
Abstract
Effects of ammonia on astrocytes play a major role in hepatic encephalopathy, acute liver failure and other diseases caused by increased arterial ammonia concentrations (e.g., inborn errors of metabolism, drug or mushroom poisoning). There is a direct correlation between arterial ammonia concentration, brain ammonia level and disease severity. However, the pathophysiology of hyperammonemic diseases is disputed. One long recognized factor is that increased brain ammonia triggers its own detoxification by glutamine formation from glutamate. This is an astrocytic process due to the selective expression of the glutamine synthetase in astrocytes. A possible deleterious effect of the resulting increase in glutamine concentration has repeatedly been discussed and is supported by improvement of some pathologic effects by GS inhibition. However, this procedure also inhibits a large part of astrocytic energy metabolism and may prevent astrocytes from responding to pathogenic factors. A decrease of the already low glutamate concentration in astrocytes due to increased synthesis of glutamine inhibits the malate–aspartate shuttle and energy metabolism. A more recently described pathogenic factor is the resemblance between NH4 + and K+ in their effects on the Na+,K+-ATPase and the Na+,K+, 2 Cl− and water transporter NKCC1. Stimulation of the Na+,K+-ATPase driven NKCC1 in both astrocytes and endothelial cells is essential for the development of brain edema. Na+,K+-ATPase stimulation also activates production of endogenous ouabains. This leads to oxidative and nitrosative damage and sensitizes NKCC1. Administration of ouabain antagonists may accordingly have therapeutic potential in hyperammonemic diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hyperammonemia causes diseases, among which hepatic encephalopathy and acute liver failure (ALF) are the quantitatively most important. Most studies of these diseases focus on a single pathological feature which is important for understanding the pathogenesis, symptomatology and treatment of the disease(s). This review takes a different approach by describing hyperammonemic diseases as multifactorial. It discusses several of these factors, as determined by a multitude of authors, including ourselves. It begins by describing the increases in magnitude of ammonia concentrations in blood and its fluxes from there to brain in hyperammonemic patients determined by Susanne Keiding and her co-workers. In this connection it points out that much larger amounts of ammonia turn over under physiological conditions in the glutamate–glutamine cycle, which is essential for supply of neurons with transmitter glutamate and GABA. These transmitters are synthesized in astrocytes because neurons are unable to do so. However, since the ammonia which is released from neurons in the glutamate–glutamine cycle (during glutamate synthesis from glutamine) is used in identical amounts in astrocytes (for synthesis of glutamine from glutamate) there is no net production of ammonia during the operation of this cycle. This is an important difference from hyperammonemic diseases, where excess ammonia is detoxified in astrocytes, almost exclusively by glutamine production from ammonia.
The possibility that glutamate conversion to glutamine during ammonia detoxification may have deleterious effects due to either an increase of glutamine or a decrease of glutamate is subsequently discussed. Conversion rates are described together with the increase in glutamine content and its possible adverse consequences. It is also discussed that reduction of glutamate has adverse functional consequences, although there probably is a small increase in glutamate synthesis.
Another reason why hyperammonemia exerts deleterious effects is the similarity between the ammonia ion, NH4 +, and K+. This is the reason for the most dreaded consequence of ALF, cerebral edema, which is due to increased uptake of ions and water mediated by operation of both the Na+,K+-ATPase and NKCC1, a cotransporter of Na+,K+, 2 Cl− and water. A similar edema occurs when the extracellular K+ concentration is highly increased during brain ischemia, and in both cases concomitant effects on endothelial cells are crucial for cell swelling. An important difference between the two ions is that NH4 + is more potent than K+. This difference is accentuated by inflammatory events. The Norenberg and Albrecht groups have thoroughly studied these events, which are accompanied by increases in compounds like reactive oxygen species (ROS), nuclear factor kappa B (NF-κB) and nitric oxide (NO). The inflammation is probably also a result of the similarity between NH4 + and K+, since they both stimulate the Na+,K+-ATPase. The catalytic effect of this ATPase is essential for the ion fluxes creating the gradients driving NKCC1 and leading to the edema, but the stimulation of the Na+,K+-ATPase has also signaling effects. This is because it activates a pathway initiated by nanomolar concentrations of endogenous ouabains, as shown by Liang Peng and her coworkers. One branch of this pathway is essential for the catalytic function of the ATPase, and the other leads to the inflammation.
The similarity between NH4 + and K+ is also the reason for stimulation of a specific glycolytic enzyme and thus of glycolysis, but it is unknown if the increased lactate production has any adverse effects. It is also not known whether the similarity between NH4 + and K+ could be associated with a decrease in cyclic GMP not only in neurons but also in astrocytes, which is associated with impairment of memory in hepatic encephalopathy, as demonstrated by the groups of Vincente Felipo and Jan Albrecht.
Ammonia Fluxes in Hepatic Encephalopathy Compared to Those in the Glutamate–Glutamine Cycle
Hepatic Encephalopathy
Human ammonia toxicity is generally not secondary to exposure to exogenous ammonia but to failure of normally occurring hepatic detoxification of ammonia generated in the gut [1]. This leads to arterial hyperammonia and cerebral ammonia uptake. Most, but not all, patients with liver disease (fibrosis, cirrhosis) develop neurological abnormalities referred to as hepatic encephalopathy. Chronic hepatic encephalopathy is the most common form of this condition and is characterized by changes in personality, altered mood, declining intellectual capacity, and abnormal muscle tone [2]. Encephalopathy caused by ALF is generally a result of drug toxicity and presents with an abrupt decline in mental function, systemic inflammation and ultimately multi-organ failure and coma [3]. A frequent, although recently less common neuropathological finding is severe brain edema, which leads to increased intracranial pressure and is associated with a high mortality rate. However, using specialized MRI imaging some degree of cerebral edema is often detectable even in less fulminant hepatic encephalopathy [4].
Ammonia Concentrations and Fluxes in Hyperammonic Diseases
Arterial ammonia concentrations and blood brain fluxes of ammonia have been measured in patients with liver fibrosis with and without hepatic encephalopathy [5]. An arterial ammonia concentration above the normal level of <30 nM causes hepatic encephalopathy due to uptake in the brain from the systemic circulation, and both arterial concentrations and uptake rates are higher in patients with liver fibrosis who show sign of encephalopathy than in those who do not (Fig. 1). However, as shown in Fig. 1 even in the latter group the arterial concentration does not exceed 100 µM and the ammonia flux into brain is generally not above 15 nmol/min/ml(~g) brain [5].

Net metabolic flux of ammonia from blood to brain cortex as a function of arterial blood ammonia concentration in patients with cirrhosis with hepatic encephalopathy (closed triangles), patients with liver cirrhosis without hepatic encephalopathy (open circles), and healthy controls (open triangles). From Sørensen and Keiding [5]
Higher ammonia levels are found in ALF and in children (and some adults) suffering from inborn errors of metabolism for example in the urea cycle [6], where the arterial ammonia concentration can exceed 1000 µM [7]. High arterial ammonia concentrations are also seen in patients with Reye’s syndrome [8] and in patients with ALF precipitated by intake of certain drugs or foods. These include normal doses of valproic acid in some patients, who may have suffered from undiagnosed inborn errors of metabolism [9–12]. Overdoses of acetaminophen [13, 14] or intake of certain mushrooms [15] can also lead to ALF. It is likely that the correlation between arterial ammonia concentration and brain uptake shown in Fig. 1 can be extrapolated to these levels.
An exciting and potentially quantitatively very important reason for ammonia-induced encephalopathy is the recently suggested possibility [16] that cerebral malaria could be due to elevated ammonia content specifically in blood vessels in brain. Plasmodium falciparum generates substantial amounts of ammonia but lacks detoxification mechanisms. It can therefore cause localized brain ammonia elevation and subsequent neurotoxic effects [16], including severe brain swelling [17]. Moreover, during Plasmodium yoelii infection in mice there is an increase in cerebral ammonia and lactate contents, and in glutamine synthetase (GS), phosphofructokinase and monoamine oxidase activities [18]. Many of these changes resemble hepatic encephalopathy as will become evident later in the review. Confirmation that cerebral malaria is a hyperammonemic brain disease would add a large number of cases without liver disease to the group of diseases presently known as hepatic encephalopathy and ALF.
In rats, which frequently are used to study mechanisms of hyperammonemic diseases, the normal arterial level of ammonia is higher (~170 nM) than in humans, and it rises to about 500 nM after portocaval anastomosis [19] and to >4 mM in animals given a large amount of ammonia i.p [20]. In both of these studies almost identical ammonia concentrations were reported in arteries and in brain. However, because ammonia crosses the blood–brain barrier mainly as NH3 and pH is lower in brain (7.1) than in blood (7.4), brain ammonia level at equilibrium is normally 1.5–3 times higher in brain than in blood [21, 22].
In animal studies of hepatic encephalopathy it is important how the human disease can best be mimicked. The International Society for Hepatic Encephalopathy and Nitrogen Metabolism recommends portocaval anastomosis or bile duct ligation as animal models for hepatic encephalopathy and hepatic devascularization or thioacetamide treatment for simulation of ALF [23]. Exposure of brain slices, cell cultures, astrocytic-neuronal co-cultures or models of the neurovascular unit to different toxins are endorsed for in vitro studies. Consideration of the neurovascular unit may be especially important due to the fact that brain edema cannot develop on the basis of effects on neural cells alone but also requires an enhanced influx of water across the blood-brain barrier. This will be discussed in more detail together with brain swelling.
Much larger amounts of ammonia are generated in the brain during the operation of the glutamate–glutamine cycle. This is a physiological metabolic pathway transporting glutamate via glutamine to neurons from astrocytes where it is generated (and eventually degraded in similar amounts) or accumulated after its neuronal release [24, 25]. Flux in the glutamate–glutamine cycle corresponds to the total neuronal rate of glucose uptake [26] or ~75 % of the total glucose uptake rate, which amounts to 0.7 µmol/min per g wet wt. in rat brain [27] and to 0.3 µmol/min per g wet wt. in human brain [28]. In human brain the rate of ammonia formation and degradation in the glutamate–glutamine cycle accordingly corresponds to 0.2–0.25 µmol/min per g wet wt. It is thus normally at least ten times larger than the rate of glutamine production due to ammonia uptake and subsequent detoxification even during hepatic encephalopathy (Fig. 1). In contrast to excess ammonia entering the brain due to an increased arterial concentration of ammonia, that generated during conversion of glutamine to glutamate in the glutamate–glutamine cycle is re-utilized in astrocytes during formation of glutamine from glutamate. There is accordingly no increase in total brain ammonia or glutamine concentrations. This is a very important difference from hyperammonemic brain diseases.
