA, Robin ED, Forkner CE Jr (1960) The existence of ammonia in blood in vivo with observations on the significance of the NH4 plus minus NH3 system. J Clin Invest 39:332–341
Chan H, Zwingmann C, Pannunzio M, Butterworth RF (2003) Effects of ammonia on high affinity glutamate uptake and glutamate transporter EAAT3 expression in cultured rat cerebellar granule cells. Neurochem Int 43:137–146
Clemmesen JO, Larsen FS, Kondrup J, Hansen BA, Ott P (1999) Cerebral herniation in patients with acute liver failure is correlated with arterial ammonia concentration. Hepatology 29:648–653
Coles JA, Marcaggi P, Véga C, Cotillon N (1996) Effects of photoreceptor metabolism on interstitial and glial cell pH in bee retina: evidence of a role for NH4+. J Physiol 495:305–318
Cooper AJ, Lai JC (1987) Cerebral ammonia metabolism in normal and hyperammonemic rats. Neurochem Pathol 6:67–95
Cooper AJ, Plum F (1987) Biochemistry and physiology of brain ammonia. Physiol Rev 67:440–519
Fan P, Szerb JC (1993) Effects of ammonium ions on synaptic transmission and on responses to quisqualate and N-methyl-D-aspartate in hippocampal CA1 pyramidal neurons in vitro. Brain Res 632:225–231
Felipo V, Butterworth RF (2002) Neurobiology of ammonia. Prog Neurobiol 67:259–279
Jalan R, Olde Damink SW, Hayes PC, Deutz NE, Lee A (2004) Pathogenesis of intracranial hypertension in acute liver failure: inflammation, ammonia and cerebral blood flow. J Hepatol 41:613–620
Jover R, Rodrigo R, Felipo V, Insausti R, Sáez-Valero J, García-Ayllón MS, Suárez I, Candela A, Compañ A, Esteban A, Cauli O, Ausó E, Rodríguez E, Gutiérrez A, Girona E, Erceg S, Berbel P, Pérez-Mateo M (2006) Brain edema and inflammatory activation in bile duct ligated rats with diet-induced hyperammonemia: a model of hepatic encephalopathy in cirrhosis. Hepatology 43:1257–1266
Kelly T, Roderigo C, Rose CR (2008) Ammonium-evoked alterations in intracellular sodium and pH homeostasis inhibit glutamate transport activity in cultured astrocytes. ISHEN abstract, 2008
Llansola M, Rodrigo R, Monfort P, Montoliu C, Kosenko E, Cauli O, Piedrafita B, El Mlili N, Felipo V (2007) NMDA receptors in hyperammonemia and hepatic encephalopathy. Metab Brain Dis 22:321–335
Mayer ML, Westbrook GL, Guthrie PB (1984) Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature 309:261–263
Moser H (1987) Electrophysiological evidence for ammonium as a substitute for potassium in activating the sodium pump in a crayfish sensory neuron. Can J Physiol Pharmacol 65:141–145
Nowak L, Bregestovski P, Ascher P, Herbet A, Prochiantz A (1984) Magnesium gates glutamate-activated channels in mouse central neurones. Nature 307:462–465
Rama Rao KV, Jayakumar AR, Norenberg DM (2003) Ammonia neurotoxicity: role of the mitochondrial permeability transition. Metab Brain Dis 18:113–127
Rose C, Kresse W, Kettenmann H (2005) Acute insult of ammonia leads to calcium-dependent glutamate release from cultured astrocytes, an effect of pH. J Biol Chem 280:20937–20944
Rose C, Michalak A, Rao KV, Quack G, Kircheis G, Butterworth RF (1999) L-ornithine-L-aspartate lowers plasma and cerebrospinal fluid ammonia and prevents brain edema in rats with acute liver failure. Hepatology 30:636–640
Sánchez-Pérez AM, Felipo V (2006) Chronic exposure to ammonia alters basal and NMDA-induced phosphorylation of NMDA receptor-subunit NR1. Neuroscience 140:1239–1244
Saparov SM, Liu K, Agre P, Pohl P (2007) Fast and selective ammonia transport by aquaporin-8. J Biol Chem 282:5296–301
Sen S, Rose C, Ytrebø LM, Davies NA, Nedredal GI, Drevland SS, Kjønnø M, Prinzen FW, Hodges SJ, Deutz NE, Williams R, Butterworth RF, Revhaug A, Jalan R (2006) Effect of albumin dialysis on intracranial pressure increase in pigs with acute liver failure: a randomized study. Crit Care Med 34:158–164
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bosoi, C.R., Rose, C.F. Identifying the direct effects of ammonia on the brain. Metab Brain Dis 24, 95–102 (2009). https://doi.org/10.1007/s11011-008-9112-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11011-008-9112-7