Effects of Increased Ammonia Detoxification via Glutamine Synthesis
Glutamate and Glutamine
Berl et al. [29] studied metabolism of excess ammonia entering the brain from the circulation during a 25-min period. They concluded that (a) glutamine was the only cerebral amino acid that showed a considerable increase; (b) this increase occurred without a corresponding decrease in glutamate (which accordingly must have been synthesized in corresponding amounts intracerebrally); and (c) the synthesized glutamine was formed from a small pool of glutamate that was both rapidly turning over and distinct from a larger tissue glutamate pool (which must have been the glutamate associated with the glutamate–glutamine cycle). Cooper et al. [30] expanded this information by demonstrating that infusion of physiological concentrations of [13N]ammonia led to a rapid increase in the specific activity of the amide nitrogen in glutamine. The simultaneous demonstration by Norenberg and Martinez-Hernandez [31] that GS is an astrocyte-specific enzyme in brain established hepatic encephalopathy as a primarily astrocytic disease. Other authors have presented evidence that GS is also expressed in oligodendrocytes, but the original astrocyte-specific localization has been confirmed by Anlauf and Derouiche [32].
Cudalbu et al. [22] simultaneously measured [5-15N]glutamine and [2-15N]glutamine, glutamate content and net glutamine accumulation in the brains of rats exposed to an arterial ammonia concentration of 1 mM for 7 h. Mathematical modeling of the data provided reliable determination of both glutamate–glutamine cycle flux (0.26 μmol/g per min), and net glutamine accumulation (0.033 μmol/g per min). The results show an increase in glutamine accumulation under hyperammonemia, which amounted to 70 nmol/g per minute or about one quarter of the glutamine formation rate in the glutamate–glutamine cycle. This is consistent with the conclusion above based on ammonia fluxes that the rate of ammonia detoxification in hepatic encephalopathy becomes closer to that of ammonia turnover in the glutamate–glutamine cycle. That the demonstrated glutamate–glutamine cycle flux is similar to that described above in human brain in spite of a higher respiratory rate in the rat brain is probably because the experiments were performed in brain slices.
Huang et al. [33] had previously shown in cultured astrocytes that chronic (3 days) exposure to 3 mM ammonia (which is similar to the final brain concentration measured by Cudalbu et al. [22]) significantly increased glutamine formation from glutamate from 2.1 to 3.35 nmol/min per mg protein. With 200 mg protein per g wet wt. [34]. this increase corresponds to 250 nmol/min per g astrocytic wet wt., which with astrocytes accounting for ~25 % of cortical volume [25] equals 60–70 nmol/min per g brain wet wt., i.e., a similar effect of ammonia as that found in brain slices [23]. This similarity supports the validity of the use of well differentiated cultured astrocytes for study of ammonia toxicity.
Due to the increased flux from glutamate to glutamine during hyperammonemia brain glutamine is increased in hyperammonemic states [21, 22, 35–39]. This applies also to patients with hepatic encephalopathy [40, 41]. It has been suggested that the increased glutamine content might contribute to ammonia toxicity and ammonia-induced brain swelling [42–45], which in hyperammonemic rats is prevented by inhibition of GS [46]. The content of glutamine in normal human brain cortex is ~3 mM [47, 48], but a slightly higher value was found in rats [49]. Most of this glutamine must be astrocytic, since the glutamine content is higher in astrocytes than in neurons [50]. The L system-dependent exchange of systemic tryptophan with brain glutamine is increased in cerebral capillary endothelial cells treated with ammonia, or isolated from rats with hepatic encephalopathy [51]. This prevents a continuous rise in cerebral glutamine. The change in osmolarity induced by glutamine accumulation can be calculated to be about 3 % from the culture data by Huang et al. [33]. It can therefore not be expected to cause significant swelling. Moreover, myo-inositol and other osmolytes are decreased [39], and mild hypothermia delays the development of brain edema in portacaval-shunted rats without any effect on cerebral glutamine level [52].
A different possible reason for a deleterious role of glutamine accumulation in astrocytic edema is that glutamine uptake in mitochondria might function as a “Trojan horse” introducing ammonia into mitochondria with subsequent free radical production and mitochondrial damage in astrocytes [44], but not in neurons [53, 54]. Intramitochondrial release of ammonia causes mitochondrial permeability transition and free radical production [53–57]. The “Trojan horse” theory is supported by the finding that histidine, which inhibits glutamine uptake in mitochondria, prevents swelling in ammonia-exposed astrocytes [57]. However, this observation must be interpreted with caution because histidine also exerts direct effects against oxidative stress [58–60]. Prevention of glutamine formation also inhibits the glutamate–glutamine cycle, and since most ATP produced in astrocytes during and after formation of glutamine is derived when glutamate subsequently is oxidized in astrocytes [61–63], it will severely reduce astrocytic energy metabolism. Energy metabolism is required for brain swelling, which during complete medial cerebral artery occlusion in rats (MCAO) does not become significant until after re-perfusion [64].
Glutathione, Malate–Aspartate Shuttle, Tricarboxylic Acid (TCA) Cycle, Pyruvate Carboxylase and Metabolic Rate
A drastic reduction in glutathione content in the thioacetamide model of hepatic encephalopathy might be a result of reduced glutamate content due to conversion of glutamate to glutamine, since glutamate is an essential precursor for glutathione [65, 66]. In rats made chronically hyperammonemic by portal-systemic shunting intraperitoneal injection of ammonium acetate induces a brief period of pre-coma (10–15 min), associated with a decreased glutamate content, and followed by a deep coma and high mortality [67]. In brain tissue from cirrhotic patients with hepatic encephalopathy cerebral glutamate content is similarly reduced by about 20 % [68].
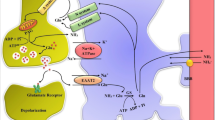
Another consequence of decreased glutamate content might be interference with the function of the malate–aspartate shuttle (MAS) (Fig. 2), which is needed for normal glucose metabolism in order to transfer reducing equivalents from the cytoplasm to mitochondria. This function is essential because one oxidative reaction occurs during glycolysis and the generated NADH is not able to traverse the mitochondrial membrane (e.g. [69]). Kosenko et al. [70] found a 20 % decrease in MAS activity in non-synaptic rat brain mitochondria after injection of ammonia acetate in rats. Lai et al. [71], studying glutamate metabolism and contents of glutamate, aspartate and glutamine in astrocyte cultures with or without 3 mM ammonium chloride similarly concluded that ammonia inhibits MAS in astrocytes. Based on levels of metabolic enzymes in mitochondria, synaptosomes and cytosol isolated from brains of normal rats and rats injected with ammonium acetate Ratnakumari and Murthy [72] reached an identical conclusion.

In the malate–aspartate shuttle (MAS) cytosolic malate dehydrogenase (MDHc) oxidizes NADH and converts oxaloacetate (OAA) to malate (top right of figure), which enters the mitochondria in exchange with α-ketoglutarate (α-KG). The mitochondrial malate dehydrogenase (MDHm) re-oxidizes malate to OAA, which is transaminated to aspartate by the mitochondrial aspartate aminotransferase (AATm). Aspartate leaves the mitochondria in exchange with glutamate. In the mitochondria glutamate conversion to α-KG is essential for AATm activity forming aspartate from OAA and delivering α-KG for mitochondrial export. The glutamate imported into the mitochondria had been formed by cytosolic aspartate aminotransferase (AATc) from α-KG after its entry into the cytosol. Without MAS activity NADH formed in the cytosol during glycolysis would have been unable to enter the mitochondria for oxidation. From Hertz and Dienel [69]
Depletion of the very small glutamate pool in astrocytes [73] during ammonia-induced glutamine formation must in the long run lead to an increase in glutamate synthesis in order to maintain glutamine synthesis. However, in non-synaptic mitochondria from thioacetamide-treated rats and normal non-synaptic mitochondria treated with 3 mM ammonia there is a reduction of the activity of pyruvate carboxylase [74], the enzyme necessary for increased production of TCA cycle intermediates and glutamate. As could be expected, the activity was negligible in synaptic mitochondria where an elevated ammonia concentration had no effect. However, Leke et al. [75, 76] found a significant increase in GABA formation from labeled glucose during hyperammonemic conditions in both co-cultures of cerebral neurons and astrocytes and in rats with liver cirrhosis. Increased anaplerosis has also been observed in cultured astrocytes in the presence of ammonia by Lapidot and Gopher [77] and in additional studies using cultured cells or animal models of hyperammonemia [78–80]. Zwingmann [81] has pointed out that the activity of an enzyme measured in tissue homogenates does not necessarily reflect the actual metabolic flux through the enzyme and reviewed additional studies indicating that ammonia increases anaplerosis.
A stimulatory effect of aspartate on glutamate and glutamine synthesis in astrocytes, probably by serving as an amino group donor, has been shown by Pardo et al. [82]. It revived the concept that α-ketoglutarate formation from glutamate during its degradation in the brain in vivo mainly is catalyzed by amino aspartate transferase [83, 84]. It also led to the concept that glutamate formation and eventual oxidation via the glutamate–glutamine cycle might be metabolically coupled [24, 25]. A potential reduction of glutamate oxidation by its enhanced use for glutamine formation under hyperammonemic conditions might accordingly also impair glutamate synthesis in the glutamate–glutamine cycle. This could explain an ammonia-induced inhibition of oxidation of both [2-14C]pyruvate [85] and [1-14C]pyruvate [86] and the prevention of this inhibition by addition of glutamate to the medium [86]. Reduced pyruvate-supported oxygen consumption has also been found in astrocytes obtained from rats with acute toxic liver damage [87]. Oxidative metabolism of glutamate in cultured astrocytes is also potently decreased by ammonia (Fig. 3) [71, 88].

Rate of 14CO2 formation from [1-14C]glutamate in primary cultures of astrocytes grown in tissue culture medium supplemented with 0.25 % dBcAMP from the age of 2 weeks and acutely exposed to 0, 0.1, 0.3, 1.0, or 3.0 mM ammonia when they were at least 3 weeks old. CO2 production rates were determined in an air-tight chamber during a 30-min period, at the end of which injections were made of perchloric acid to acidify the medium and of hyamine hydroxide into a suspended beaker for quantitative trapping of 14CO2 within the chamber. From the measured activity in the trapped CO2, the specific activity of [1-14C]glutamate in the medium and the protein content of the culture rates of 14CO2 formation were determined. Results are means of six to nine experiments and SEM values are shown by vertical bars. From Yu et al. [88]
15O-oxygen positron emission tomography (PET) studies by Iversen et al. [89] established a decrease in cerebral oxygen consumption in patients with cirrhosis and an acute episode of hepatic encephalopathy. Dam et al. [90], analogously showed a decrease in oxidative metabolism in patients with hepatic encephalopathy and a clear increase after recovery to control values. Keiding and Pavese [91] reviewed studies on brain metabolism in human patients with different degrees of encephalopathy and concluded that cerebral oxygen uptake is reduced to 2/3 in cirrhotic patients with clinically overt hepatic encephalopathy. Iversen et al. [92] examined specifically astrocytic metabolism with [11C]acetate PET in patients with liver cirrhosis with and without hepatic encephalopathy. Although ammonia evoked no significant decrease it seemed to reduce astrocytic metabolism by between 10 and 15 % in both types of patients.
Branched Chain Amino Acids and Glutamate Dehydrogenase
Other processes than pyruvate carboxylation might also contribute to glutamate formation in hyperammonemic brain. Some branched-chain amino acids (BCAAs) are transported rapidly across the blood–brain barrier [93], and valine and isoleucine are metabolized in the TCA cycle (via respectively acetyl CoA and succinyl CoA). A role of BCAAs, especially isoleucine, in production of glutamate and glutamine in brain and muscle was therefore suggested by Bak et al. [94]. Such a production might reduce the formation of CO2 from these amino acids and their incorporation into protein. It may therefore be of interest that both of these processes were strongly inhibited by acute exposure to 3 mM ammonia in cultured astrocytes, although they were less affected by chronic exposure [95, 96]. There is also clinical evidence that a diet rich in BCAA increases event-free survival in cirrhotic patients, but this might be on account of increased ammonia detoxification in muscle [97].
The enzyme glutamate dehydrogenase (GDH) produces glutamate from α-ketoglutarate plus ammonia with concomitant oxidation of NAD(P)H. This process is stimulated by an increase in the ammonia concentration [98], and it might catalyze the increased glutamate production required for ammonia detoxification by glutamine production. The thermodynamic equilibrium constant of GDH favors glutamate formation, but in normal brain a high NAD+/NADH ratio and a low ammonia concentration enables glutamate oxidation [99]. The involvement of GDH in glutamate oxidation is obvious in cultured astrocytes [100, 101]. However, it was mentioned above that in normal, non-hyperammonemic brain astrocytic glutamate formation and degradation might be metabolically coupled and to a large extent catalyzed by aspartate aminotransferase [24, 25]. Even if that is the case GDH might also play a role, possibly by supporting glutamate’s cellular uptake, mediated by ion gradient-driven cotransport with Na+, which in the long run requires energy-consuming Na+,K+-ATPase-mediated Na+ extrusion. This is in agreement with the observation by Robinson and Jackson [102] that astrocytic glutamate transporters, mitochondrial enzymes and Na+,K+-ATPase are co-localized and co-precipitate. These authors also suggest that glutamate transport (and thus presumably its subsequent intense metabolism [63]) plays an essential role in regulation of brain energetics. The inhibition of glutamate metabolism shown in Fig. 3 might therefore have considerable adverse effects.
Ammonia Effects on Lactate and Pyruvate
Brain glucose consumption is increased in brains of acutely hyperammonemic animals [103–105]. An ammonia-induced stimulation of glycolysis has been demonstrated in brain slices [106, 107], rats with ALF [108] and bile duct-ligated rats [109]. Production of lactate is increased in cultured astrocytes (Fig. 4), but not in cultured neurons [110]. In contrast the rate of pyruvate production is decreased, leading to a reduced pyruvate/lactate ratio, consistent with the decreased cytosolic [NAD+/NADH] ratio observed by Hindfelt and Siesjo [104] and Gjedde et al. [111]. The effect of ammonia on pyruvate/lactate ratio is more potent in cells that had not been treated with dBcAMP, where the pyruvate/lactate ratio progressively decreases within the entire concentration range 0.1–3.0 mM [110]. Addition of glutamate to the incubation medium diminished the reduced pyruvate/lactate ratio, consistent with the previously discussed inhibition of MAS and of glutathione synthesis due to enhanced glutamate utilization for glutamine production during hyperammonemic conditions. However, addition of glutamate had no effect on the increased lactate production (Fig. 4), suggesting that the main reason for this is not reduced oxidative metabolism. Ammonia activates brain phosphofructokinase, a rate-limiting and highly regulated enzyme in glycolysis [112–116]. This may be due to the similarity between NH4 + and K+ (see below), since highly elevated K+ concentrations have a similar, although less marked effect and prevent an additional effect of ammonia [115].

Accumulation of lactate in the incubation medium of 3–4-week-old primary cultures of mouse astrocytes as a function of the length of the incubation period under control conditions (open symbols), i.e., incubation in normal serum-free tissue culture medium with addition of 3 mM sodium chloride, and after incubation in normal serum-free tissue culture medium with addition of 3 mM ammonium chloride (filled-in symbols). The cells had either been grown in tissue culture medium supplemented with 0.25 % dBcAMP from the age of 2 weeks and were accordingly morphologically differentiated (squares), or they had been grown for 3–4 weeks without dBcAMP supplementation and were accordingly morphologically undifferentiated (circles). Results are means ± SEM values (if extending beyond the symbols) of 4–20 individual experiments using cultures obtained from at least two different batches. All values obtained in the presence of ammonia are significantly different (P < 0.05 or better) from corresponding control values. From Kala and Hertz [110]
The pathophysiological importance of the ammonia-induced increase in lactate production may be minor since lactate can be exported from brain during hyperammonemic conditions [105], but it occurs early during the disease [117], and its magnitude is proportional to the severity of the condition [108].
Ammonia Effects on Cyclic GMP
Cyclic GMP and Nitric Oxide
Lymphocytes from patients with liver cirrhosis show reduction of intracellular cyclic guanosine monophosphate (cGMP) and less than normal activation of soluble guanylate cyclase (sGC) by NO, which correlates with the degree of encephalopathy [118]. cGMP modulates some forms of learning and memory by activating cGMP-dependent protein kinase and phosphorylation of the glutamate receptor GluR1, which results in insertion of AMPA receptors in the synaptic membrane and increased magnitude of long-term potentiation (LTP) [119]. At least two pathways modulate cGMP levels in brain. One of these is the glutamate-NO-cGMP pathway which is impaired by ammonia due to reduced activation of sGC by NO [120–123]. Impairment of this pathway in brains and neurons of rats with hyperammonemia or hepatic encephalopathy may be partly responsible for their reduced ability to learn [124, 125]. Moreover, restoration of cGMP levels in brain by administering phosphodiesterase inhibitors, cGMP or anti-inflammatory drugs improves learning ability in patients with hepatic encephalopathy [118].
Cyclic GMP and Natriuretic Peptides
cGMP synthesis is also elicited by stimulation of the natriuretic peptide receptor 2 (NPR-2) with its natural ligand, C-type natriuretic peptide [126, 127]. NPR-2 is expressed both in neurons and astrocytes [128, 129]. Atrial natriuretic peptide seems to be enriched in astrocytes [130], and although it is also expressed in neurons it is possible that natriuretic peptides modulate neuronal activity via their effect on glial cells [131]. It is consistent with this possibility that Zielinska et al. [125] showed that hyperammonemia interferes with this pathway in rat cerebral cortical slices and that it does so in astrocytes. In slices from control animals CNP stimulated cGMP synthesis to a similar extent as the NO donor, S-nitroso-N-acetylpenicillamine (SNAP) used at optimal concentrations. Inhibition of specifically astrocytic oxidative metabolism with fluoroacetate reduced cGMP synthesis by ~50 %, and in slices from animals with ammonium acetate-induced hepatic encephalopathy it was decreased by 68 %. This inhibition was absent after treatment with fluoroacetate indicating that the CNP-dependent cGMP synthesis occurred in the fluoroacetate-inhibitable astrocytic compartment, not in the fluoroacetate-resistant neurons.
Effects of Hyperammonemia Due to Similarity with Catalytic and Signaling Effects of K+ on the Na+,K+-ATPase
Brain Edema, Na+,K+-ATPase, NKCC1 and Inflammation
Brain edema is a serious and often fatal complication in ALF [109, 132, 133] and in inborn errors of metabolism [134]. A major reason for the development of brain edema is the similarity between NH4 + and K+. NH4 + can potently replace K+ in stimulation of the Na+,K+-ATPase and active Na+ transport [135, 136] as well as the associated increase in oxygen consumption [137, 138]. Expression and phosphorylation of NKCC1, a cotransporter of Na+, K+, 2 Cl− and water [139–141] are increased in ammonia-treated cultured astrocytes and in brain slices from thioacetamide-rats with a delay of 3–6 h (Fig. 5) [142, 143]. Similarly, Dai et al. [144] found an increase in cell volume of cultured astrocytes after 12 h’ exposure to 3 mM ammonia. Kelly and Rose [145] showed in hippocampal slices that intracellular Na+ in astrocytes but not in neurons rapidly increased by 25–30 mM after exposure to a high concentration of NH4Cl (5 mM). This increase was prevented by bumetanide, a specific NKCC1 inhibitor. As discussed by Hertz et al. [146] these findings all indicate that the ammonia induced swelling is secondary to stimulation of NKCC1 and that ammonia acts more potently than K+, which needs to be increased by 10 mM to activate the cotransporter.

Similarity between effects of ammonia (5 mM) on protein expression of both NKCC1 and phosphorylated NKCC1 (p-NKCC1) in cultured rat astrocytes (a1, a2) and the effect of thioacetamide-induced acute liver failure in rats in vivo, leading to hyperammonemia (b1, b2). The cultures had been grown in almost the same manner as the cultures used by us and were 98 % GFAP- and glutamine synthetase-positive with microglia constituting the rest of the cells. After solubilization in lysis buffer, cellular protein levels were measured and equal amounts of protein were subjected to gel electrophoresis and transferred to nitrocellulose membranes, which after blocking with non-fat dry milk were incubated with respective antibodies. Primary antibodies were used at 1:1000 to detect total NKCC1 (a1) and R5-phosphorylated NKCC1 (a2). Antibody to α-tubulin was used as housekeeping gene and results expressed as the ratios between the NKCC1 genes and tubulin after optical density of the bands had been determined with Chemi-Imager digital imaging system. In the cultures both total NKCC1 and p-NKCC1 show a significant (P < 0.05) increase after 3 h of ammonia exposure (asterisk) and maximum response is perhaps reached slightly earlier for p-NKCC1 (6 h) than for total NKCC1 (12 h). The reason that the response is not immediate is that it is dependent upon oxidative and nitrosative damage of NKCC1. In the rat experiments thioacetamide (300 mg/kg body wt) was given daily for between 1 and 3 days and protein expression of NKCC1 (b1) and p-NKCC1 (b2) determined as in a1, a2. Note significant (P < 0.05) increases after 2 days (asterisk) and a further significant increase (dagger symbol) after 3 days. The responses are identical in a, b (a maximum~threefold increase), but it occurs faster in (a) than in (b). a From Jayakumar et al. [142] and b from Jayakumar et al. [143]
The increase in NKCC1 activity is secondary to ion-induced astrocytic depolarization [147, 148] and opening of L-channels for Ca2+ [149]. An ammonia-induced increase in [Ca2+]i and in expression of the gene for an L-type Ca2+ channel Cav1.2 in cultured astrocytes treated with 3 mM ammonia [150–152] is consistent with the operation of this pathway. Increased [Ca2+]i also occurs in brain slices exposed to as little as 1–2 mM ammonia, and inhibition of GS results in a significantly larger [Ca2+] increases [153], perhaps due to impaired ammonia detoxification. The reason for the delayed response at relative low ammonia concentrations is a gradually developing, marked nitrosative and oxidative damage after ammonia exposure [154–157] which lowers the threshold for NKCC1 activation. The presence of inflammation is in agreement with a good correlation between arterial pro-inflammatory cytokines and intracranial pressure in patients with ALF [158].
Re-distribution of water between extracellular and intracellular spaces alone cannot cause brain edema, which in addition requires uptake from the systemic circulation. Effects on endothelial cells in the blood–brain barrier are necessary for this component of the swelling. This is similar to the situation during brain ischemia/reperfusion, where NKCC1 plays a major role in the pathway importing ions and water into brain parenchyma [159]. In rats subjected to permanent middle cerebral artery occlusion immune-electron microscopy has demonstrated a predominant expression of NKCC1 at the luminal membrane of the blood-brain barrier [160]. It is therefore important that cultured cerebral endothelial cells treated with ammonia also react with oxidative/nitrosative stress [161] and that the transcription factor NF-κB is activated in cortical endothelial cells from thioacetamide-treated animals [162]. A multitude of inflammatory mediators are increased in hepatic encephalopathy and microglia is activated by ammonia [157]. In liver cirrhosis patients with and without hepatic encephalopathy expression levels are altered for more than 1000 genes related to oxidative stress, microglia activation, receptor signaling, inflammatory and anti-inflammatory pathways, cell proliferation, and apoptosis [163]. As a consequence of the role of inflammation in hepatic encephalopathy anti-inflammatory therapy is becoming of major importance in treatment of liver failure [164].
NKCC1 is also expressed in GABAergic neurons. Using very high plasma ammonia concentrations in non-anesthetized mice Rangroo Thrane et al. [20] concluded that over-activation of NKCC1 in these neurons compromised inhibitory neurotransmission, which could be prevented by bumetanide. The plasma ammonia concentrations (4 mM) were one order of magnitude larger than those seen in hepatic encephalopathy, all animals died within 1 h, and death was only postponed by ~10 min by bumetanide treatment. This study is accordingly relevant for the acute and deadly toxicity caused by ingestion of very high concentrations of ammonia. This does not exclude that neuronal NKCC1 stimulation may contribute to the pathophysiology of hepatic encephalopathy, although the decreased inhibitory transmission is in disagreement with the increased GABA formation demonstrated by Leke et al. [75, 76]. However, the concern expressed by Hadjihambi et al. [165] that the expression of NKCC1 also on astrocytes and endothelial cells may produce off-target actions of bumetanide is unjustified and in complete disagreement with the beneficial effects of NKCC1 inhibition described above.
Endogenous Ouabains
The role of endogenous ouabains in Na+,K+-ATPase-mediated K+ uptake in astrocytes will be briefly discussed, since (a) Na+,K+-ATPase is stimulated by both NH4 + and K+; (b) Na+,K+-ATPase activity is required to create the ion gradients driving NKCC1 [166]; and (c) stimulation of the Na+,K+-ATPase by an increased extracellular K+ concentration opens a pathway mediated by endogenous ouabains [167]. Because simultaneous stimulation of the intracellular Na+-stimulated site of the Na+,K+-ATPase is required for its stimulation by elevation of the extracellular K+ concentration the K+-induced stimulation requires a concomitant increase in intracellular Na+ [146, 168–170]. Astrocytes are non-excitable cells and their intracellular Na+ concentration is therefore not increased when neuronal excitation raises extracellular K+, except as a consequence of glutamate uptake [171]. Experiments in cultured astrocytes known to mimic other signaling events and gene expression in astrocytes in situ [172] have shown that signaling mediated by nanomolar concentrations of ouabain opens an astrocytic Na+ channel and thereby enables K+-stimulated K+ uptake in astrocytes [146, 169, 173]. This effect is probably essential for an initial Na+,K+-ATPase-mediated K+ uptake in astrocytes during clearance of elevated extracellular K+ following neuronal activity, at least in astrocytes that have not accumulated Na+ during glutamate uptake. That elevated extracellular K+ initially stimulates astrocytic K+ uptake in brain tissue and optic nerve is now well established [174–179]. Subsequently the accumulated K+ is released [180], probably after re-distribution over a wider area [181] which prevents a second rise in extracellular K+ and allows neuronal uptake.
Na+,K+-ATPase activity is increased in primary cultures of rat astrocytes exposed acutely or chronically to ammonia [182, 183] and in astrocytes from animals treated chronically with thioacetamide [184]. Kala et al. [183] investigated if this could represent an up-regulation of the Na+,K+-ATPase, compensating for exposure to an increased concentration of endogenous ouabains. They found an increase of ouabain(s) in the incubation media of cultured astrocytes exposed to 3 mM ammonia for 4 days. This is accompanied by increased expression of the astrocytic α2 subunit of the Na+,K+-ATPase [185], which is abolished by AG1478, an inhibitor of the epidermal growth factor receptor (EGFR). This receptor is a key component of a pathway mediated by endogenous ouabain(s) in cultured astrocytes [144, 152, 169]. This astrocytic pathway [169] leads like many other signaling pathways initiated by G-protein-coupled receptors [186, 187] to metalloproteinase-mediated release of a growth factor and ‘transactivation’ and phosphorylation of the EGFR [188]. Stimulation of the EGFR activates two pathways, one leading to Ca2+ release and glycogenolysis which is needed for uptake of K+, and the other to phosphorylation of extracellular-regulated kinases one and two (ERK1/2) via Ras, Raf and MEK. Phosphorylation of ERK, increased ROS production and swelling in ammonia-exposed cultured astrocytes is abolished by the ouabain antagonist canrenone (Fig. 6) [144]. This is consistent with observations in cardiac myocytes that ouabain increases ROS, an effect that was antagonized by a dominant negative Ras, suggesting Ras involvement in ROS generation [189]. Ouabain-induced nitrosative damage is also likely in cardiac myocytes, since iNOS, the form expressed in astrocytes is increased by ouabain [190].

Ammonia-induced ERK phosphorylation, ROS production and cell swelling is inhibited by canrenone, an inhibitor of ouabain. a Astrocyte cultures grown as described in legend of in Fig. 3 were incubated with 0 or 3 mM NH4Cl in the absence (control: no NH4Cl, no canrenone) or presence of 100 µM canrenone for 20 min. a1 Immunoblots from a representative experiment. Similar results were obtained from three independent experiments using cultures from three different batches. Bands of 44 and 42 kDa represent p-ERK1 (phosphorylated ERK1) and p-ERK2 (phosphorylated ERK2), respectively (upper rows), or total ERK1 and ERK2 (lower rows). Average ERK phosphorylation was quantitated as ratios between p-ERK1/2 and ERK1/2 (a2). The ratio between p-ERK1/2 and ERK1/2 in control group was designated a value of one. S.E.M. values are indicated by vertical bars. b Cells were incubated as in (a) for 2 h. ROS was determined as fluorescence intensity of oxidized carboxy-H2DCFDA. Fifteen and twenty cells were selected in each coverslip, and three coverslips were used in each experimental group. c Cells were incubated as in (a) for 12 h Cell volume was determined as fluorescence intensity of calcein. Fifteen and twenty cells were selected in each coverslip, and three coverslips were used in each experimental group. *Statistically significant (P < 0.05) difference from control group. From Dai et al. [144]
Two endogenous steroids with ouabain-like activity, the cardenolide, endogenous ouabain and the bufadienolide, marinobufagenin are found in small but equal amounts in brain cortex [191]. Synthesis of marinobufagenin has recently been studied by Fedorova et al. [192] in adrenocortical and placental cells and found to occur from cholesterol via the acidic bile acid pathway, which is controlled by enzyme mitochondrial sterol 27-hydroxylase (CYP27A1). In macrophages the needed cholesterol trafficking from the outer to the inner mitochondrial membrane is mediated by a highly regulated multimeric protein complex. This complex comprises mitochondrial TSPO (translocator protein 18 KDa or mitochondrial peripheral benzodiazepine receptor) and VDAC (voltage-dependent anion channel) together with additional proteins [193, 194]. TSPO is expressed in both astrocytes and microglia [195]. In cultured retinal microglia its antagonists Ro5-4864 and PK11195 both decrease ROS production, whereas only Ro5-4864 decreases TNFα [196]. TSPO is activated in hyperammonemic animals and in brain tissue from patients with hepatic encephalopathy, resulting in increased synthesis of allopregnanolone and tetrahydrodeoxycorticosterone, neurosteroids which have positive modulatory effect on the GABAA receptor complex [197]. The induced sedation may be one reason for a therapeutic effect of partial inverse agonists of the benzodiazepine receptor in hepatic encephalopathy [198], but a potential reduction of endogenous ouabains might be even more important. This might not only apply to such a serious effect as brain swelling but also to relatively early manifestations of hepatic encephalopathy like the memory impairment found by Zielinska et al. [125] to be caused by deficient cGMP simulation by NPR-2. Since marinobufagonin stimulates collagen synthesis [199] it is possible that endogenous ouabains even play a role in the development of liver cirrhosis.
Concluding Remarks
The symptoms in hyperammonemic diseases depend on the concentration of ammonia in the systemic circulation and as a consequence in the brain. Ammonia detoxification increases brain glutamine and decreases glutamate, which has metabolic consequences. Another major reason for ammonia toxicity is the resemblance between NH4 + and K+, which both stimulate NKCC1 and the Na+,K+-ATPase in astrocytes and endothelial cells. In ALF these effects lead to brain swelling, which is generally delayed because it depends upon additional oxidative and nitrosative damage of NKCC1, induced via signaling by nanomolar concentrations of endogenous ouabains. These effects can be prevented by an ouabain antagonist which might become useful in the therapy of hyperammonemic disorders.
References
Hahn M, Massen O, Nencki M, Pavlov I (1893) Die Eck’sche fistel zwischen der unteren hohlvene und der pfortader und ihre folgen fur den organismus. Arch Exp Pathol Pharm 32:161–210
Jones EA, Weissenborn K (1997) Neurology and the liver. J Neurol Neurosurg Psychiatry 63:279–293
Bernal W, Lee WM, Wendon J, Larsen FS, Williams R (2015) Acute liver failure: a curable disease by 2024? J Hepatol 62:S112–S120
Lee GH (2015) Hepatic encephalopathy in acute-on-chronic liver failure. Hepatol Int 9:520–526
Sorensen M, Keiding S (2007) New findings on cerebral ammonia uptake in HE using functional (13)N-ammonia PET. Metab Brain Dis 22:277–284
Kojic J, Robertson PL, Quint DJ, Martin DM, Pang Y, Sundgren PC (2005) Brain glutamine by MRS in a patient with urea cycle disorder and coma. Pediatr Neurol 32:143–146
Burton BK (1998) Inborn errors of metabolism in infancy: a guide to diagnosis. Pediatrics 102:E69
Shannon DC, De Long R, Bercu B, Glick T, Herrin JT, Moylan FM, Todres ID (1975) Studies on the pathophysiology of encephalopathy in Reye’s syndrome; hyperammonemia in Reye’s syndrome. Pediatrics 56:999–1004
Coulter DL, Allen RJ (1980) Secondary hyperammonaemia: a possible mechanism for valproate encephalopathy. Lancet 1:1310–1311
Coulter DL, Allen RJ (1981) Hyperammonemia with valproic acid therapy. J Pediatr 99:317–319
Baganz MD, Dross PE (1994) Valproic acid-induced hyperammonemic encephalopathy: MR appearance. AJNR Am J Neuroradiol 15:1779–1781
Ziyeh S, Thiel T, Spreer J, Klisch J, Schumacher M (2002) Valproate-induced encephalopathy: assessment with MR imaging and 1 H MR spectroscopy. Epilepsia 43:1101–1105
Brusilow SW, Cooper AJ (2011) Encephalopathy in acute liver failure resulting from acetaminophen intoxication: new observations with potential therapy. Crit Care Med 39:2550–2553
Manakkat Vijay GK, Ryan JM, Abeles RD, Ramage S, Patel V, Bernsmeier C, Riva A, McPhail MJ, Tranah TH, Markwick LJ, Taylor NJ, Bernal W, Auzinger G, Willars C, Chokshi S, Wendon JA, Ma Y, Shawcross DL (2016) Neutrophil toll-like receptor 9 expression and the systemic inflammatory response in acetaminophen-induced acute liver failure. Crit Care Med 44:43–53
Soldo I, Kucan Z, Timarac J, Mihaljevic I, Matijevic M, Peric L, Lisnjic D, Sesar Z, Kadojic D, Vcev A, Micunovic N (2007) Mushroom poisoning. Coll Antropol 31:1099–1103
Kimoloi S, Rashid K (2015) Potential role of plasmodium falciparum-derived ammonia in the pathogenesis of cerebral malaria. Front Neurosci 9:234
Seydel KB, Kampondeni SD, Valim C, Potchen MJ, Milner DA, Muwalo FW, Birbeck GL, Bradley WG, Fox LL, Glover SJ, Hammond CA, Heyderman RS, Chilingulo CA, Molyneux ME, Taylor TE (2015) Brain swelling and death in children with cerebral malaria. N Engl J Med 372:1126–1137
Sharma MC, Tripathi LM, Sagar P, Dutta GP, Pandey VC (1992) Cerebral ammonia levels and enzyme changes during plasmodium yoelii infection in mice. J Trop Med Hyg 95:410–415
Butterworth RF, Giguere JF, Michaud J, Lavoie J, Layrargues GP (1987) Ammonia: key factor in the pathogenesis of hepatic encephalopathy. Neurochem Pathol 6:1–12
Rangroo Thrane V, Thrane AS, Wang F, Cotrina ML, Smith NA, Chen M, Xu Q, Kang N, Fujita T, Nagelhus EA, Nedergaard M (2013) Ammonia triggers neuronal disinhibition and seizures by impairing astrocyte potassium buffering. Nat Med 19:1643–1648
Cooper AJ, Plum F (1987) Biochemistry and physiology of brain ammonia. Physiol Rev 67:440–519
Cudalbu C, Lanz B, Duarte JM, Morgenthaler FD, Pilloud Y, Mlynarik V, Gruetter R (2012) Cerebral glutamine metabolism under hyperammonemia determined in vivo by localized (1)H and (15)N NMR spectroscopy. J Cereb Blood Flow Metab 32:696–708
Butterworth RF, Norenberg MD, Felipo V, Ferenci P, Albrecht J, Blei AT, Members of the ICoEMoHE (2009) Experimental models of hepatic encephalopathy: ISHEN guidelines. Liver Int 29:783–788
Hertz L (2013) The glutamate–glutamine (GABA) cycle: importance of late postnatal development and potential reciprocal interactions between biosynthesis and degradation. Front Endocrinol (Lausanne) 4:59
Hertz L, Rothman DL (2016) Glucose, lactate, β-hydroxybutyrate, acetate, GABA, and succinate as substrates for synthesis of glutamate and GABA in the glutamine-glutamate/GABA cycle. In: Sonnewald U, Schousboe A (eds) Advances in Neurobiology. Springer, Berlin (in press)
Sibson NR, Dhankhar A, Mason GF, Rothman DL, Behar KL, Shulman RG (1998) Stoichiometric coupling of brain glucose metabolism and glutamatergic neuronal activity. Proc Natl Acad Sci USA 95:316–321
Sokoloff L, Reivich M, Kennedy C, Des Rosiers MH, Patlak CS, Pettigrew KD, Sakurada O, Shinohara M (1977) The [14C]deoxyglucose method for the measurement of local cerebral glucose utilization: theory, procedure, and normal values in the conscious and anesthetized albino rat. J Neurochem 28:897–916
Duara R, Grady C, Haxby J, Ingvar D, Sokoloff L, Margolin RA, Manning RG, Cutler NR, Rapoport SI (1984) Human brain glucose utilization and cognitive function in relation to age. Ann Neurol 16:703–713
Berl S, Takagaki G, Clarke DD, Waelsch H (1962) Metabolic compartments in vivo. Ammonia and glutamic acid metabolism in brain and liver. J Biol Chem 237:2562–2569
Cooper AJ, McDonald JM, Gelbard AS, Gledhill RF, Duffy TE (1979) The metabolic fate of 13N-labeled ammonia in rat brain. J Biol Chem 254:4982–4992
Norenberg MD, Martinez-Hernandez A (1979) Fine structural localization of glutamine synthetase in astrocytes of rat brain. Brain Res 161:303–310
Anlauf E, Derouiche A (2013) Glutamine synthetase as an astrocytic marker: its cell type and vesicle localization. Front Endocrinol (Lausanne) 4:144
Huang R, Kala G, Murthy RK, Hertz L (1994) Effects of chronic exposure to ammonia on glutamate and glutamine interconversion and compartmentation in homogeneous primary cultures of mouse astrocytes. Neurochem Res 19:257–265
Chen Y, McNeill JR, Hajek I, Hertz L (1992) Effect of vasopressin on brain swelling at the cellular level: do astrocytes exhibit a furosemide–vasopressin-sensitive mechanism for volume regulation? Can J Physiol Pharmacol 70 Suppl:S367–373
Hindfelt B (1972) The effect of sustained hyperammonemia upon the metabolic state of the brain. Scand J Clin Lab Invest 30:245–255
Hindfelt B (1975) On mechanisms in hyperammonemic coma–with particular reference to hepatic encephalopathy. Ann N Y Acad Sci 252:116–123
Cremer JE, Heath DF, Patel AJ, Balázs R, Cavanagh JB (1975) An experimental model of CNS changes associated with chronic liver disease; portocaval anastomosis in the rat. In: Berl S, Clarke DD, Schneider D (eds) Metabolic compartmentation and neurotransmission. Plenum, New York, pp 461–478
Sadasivudu B, Radha Krishna Murthy C (1978) Effects of ammonia on monoamine oxidase and enzymes of GABA metabolism in mouse brain. Arch Int Physiol Biochim 86:67–82
Zwingmann C (2007) Nuclear magnetic resonance studies of energy metabolism and glutamine shunt in hepatic encephalopathy and hyperammonemia. J Neurosci Res 85:3429–3442
Kreis R, Farrow N, Ross BD (1990) Diagnosis of hepatic encephalopathy by proton magnetic resonance spectroscopy. Lancet 336:635–636
Laubenberger J, Haussinger D, Bayer S, Gufler H, Hennig J, Langer M (1997) Proton magnetic resonance spectroscopy of the brain in symptomatic and asymptomatic patients with liver cirrhosis. Gastroenterology 112:1610–1616
Norenberg MD (1977) A light and electron microscopic study of experimental portal-systemic (ammonia) encephalopathy. Progression and reversal of the disorder. Lab Invest 36:618–627
Traber PG, Dal Canto M, Ganger DR, Blei AT (1987) Electron microscopic evaluation of brain edema in rabbits with galactosamine-induced fulminant hepatic failure: ultrastructure and integrity of the blood–brain barrier. Hepatology 7:1272–1277
Albrecht J, Norenberg MD (2006) Glutamine: a Trojan horse in ammonia neurotoxicity. Hepatology 44:788–794
Swain MS, Blei AT, Butterworth RF, Kraig RP (1991) Intracellular pH rises and astrocytes swell after portacaval anastomosis in rats. Am J Physiol 261:R1491–R1496
Takahashi H, Koehler RC, Brusilow SW, Traystman RJ (1991) Inhibition of brain glutamine accumulation prevents cerebral edema in hyperammonemic rats. Am J Physiol 261:H825–H829
Petroff OA, Errante LD, Rothman DL, Kim JH, Spencer DD (2002) Neuronal and glial metabolite content of the epileptogenic human hippocampus. Ann Neurol 52:635–642
Mangia S, Tkac I, Gruetter R, Van De Moortele PF, Giove F, Maraviglia B, Ugurbil K (2006) Sensitivity of single-voxel 1 H-MRS in investigating the metabolism of the activated human visual cortex at 7 T. Magn Reson Imaging 24:343–348
Albrecht J, Zielinska M, Norenberg MD (2010) Glutamine as a mediator of ammonia neurotoxicity: a critical appraisal. Biochem Pharmacol 80:1303–1308
Zhang NH, Laake J, Nagelhus E, Storm-Mathisen J, Ottersen OP (1991) Distribution of glutamine-like immunoreactivity in the cerebellum of rat and baboon (Papio anubis) with reference to the issue of metabolic compartmentation. Anat Embryol (Berl) 184:213–223
Zielinska M, Popek M, Albrecht J (2014) Roles of changes in active glutamine transport in brain edema development during hepatic encephalopathy: an emerging concept. Neurochem Res 39:599–604
Cordoba J, Crespin J, Gottstein J, Blei AT (1999) Mild hypothermia modifies ammonia-induced brain edema in rats after portacaval anastomosis. Gastroenterology 116:686–693
Jayakumar AR, Rama Rao KV, Schousboe A, Norenberg MD (2004) Glutamine-induced free radical production in cultured astrocytes. Glia 46:296–301
Rama Rao KV, Jayakumar AR, Norenberg MD (2005) Role of oxidative stress in the ammonia-induced mitochondrial permeability transition in cultured astrocytes. Neurochem Int 47:31–38
Rao KV, Norenberg MD (2001) Cerebral energy metabolism in hepatic encephalopathy and hyperammonemia. Metab Brain Dis 16:67–78
Rama Rao KV, Jayakumar AR, Norenberg MD (2003) Induction of the mitochondrial permeability transition in cultured astrocytes by glutamine. Neurochem Int 43:517–523
Rama Rao KV, Reddy PV, Tong X, Norenberg MD (2010) Brain edema in acute liver failure: inhibition by l-histidine. Am J Pathol 176:1400–1408
Pichili VB, Rao KV, Jayakumar AR, Norenberg MD (2007) Inhibition of glutamine transport into mitochondria protects astrocytes from ammonia toxicity. Glia 55:801–809
Ruszkiewicz J, Fresko I, Hilgier W, Albrecht J (2013) Decrease of glutathione content in the prefrontal cortical mitochondria of rats with acute hepatic encephalopathy: prevention by histidine. Metab Brain Dis 28:11–14
Ruszkiewicz J, Albrecht J (2015) Changes of the thioredoxin system, glutathione peroxidase activity and total antioxidant capacity in rat brain cortex during acute liver failure: modulation by l-histidine. Neurochem Res 40:293–300
Hertz L, Peng L, Dienel GA (2007) Energy metabolism in astrocytes: high rate of oxidative metabolism and spatiotemporal dependence on glycolysis/glycogenolysis. J Cereb Blood Flow Metab 27:219–249
Whitelaw BS, Robinson MB (2013) Inhibitors of glutamate dehydrogenase block sodium-dependent glutamate uptake in rat brain membranes. Front Endocrinol (Lausanne) 4:123
McKenna MC (2013) Glutamate pays its own way in astrocytes. Front Endocrinol (Lausanne) 4:191
Song D, Xu J, Du T, Yan E, Hertz L, Walz W, Peng L (2014) Inhibition of brain swelling after ischemia-reperfusion by beta-adrenergic antagonists: correlation with increased K+ and decreased Ca2+ concentrations in extracellular fluid. Biomed Res Int 2014:873590
Dringen R, Gutterer JM, Hirrlinger J (2000) Glutathione metabolism in brain metabolic interaction between astrocytes and neurons in the defense against reactive oxygen species. Eur J Biochem 267:4912–4916
Hertz L, Zielke HR (2004) Astrocytic control of glutamatergic activity: astrocytes as stars of the show. Trends Neurosci 27:735–743
Hindfelt B, Plum F, Duffy TE (1977) Effect of acute ammonia intoxication on cerebral metabolism in rats with portacaval shunts. J Clin Invest 59:386–396
Lavoie J, Giguere JF, Layrargues GP, Butterworth RF (1987) Amino acid changes in autopsied brain tissue from cirrhotic patients with hepatic encephalopathy. J Neurochem 49:692–697
Hertz L, Dienel GA (2002) Energy metabolism in the brain. Int Rev Neurobiol 51:1–102
Kosenko E, Felipo V, Montoliu C, Grisolia S, Kaminsky Y (1997) Effects of acute hyperammonemia in vivo on oxidative metabolism in nonsynaptic rat brain mitochondria. Metab Brain Dis 12:69–82
Lai JC, Murthy CR, Cooper AJ, Hertz E, Hertz L (1989) Differential effects of ammonia and beta-methylene-dl-aspartate on metabolism of glutamate and related amino acids by astrocytes and neurons in primary culture. Neurochem Res 14:377–389
Ratnakumari L, Murthy CR (1989) Activities of pyruvate dehydrogenase, enzymes of citric acid cycle, and aminotransferases in the subcellular fractions of cerebral cortex in normal and hyperammonemic rats. Neurochem Res 14:221–228
Ottersen OP, Zhang N, Walberg F (1992) Metabolic compartmentation of glutamate and glutamine: morphological evidence obtained by quantitative immunocytochemistry in rat cerebellum. Neuroscience 46:519–534
Faff-Michalak L, Albrecht J (1991) Aspartate aminotransferase, malate dehydrogenase, and pyruvate carboxylase activities in rat cerebral synaptic and nonsynaptic mitochondria: effects of in vitro treatment with ammonia, hyperammonemia and hepatic encephalopathy. Metab Brain Dis 6:187–197
Leke R, Bak LK, Anker M, Melo TM, Sorensen M, Keiding S, Vilstrup H, Ott P, Portela LV, Sonnewald U, Schousboe A, Waagepetersen HS (2011) Detoxification of ammonia in mouse cortical GABAergic cell cultures increases neuronal oxidative metabolism and reveals an emerging role for release of glucose-derived alanine. Neurotox Res 19:496–510
Leke R, Bak LK, Iversen P, Sorensen M, Keiding S, Vilstrup H, Ott P, Portela LV, Schousboe A, Waagepetersen HS (2011) Synthesis of neurotransmitter GABA via the neuronal tricarboxylic acid cycle is elevated in rats with liver cirrhosis consistent with a high GABAergic tone in chronic hepatic encephalopathy. J Neurochem 117:824–832
Lapidot A, Gopher A (1997) Quantitation of metabolic compartmentation in hyperammonemic brain by natural abundance 13C-NMR detection of 13C-15N coupling patterns and isotopic shifts. Eur J Biochem 243:597–604
Zwingmann C, Brand A, Richter-Landsberg C, Leibfritz D (1998) Multinuclear NMR spectroscopy studies on NH4Cl-induced metabolic alterations and detoxification processes in primary astrocytes and glioma cells. Dev Neurosci 20:417–426
Kanamatsu T, Tsukada Y (1999) Effects of ammonia on the anaplerotic pathway and amino acid metabolism in the brain: an ex vivo 13C NMR spectroscopic study of rats after administering [2–13C] glucose with or without ammonium acetate. Brain Res 841:11–19
Sibson NR, Mason GF, Shen J, Cline GW, Herskovits AZ, Wall JE, Behar KL, Rothman DL, Shulman RG (2001) In vivo (13)C NMR measurement of neurotransmitter glutamate cycling, anaplerosis and TCA cycle flux in rat brain during [2-13C]glucose infusion. J Neurochem 76:975–989
Zwingmann C (2007) The anaplerotic flux and ammonia detoxification in hepatic encephalopathy. Metab Brain Dis 22:235–249
Pardo B, Rodrigues TB, Contreras L, Garzon M, Llorente-Folch I, Kobayashi K, Saheki T, Cerdan S, Satrustegui J (2011) Brain glutamine synthesis requires neuronal-born aspartate as amino donor for glial glutamate formation. J Cereb Blood Flow Metab 31:90–101
Balazs R (1965) Control of glutamate oxidation in brain and liver mitochondrial systems. Biochem J 95:497–508
Dennis SC, Lai JC, Clark JB (1977) Comparative studies on glutamate metabolism in synpatic and non-synaptic rat brain mitochondria. Biochem J 164:727–736
Fitzpatrick SM, Cooper AJ, Hertz L (1988) Effects of ammonia and beta-methylene-dl-aspartate on the oxidation of glucose and pyruvate by neurons and astrocytes in primary culture. J Neurochem 51:1197–1203
Hertz L, Murthy CR, Lai JC, Fitzpatrick SM, Cooper AJ (1987) Some metabolic effects of ammonia on astrocytes and neurons in primary cultures. Neurochem Pathol 6:97–129
Albrecht J, Wysmyk-Cybula U, Rafalowska U (1987) Cerebral oxygen consumption in experimental hepatic encephalopathy: different responses in astrocytes, neurons, and synaptosomes. Exp Neurol 97:418–422
Yu AC, Schousboe A, Hertz L (1984) Influence of pathological concentrations of ammonia on metabolic fate of 14C-labeled glutamate in astrocytes in primary cultures. J Neurochem 42:594–597
Iversen P, Sorensen M, Bak LK, Waagepetersen HS, Vafaee MS, Borghammer P, Mouridsen K, Jensen SB, Vilstrup H, Schousboe A, Ott P, Gjedde A, Keiding S (2009) Low cerebral oxygen consumption and blood flow in patients with cirrhosis and an acute episode of hepatic encephalopathy. Gastroenterology 136:863–871
Dam G, Keiding S, Munk OL, Ott P, Vilstrup H, Bak LK, Waagepetersen HS, Schousboe A, Sorensen M (2013) Hepatic encephalopathy is associated with decreased cerebral oxygen metabolism and blood flow, not increased ammonia uptake. Hepatology 57:258–265
Keiding S, Pavese N (2013) Brain metabolism in patients with hepatic encephalopathy studied by PET and MR. Arch Biochem Biophys 536:131–142
Iversen P, Mouridsen K, Hansen MB, Jensen SB, Sorensen M, Bak LK, Waagepetersen HS, Schousboe A, Ott P, Vilstrup H, Keiding S, Gjedde A (2014) Oxidative metabolism of astrocytes is not reduced in hepatic encephalopathy: a PET study with [(11)C]acetate in humans. Front Neurosci 8:353
Smith QR, Takasato Y, Sweeney DJ, Rapoport SI (1985) Regional cerebrovascular transport of leucine as measured by the in situ brain perfusion technique. J Cereb Blood Flow Metab 5:300–311
Bak LK, Waagepetersen HS, Sorensen M, Ott P, Vilstrup H, Keiding S, Schousboe A (2013) Role of branched chain amino acids in cerebral ammonia homeostasis related to hepatic encephalopathy. Metab Brain Dis 28:209–215
Murthy CR, Hertz L (1987) Acute effect of ammonia on branched-chain amino acid oxidation and incorporation into proteins in astrocytes and in neurons in primary cultures. J Neurochem 49:735–741
Murthy CR, Hertz L (1987) Comparison between acute and chronic effects of ammonia on branched-chain amino acid oxidation and incorporation into protein in primary cultures of astrocytes and of neurons. J Neurosci Res 17:271–276
Muto Y, Sato S, Watanabe A, Moriwaki H, Suzuki K, Kato A, Kato M, Nakamura T, Higuchi K, Nishiguchi S, Kumada H, Long-Term Survival Study G (2005) Effects of oral branched-chain amino acid granules on event-free survival in patients with liver cirrhosis. Clin Gastroenterol Hepatol 3:705–713
Zaganas I, Waagepetersen HS, Georgopoulos P, Sonnewald U, Plaitakis A, Schousboe A (2001) Differential expression of glutamate dehydrogenase in cultured neurons and astrocytes from mouse cerebellum and cerebral cortex. J Neurosci Res 66:909–913
Schousboe A, Scafidi S, Bal LK, Waagepetersen HS, McKenna MC (2015) Glutamate metabolism in the brain focusing ion astrocytes. In: Parpura V, Schousboe A, Verkhratsky A (eds) Glutamate and ATP at the interface of metabolism and signaling in brain. Springer, Berlin, pp 13–30
Yu AC, Schousboe A, Hertz L (1982) Metabolic fate of 14C-labeled glutamate in astrocytes in primary cultures. J Neurochem 39:954–960
McKenna MC, Sonnewald U, Huang X, Stevenson J, Zielke HR (1996) Exogenous glutamate concentration regulates the metabolic fate of glutamate in astrocytes. J Neurochem 66:386–393
Robinson MB, Jackson JG (2016) Astroglial glutamate transporters coordinate excitatory signaling and brain energetics. Neurochem Int. doi:10.1016/j.neuint.2016.03.014
Schenker S, McCandless DW, Brophy E, Lewis MS (1967) Studies on the intracerebral toxicity of ammonia. J Clin Invest 46:838–848
Hindfelt B, Siesjo BK (1971) Cerebral effects of acute ammonia intoxication. II. The effect upon energy metabolism. Scand J Clin Lab Invest 28:365–374
Hawkins RA, Miller AL, Nielsen RC, Veech RL (1973) The acute action of ammonia on rat brain metabolism in vivo. Biochem J 134:1001–1008
Ashford CA, Dixon KC (1935) The effect of potassium on the glucolysis of brain tissue with reference to the Pasteur effect. Biochem J 29:157–168
McKhann GM, Tower DB (1961) Ammonia toxicity and cerebral oxidative metabolism. Am J Physiol 200:420–424
Zwingmann C, Chatauret N, Leibfritz D, Butterworth RF (2003) Selective increase of brain lactate synthesis in experimental acute liver failure: results of a [H-C] nuclear magnetic resonance study. Hepatology 37:420–428
Bosoi CR, Rose CF (2013) Brain edema in acute liver failure and chronic liver disease: similarities and differences. Neurochem Int 62:446–457
Kala G, Hertz L (2005) Ammonia effects on pyruvate/lactate production in astrocytes––interaction with glutamate. Neurochem Int 47:4–12
Gjedde A, Lockwood AH, Duffy TE, Plum F (1978) Cerebral blood flow and metabolism in chronically hyperammonemic rats: effect of an acute ammonia challenge. Ann Neurol 3:325–330
Lowry OH, Passonneau JV (1966) Kinetic evidence for multiple binding sites on phosphofructokinase. J Biol Chem 241:2268–2279
Sugden PH, Newsholme EA (1975) The effects of ammonium, inorganic phosphate and potassium ions on the activity of phosphofructokinases from muscle and nervous tissues of vertebrates and invertebrates. Biochem J 150:113–122
Ratnakumari L, Murthy CR (1993) Response of rat cerebral glycolytic enzymes to hyperammonemic states. Neurosci Lett 161:37–40
Marcaggi P, Coles JA (2001) Ammonium in nervous tissue: transport across cell membranes, fluxes from neurons to glial cells, and role in signalling. Prog Neurobiol 64:157–183
Mehrotra A, Trigun SK (2013) Moderate grade hyperammonemia activates lactate dehydrogenase-4 and 6-phosphofructo-2-kinase to support increased lactate turnover in the brain slices. Mol Cell Biochem 381:157–161
Papenberg J, Lanzinger G, Kommerell B, Hoyer S (1975) Comparative studies of the electroencephalogram and the cerebral oxidative metabolism in patients with liver cirrhosis. Klin Wochenschr 53:1107–1113
Montoliu C, Rodrigo R, Monfort P, Llansola M, Cauli O, Boix J, Elmlili N, Agusti A, Felipo V (2010) Cyclic GMP pathways in hepatic encephalopathy. Neurological and therapeutic implications. Metab Brain Dis 25:39–48
Monfort P, Gomez-Gimenez B, Llansola M, Felipo V (2015) Gender differences in spatial learning, synaptic activity, and long-term potentiation in the hippocampus in rats: molecular mechanisms. ACS Chem Neurosci 6:1420–1427
Hermenegildo C, Montoliu C, Llansola M, Munoz MD, Gaztelu JM, Minana MD, Felipo V (1998) Chronic hyperammonemia impairs the glutamate-nitric oxide-cyclic GMP pathway in cerebellar neurons in culture and in the rat in vivo. Eur J Neurosci 10:3201–3209
Hermenegildo C, Monfort P, Felipo V (2000) Activation of N-methyl-d-aspartate receptors in rat brain in vivo following acute ammonia intoxication: characterization by in vivo brain microdialysis. Hepatology 31:709–715
Hilgier W, Oja SS, Saransaari P, Albrecht J (2004) A novel glycine site-specific N-methyl-d-aspartate receptor antagonist prevents activation of the NMDA/NO/CGMP pathway by ammonia. Brain Res 1015:186–188
Felipo V (2006) Contribution of altered signal transduction associated to glutamate receptors in brain to the neurological alterations of hepatic encephalopathy. World J Gastroenterol 12:7737–7743
Rodrigo R, Erceg S, Rodriguez-Diaz J, Saez-Valero J, Piedrafita B, Suarez I, Felipo V (2007) Glutamate-induced activation of nitric oxide synthase is impaired in cerebral cortex in vivo in rats with chronic liver failure. J Neurochem 102:51–64
Zielinska M, Fresko I, Konopacka A, Felipo V, Albrecht J (2007) Hyperammonemia inhibits the natriuretic peptide receptor 2 (NPR-2)-mediated cyclic GMP synthesis in the astrocytic compartment of rat cerebral cortex slices. Neurotoxicology 28:1260–1263
Herman JP, Dolgas CM, Rucker D, Langub MC Jr (1996) Localization of natriuretic peptide-activated guanylate cyclase mRNAs in the rat brain. J Comp Neurol 369:165–187
Koller KJ, Lowe DG, Bennett GL, Minamino N, Kangawa K, Matsuo H, Goeddel DV (1991) Selective activation of the B natriuretic peptide receptor by C-type natriuretic peptide (CNP). Science 252:120–123
Deschepper CF (1998) Peptide receptors on astrocytes. Front Neuroendocrinol 19:20–46
Sumners C, Tang W (1992) Atrial natriuretic peptide receptor subtypes in rat neuronal and astrocyte glial cultures. Am J Physiol 262:C1134–C1143
McKenzie JC, Berman NE, Thomas CR, Young JK, Compton LY, Cothran LN, Liu WL, Klein RM (1994) Atrial natriuretic peptide-like (ANP-LIR) and ANP prohormone immunoreactive astrocytes and neurons of human cerebral cortex. Glia 12:228–243
Hodes A, Lichtstein D (2014) Natriuretic hormones in brain function. Front Endocrinol (Lausanne) 5:201
Vaquero J, Chung C, Blei AT (2003) Brain edema in acute liver failure. A window to the pathogenesis of hepatic encephalopathy. Ann Hepatol 2:12–22
Rama Rao KV, Jayakumar AR, Norenberg MD (2014) Brain edema in acute liver failure: mechanisms and concepts. Metab Brain Dis 29:927–936
Gropman AL (2012) Patterns of brain injury in inborn errors of metabolism. Semin Pediatr Neurol 19:203–210
Post RL, Merritt CR, Kinsolving CR, Albright CD (1960) Membrane adenosine triphosphatase as a participant in the active transport of sodium and potassium in the human erythrocyte. J Biol Chem 235:1796–1802
Moser H (1987) Electrophysiological evidence for ammonium as a substitute for potassium in activating the sodium pump in a crayfish sensory neuron. Can J Physiol Pharmacol 65:141–145
Hertz L, Schou M (1962) Univalent cations and the respiration of brain-cortex slices. Biochem J 85:93–104
Kurtz I, Balaban RS (1986) Ammonium as a substrate for Na+-K+-ATPase in rabbit proximal tubules. Am J Physiol 250:F497–502
Epstein FH, Silva P (1985) Na-K-Cl cotransport in chloride-transporting epithelia. Ann N Y Acad Sci 456:187–197
Dawson DC (1987) Cellular mechanisms for K transport across epithelial cell layers. Semin Nephrol 7:185–192
Hamann S, Herrera-Perez JJ, Zeuthen T, Alvarez-Leefmans FJ (2010) Cotransport of water by the Na+-K+-2Cl(−) cotransporter NKCC1 in mammalian epithelial cells. J Physiol 588:4089–4101
Jayakumar AR, Liu M, Moriyama M, Ramakrishnan R, Forbush B 3rd, Reddy PV, Norenberg MD (2008) Na-K-Cl Cotransporter-1 in the mechanism of ammonia-induced astrocyte swelling. J Biol Chem 283:33874–33882
Jayakumar AR, Valdes V, Norenberg MD (2011) The Na-K-Cl cotransporter in the brain edema of acute liver failure. J Hepatol 54:272–278
Dai H, Song D, Xu J, Li B, Hertz L, Peng L (2013) Ammonia-induced Na, K-ATPase/ouabain-mediated EGF receptor transactivation, MAPK/ERK and PI3K/AKT signaling and ROS formation cause astrocyte swelling. Neurochem Int 63:610–625
Kelly T, Rose CR (2010) Ammonium influx pathways into astrocytes and neurones of hippocampal slices. J Neurochem 115:1123–1136
Hertz L, Peng L, Song D (2015) Ammonia, like K+, stimulates the Na+, K+, 2Cl− cotransporter NKCC1 and the Na+,K+-ATPase and interacts with endogenous ouabain in astrocytes. Neurochem Res 40:241–257
Walz W, Wuttke W, Hertz L (1984) Astrocytes in primary cultures: membrane potential characteristics reveal exclusive potassium conductance and potassium accumulator properties. Brain Res 292:367–374
Stephan J, Haack N, Kafitz KW, Durry S, Koch D, Hochstrate P, Seifert G, Steinhauser C, Rose CR (2012) Kir4.1 channels mediate a depolarization of hippocampal astrocytes under hyperammonemic conditions in situ. Glia 60:965–978
Hertz L, Bender AS, Woodbury DM, White HS (1989) Potassium-stimulated calcium uptake in astrocytes and its potent inhibition by nimodipine. J Neurosci Res 22:209–215
Liang C, Du T, Zhou J, Verkhratsky A, Peng L (2014) Ammonium increases Ca(2+) signalling and up-regulates expression of TRPC1 gene in astrocytes in primary cultures and in the in vivo brain. Neurochem Res 39:2127–2135
Wang F, Du T, Liang C, Verkhratsky A, Peng L (2015) Ammonium increases Ca(2+) signalling and upregulates expression of Cav1.2 gene in astrocytes in primary cultures and in the in vivo brain. Acta Physiol (Oxf) 214:261–274
Song D, Du T (2014) Ammonium activates ouabain-activated signalling pathway in astrocytes: therapeutic potential of ouabain antagonist. Curr Neuropharmacol 12:334–341
Haack N, Dublin P, Rose CR (2014) Dysbalance of astrocyte calcium under hyperammonemic conditions. PLoS One 9:e105832
Norenberg MD, Rao KV, Jayakumar AR (2005) Mechanisms of ammonia-induced astrocyte swelling. Metab Brain Dis 20:303–318
Carbonero-Aguilar P, Diaz-Herrero Mdel M, Cremades O, Romero-Gomez M, Bautista J (2011) Brain biomolecules oxidation in portacaval-shunted rats. Liver Int 31:964–969
Lachmann V, Gorg B, Bidmon HJ, Keitel V, Haussinger D (2013) Precipitants of hepatic encephalopathy induce rapid astrocyte swelling in an oxidative stress dependent manner. Arch Biochem Biophys 536:143–151
Jayakumar AR, Rama Rao KV, Norenberg MD (2015) Neuroinflammation in hepatic encephalopathy: mechanistic aspects. J Clin Exp Hepatol 5:S21–S28
Wright G, Shawcross D, Olde Damink SW, Jalan R (2007) Brain cytokine flux in acute liver failure and its relationship with intracranial hypertension. Metab Brain Dis 22:375–388
Khanna A, Kahle KT, Walcott BP, Gerzanich V, Simard JM (2014) Disruption of ion homeostasis in the neurogliovascular unit underlies the pathogenesis of ischemic cerebral edema. Transl Stroke Res 5:3–16
O’Donnell ME, Tran L, Lam TI, Liu XB, Anderson SE (2004) Bumetanide inhibition of the blood-brain barrier Na-K-Cl cotransporter reduces edema formation in the rat middle cerebral artery occlusion model of stroke. J Cereb Blood Flow Metab 24:1046–1056
Skowronska M, Zielinska M, Wojcik-Stanaszek L, Ruszkiewicz J, Milatovic D, Aschner M, Albrecht J (2012) Ammonia increases paracellular permeability of rat brain endothelial cells by a mechanism encompassing oxidative/nitrosative stress and activation of matrix metalloproteinases. J Neurochem 121:125–134
Jayakumar AR, Tong XY, Ospel J, Norenberg MD (2012) Role of cerebral endothelial cells in the astrocyte swelling and brain edema associated with acute hepatic encephalopathy. Neuroscience 218:305–316
Gorg B, Bidmon HJ, Haussinger D (2013) Gene expression profiling in the cerebral cortex of patients with cirrhosis with and without hepatic encephalopathy. Hepatology 57:2436–2447
Aldridge DR, Tranah EJ, Shawcross DL (2015) Pathogenesis of hepatic encephalopathy: role of ammonia and systemic inflammation. J Clin Exp Hepatol 5:S7–S20
Hadjihambi A, Rose CF, Jalan R (2014) Novel insights into ammonia-mediated neurotoxicity pointing to potential new therapeutic strategies. Hepatology 60:1101–1103
Pedersen SF, O’Donnell ME, Anderson SE, Cala PM (2006) Physiology and pathophysiology of Na+/H+ exchange and Na+-K+-2Cl− cotransport in the heart, brain, and blood. Am J Physiol Regul Integr Comp Physiol 291:R1–R25
Xie Z, Askari A (2002) Na(+)/K(+)-ATPase as a signal transducer. Eur J Biochem 269:2434–2439
Wang F, Smith NA, Xu Q, Fujita T, Baba A, Matsuda T, Takano T, Bekar L, Nedergaard M (2012) Astrocytes modulate neural network activity by Ca(2+) -dependent uptake of extracellular K+. Sci Signal 5:ra26
Xu J, Song D, Xue Z, Gu L, Hertz L, Peng L (2013) Requirement of glycogenolysis for uptake of increased extracellular K+ in astrocytes: potential implications for K+ homeostasis and glycogen usage in brain. Neurochem Res 38:472–485
Hertz L, Peng L, Song D (2016) The astrocytic Na+,K+-ATPase: stimulation by increased extracellular K+, b-adrenergic activation, ouabain-mediated signaling, and interaction with the transporter NKCC1. In: Chakraborti S, Dhalla NS (eds) Regulation of Na+,K+- ATPase. Springer, Berlin, pp 195–221
Langer J, Rose CR (2009) Synaptically induced sodium signals in hippocampal astrocytes in situ. J Physiol 587:5859–5877
Hertz L, Chen Y, Song D (2016) Astrocyte cultures mimicking brain astrocytes in gene expression, signaling, metabolism and K+ uptake and showing astrocytic gene expression overlooked by immunohistochemistry and in situ hybridization. Neurochem Res. doi:10.1007/s11064-016-1828-x
Hertz L, Peng L, Song D (2016) The astrocytic Na+,K+-ATPase: stimulation by increased extracellular K+,β-adrenergic activation, ouabain-mediated signaling, and interaction with the transporter NKCC1. In: Chakraborti S, Dhalla NS (eds) Regulation of Na+,K+- ATPase. Springer, Berlin, pp 195–221
Ransom CB, Ransom BR, Sontheimer H (2000) Activity-dependent extracellular K+ accumulation in rat optic nerve: the role of glial and axonal Na+ pumps. J Physiol 522 Pt 3:427–442
Dufour S, Dufour P, Chever O, Vallee R, Amzica F (2011) In vivo simultaneous intra- and extracellular potassium recordings using a micro-optrode. J Neurosci Methods 194:206–217
Somjen GG, Kager H, Wadman WJ (2008) Computer simulations of neuron-glia interactions mediated by ion flux. J Comput Neurosci 25:349–365
Macaulay N, Zeuthen T (2012) Glial K(+) clearance and cell swelling: key roles for cotransporters and pumps. Neurochem Res 37:2299–2309
Larsen BR, Assentoft M, Cotrina ML, Hua SZ, Nedergaard M, Kaila K, Voipio J, MacAulay N (2014) Contributions of the Na(+)/K(+)-ATPase, NKCC1, and Kir4.1 to hippocampal K(+) clearance and volume responses. Glia 62:608–622
Larsen BR, MacAulay N (2014) Kir4.1-mediated spatial buffering of K(+): experimental challenges in determination of its temporal and quantitative contribution to K(+) clearance in the brain. Channels (Austin) 8:544–550
Bay V, Butt AM (2012) Relationship between glial potassium regulation and axon excitability: a role for glial Kir4.1 channels. Glia 60:651–660
Strohschein S, Huttmann K, Gabriel S, Binder DK, Heinemann U, Steinhauser C (2011) Impact of aquaporin-4 channels on K+ buffering and gap junction coupling in the hippocampus. Glia 59:973–980
Norenberg MD, Mozes LW, Norenberg LOB, Gregorios JB (1986) Effects of ammonia in primary astrocyte cultures: morphological and biochemical considerations. In: Grisar T, Franck G, Hertz L, Norton WT, Sensenbrenner M, Woodbury DM (eds) Dynamic properties of glia cells II: cellular and molecular aspects advances in the biosciences. Pergamon Press, Oxford, pp 353–362
Kala G, Kumarathasan R, Peng L, Leenen FH, Hertz L (2000) Stimulation of Na+,K+-ATPase activity, increase in potassium uptake, and enhanced production of ouabain-like compounds in ammonia-treated mouse astrocytes. Neurochem Int 36:203–211
Albrecht J, Wysmyk-Cybula U, Rafalowska U (1985) Na+/K+-ATPase activity and GABA uptake in astroglial cell-enriched fractions and synaptosomes derived from rats in the early stage of experimental hepatogenic encephalopathy. Acta Neurol Scand 72:317–320
Xue Z, Li B, Gu L, Hu X, Li M, Butterworth RF, Peng L (2010) Increased Na, K-ATPase alpha2 isoform gene expression by ammonia in astrocytes and in brain in vivo. Neurochem Int 57:395–403
Luttrell LM, Daaka Y, Lefkowitz RJ (1999) Regulation of tyrosine kinase cascades by G-protein-coupled receptors. Curr Opin Cell Biol 11:177–183
Lin HH (2013) G-protein-coupled receptors and their (Bio) chemical significance win 2012 Nobel Prize in Chemistry. Biomed J 36:118–124
Peng L, Du T, Xu J, Song D, Li B, Zhang M, Hertz L (2012) Adrenergic and V1-ergic agonists/antagonists affecting recovery from brain trauma in the Lund project act on astrocytes. Curr Signal Transduct Ther 7:43–55
Liu J, Tian J, Haas M, Shapiro JI, Askari A, Xie Z (2000) Ouabain interaction with cardiac Na+/K+-ATPase initiates signal cascades independent of changes in intracellular Na+ and Ca2+ concentrations. J Biol Chem 275:27838–27844
Gan XT, Hunter JC, Huang C, Xue J, Rajapurohitam V, Javadov S, Karmazyn M (2012) Ouabain increases iNOS-dependent nitric oxide generation which contributes to the hypertrophic effect of the glycoside: possible role of peroxynitrite formation. Mol Cell Biochem 363:323–333
Fedorova OV, Zhuravin IA, Agalakova NI, Yamova LA, Talan MI, Lakatta EG, Bagrov AY (2007) Intrahippocampal microinjection of an exquisitely low dose of ouabain mimics NaCl loading and stimulates a bufadienolide Na/K-ATPase inhibitor. J Hypertens 25:1834–1844
Fedorova OV, Zernetkina VI, Shilova VY, Grigorova YN, Juhasz O, Wei W, Marshall CA, Lakatta EG, Bagrov AY (2015) Synthesis of an endogenous steroidal Na pump inhibitor marinobufagenin, implicated in human cardiovascular diseases, is initiated by CYP27A1 via bile acid pathway. Circ Cardiovasc Genet 8:736–745
Allen AM, Taylor JM, Graham A (2013) Mitochondrial (dys)function and regulation of macrophage cholesterol efflux. Clin Sci (Lond) 124:509–515
Graham A (2015) Mitochondrial regulation of macrophage cholesterol homeostasis. Free Radic Biol Med 89:982–992
Arlicot N, Tronel C, Bodard S, Garreau L, de la Crompe B, Vandevelde I, Guilloteau D, Antier D, Chalon S (2014) Translocator protein (18 kDa) mapping with [125I]-CLINDE in the quinolinic acid rat model of excitotoxicity: a longitudinal comparison with microglial activation, astrogliosis, and neuronal death. Mol Imaging 13:4–11
Wang M, Wang X, Zhao L, Ma W, Rodriguez IR, Fariss RN, Wong WT (2014) Macroglia–microglia interactions via TSPO signaling regulates microglial activation in the mouse retina. J Neurosci 34:3793–3806
Butterworth RF (2016) Neurosteroids in hepatic encephalopathy: novel insights and new therapeutic opportunities. J Steroid Biochem Mol Biol 160:94–97
Ahboucha S, Butterworth RF (2005) Role of endogenous benzodiazepine ligands and their GABA-A–associated receptors in hepatic encephalopathy. Metab Brain Dis 20:425–437
Elkareh J, Kennedy DJ, Yashaswi B, Vetteth S, Shidyak A, Kim EG, Smaili S, Periyasamy SM, Hariri IM, Fedorova L, Liu J, Wu L, Kahaleh MB, Xie Z, Malhotra D, Fedorova OV, Kashkin VA, Bagrov AY, Shapiro JI (2007) Marinobufagenin stimulates fibroblast collagen production and causes fibrosis in experimental uremic cardiomyopathy. Hypertension 49:215–224
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Hertz, L., Song, D., Peng, L. et al. Multifactorial Effects on Different Types of Brain Cells Contribute to Ammonia Toxicity. Neurochem Res 42, 721–736 (2017). https://doi.org/10.1007/s11064-016-1966-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-016-1966-1