
Abstract
Angiotensin II (Ang II) plays a profound regulatory effect on NADPH oxidase and the functional features of vascular adventitial fibroblasts, but its role in antioxidant enzyme defense remains unclear. This study investigated the effect of Ang II on expressions and activities of superoxide dismutase (SOD), catalase (CAT), and glutathione peroxidase (GPx) in adventitial fibroblasts and the possible mechanism involved. Ang II decreased the expression and activity of CAT in a dose- and time-dependent manner, but not that of SOD and GPx. The effects were abolished by the angiotensin II type 1 receptor (AT1R) blocker losartan and AT1R small-interfering RNA (siRNA). Incubation with polyethylene glycol-CAT prevented the Ang II-induced effects on reactive oxygen species (ROS) generation and myofibroblast differentiation. Moreover, Ang II rapidly induced phosphorylation of ERK1/2, which was reversed by losartan and AT1R siRNA. Pharmacological blockade of ERK1/2 improved Ang II-induced decrease in CAT protein expression. These in vitro results indicate that Ang II induces ERK1/2 activation, contributing to the downregulation of CAT as well as promoting oxidative stress and adventitial fibroblast phenotypic differentiation in an AT1R-mediated manner.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hypertension is associated with vascular changes characterized by vascular inflammation, structural remodeling, and altered vascular tone [1]. Undoubtedly, the mechanisms involved are complex, however, there is now a compelling evidence that an elevation in reactive oxygen species (ROS) and an impairment of antioxidant enzyme defense play a pivotal role in this process [2, 3]. Generation of angiotensin II (Ang II) by cells present in the adventitia could potentiate endogenous ROS production and myofibroblast formation [4], leading to a potential mechanism for vascular pathology during hypertension.
Superoxide dismutase (SOD), catalase (CAT), and glutathione peroxidase (GPx) are important endogenous antioxidant enzymes that scavenge intracellular ROS in the physiological state. Deficiencies in these can cause oxidative stress, in which uncontained ROS can inflict tissue injury and dysfunction by attacking functional and structural molecules. Evidence suggesting the role for the above antioxidant enzymes include reports that overexpression of CAT or treatment with liposome-encapsulated SOD attenuates ROS generation [5] and prevents hypertension [6]. A number of tissues and cell types have developed CAT, SOD, or GPx with their activities and/or levels varying widely [7, 8]. Ang II has been shown to reduce the activity levels and expression of SOD in cardiac fibroblasts with no influence on that of CAT and GPx [9]. However, little is known about the role of Ang II in the expression of the above antioxidant enzymes in vascular adventitial fibroblasts and their potential functions in Ang II-induced adventitial fibroblast phenotypic differentiation.
Extracellular signal-regulated kinases 1/2 (ERK1/2) are a family of serine/threonine protein kinases activated as an early response to a variety of stimuli, such as cytokines and ROS, and ultimately regulate transcription factor activation, proliferation, and differentiation [10]. Ang II can rapidly trigger the phosphorylation of ERK1/2 via ROS generation in different cells [11, 12]. Meanwhile, it has been suggested that activation of the ERK1/2 pathway is regulated through phosphoinositide-3 (PI3)-kinase by several stimuli [13], which itself is regulated by Ang II via angiotensin II type 1 receptor (AT1R) in fibroblasts of human hypertrophic scars [14].
Thus, we hypothesized that Ang II stimulates an antioxidant enzyme expression via a mechanism involving the ERK1/2 and/or PI3-kinase-dependent activation in vascular adventitial fibroblasts. To test this hypothesis, we investigated the effects of Ang II on the expressions and activities of SOD, CAT, and GPx in the presence and absence of inhibitors for ERK1/2 and PI3-kinase in adventitial fibroblasts.
Materials and methods
Cell culture and materials
Adventitial fibroblasts were isolated from thoracic aortas of 200 g male Sprague–Dawley rats [15]. Cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 20% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin in 5% CO2 at 37°C. Fibroblasts from the third to sixth passages were used for all experiments. The subconfluent cells were made quiescent by incubation in serum-free medium for 24 h before stimulation. Ang II, PD98059, PD123319, LY294002, and losartan were purchased from Sigma (St Louis, MO, USA). Actinomycin D and cycloheximide were from Beyotime Institute of Biotechnology (Beyotime, Jiangsu, China).
Western blotting
Following stimulation with different compounds, total protein was extracted, and protein concentration was measured with Protein Extraction Kit and BCA Protein Assay Kit, respectively (Beyotime, Jiangsu, China). Protein samples were separated on 10% SDS-PAGE and transferred to nitrocellulose membranes. After blocking with 5% non-fat milk, the membranes were incubated with primary antibodies against CAT, Mn-SOD, Cu/Zn-SOD, GPx (Cayman Chemical, Ann Arbor, MI, USA), phosphor-ERK1/2, ERK1/2 (Cell Signaling, Beverly, MA, USA), AT1R (Santa Cruz, CA, USA), and α-smooth muscle actin (α-SMA, Sigma, St. Louis, MO, USA) at 4°C overnight. After washing, the membranes were incubated with horseradish peroxidase-conjugated antibody for 1 h at room temperature. Western blots were developed using ECL (Roche, Mannheim, Germany) and quantified by scanning densitometry.
Reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was extracted from cultured adventitial fibroblasts using the TRIzol reagent (Invitrogen, USA), and RT-PCR was performed as described previously [16]. The primers used were as follows: CAT, 5′-GAG TCT CTC CAT CAG GTT AC-3′ (sense) and 5′-TGA TGC CCT GGT CAG TCT TG-3′ (antisense); Cu/Zn-SOD, 5′-GCA GAA GGC AAG CGG TGA AC-3′ (sense) and 5′-TAG CAG GAC AGC AGA TGA GT-3′ (antisense); Mn-SOD, 5′-TGA CCT GCC TTA CGA CTA TG-3′ (sense) and 5′-GCT GCA ATG CTC TAC ACT AC-3′ (antisense); GPx, 5′-CTC TCC GCG GTG GCA CAG-3′ (sense) and 5′-CCA CCA CCG GGT CGG ACA TAC-3′ (antisense). The temperature programme for the amplification was 32 cycles of 50 s at 94°C, 45 s at 60°C and 50 s at 72°C. RT-PCR analyses were conducted in triplicate for each sample.
Measurement of CAT, SOD and GPx activities
The CAT activity was measured with the OxiSelect™ Catalase Activity Assay Kit (Cell Biolabs, San Diego, CA, USA) according to the manufacturer,s protocol. Total SOD activity was assayed by using commercial test kit purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, China), which was presented as units per milligram of protein. The GPx activity was determined spectrophotometrically by Bioxytech GPx-340™ Assay Kit (Oxis Research, Portland, OR, USA).
Small-interfering RNA (siRNA)
A double strand siRNA oligonucleotide targeting AT1R (sense: 5′-CGU CAU CCA UGA CUG UAA ATT-3′, antisense: 5′-UUU ACA GUC AUG GAU GAC GTT-3′) and a pair of negative control siRNA (NC siRNA) (sense: 5′-UUC UCC GAA CGU GUC ACG UTT-3′, antisense: 5′-ACG UGA CAC UGG CGG AGA ATT-3′) were designed and synthesized by Shanghai GenePharma Co Ltd. For transfection, cells were plated into 6-well plates and grown to 80% confluence. AT1R siRNA or NC siRNA was then delivered to cells at 100 nM final concentration for 48 h through FuGENE® HD Transfection Reagent (Roche, Mannheim, Germany) according to the manufacturer’s instructions.
Assay of ROS production
2′,7′-Dichlorodihydrofluorescein diacetate (H2DCFH-DA, Calbiochem, Darmstadt, Germany)was utilized to detect the production of intracellular ROS as described by Baas and Berk [17]. In brief, serum-starved cells on round coverslips were incubated with H2DCFH-DA (10 μM) for 30 min at 37°C, then harvested and suspended in phosphate-buffered saline. Fluorescence was analyzed by flow cytometry (FACS Calibur, Becton–Dickinson, San Jose, CA, USA).
Statistical analysis
All values are presented as mean ± SD. Statistical analysis was done by Student’s t-test or by one-way analysis of variation (ANOVA). A value of P < 0.05 was considered to be statistically significant.
Results
Effect of Ang II on antioxidant enzymes activity and expression levels in adventitial fibroblasts
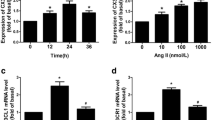
Adventitial fibroblasts were cultured with medium in the presence of different concentrations (0, 10−8, 10−7, 10−6, 10−5 M) of Ang II for 24 h or in the presence of 10−7 M Ang II for 0–24 h (0, 0.5, 2, 6, 12, and 24 h). The basic activity levels of CAT were very high, and Ang II remarkably decreased the CAT activity in a time-dependent (Fig. 1a) and dose-dependent (Fig. 1b) manner. The CAT protein and mRNA levels were also reduced time-dependently in Ang II-stimulated fibroblasts after a transient increase (Fig. 1c). Furthermore, CAT protein expression decreased dose-dependently after exposure to Ang II (Fig. 1d). However, no differences were found between the controls and the Ang II-stimulated fibroblasts with regard to the expressions and activities of SOD and GPx (see Supplementary Fig. S1–S11 online).

Effect of Ang II on CAT activity and expression in adventitial fibroblasts. a Time course of CAT activity. Cells were stimulated with Ang II (10−7 M) for the indicated time. b Dose-dependent CAT activity. Cells were incubated for 24 h with indicated Ang II concentrations. c Time course of CAT protein and mRNA expressions. Cells were stimulated with Ang II (10−7 M) for the indicated time. d Dose-dependent CAT protein expression. Cells were incubated for 24 h with indicated Ang II concentrations. e Effect of Ang II on CAT mRNA stability. Cells were incubated in the absence or presence of Ang II (10−7 M) for 24 h. Actinomycin D (5 μg/ml) was then added and incubated for an additional 2 or 4 h. f Effect of Ang II on CAT protein synthesis. Cells were preincubated for 30 min with cycloheximide (10 μg/ml), followed by Ang II (10−7 M) for 24 h. CAT activity was measured with the CAT Activity Assay Kit according to the manufacturer’s protocol. The whole cell lysates and total RNA were analyzed by Western blotting and RT-PCR. Data are means ± SD from three independent experiments. ▲ P < 0.05, *P < 0.01 versus basic value
We next examined whether de novo RNA or protein synthesis was required for CAT downregulation by Ang II. As shown in Fig. 1e, CAT mRNA levels were stable and similar in both control cells and Ang II-stimulated fibroblasts after actinomycin D pulse, a potent RNA synthesis inhibitor. In addition, the CAT protein synthesis was significantly decreased by Ang II relative to the controls. The effect of Ang II was not prevented by cycloheximide, a protein synthesis inhibitor that blocked CAT expression in the normal fibroblasts (Fig. 1f). These results suggest that RNA transcription was responsible for the decrease in CAT expression.
Ang II-induced suppression of CAT expression was mediated by AT1R in adventitial fibroblasts
To explore the receptor involved in the regulation of CAT expression by Ang II, we first examined the protein expression of CAT after preincubation with the AT1R antagonist losartan and the angiotensin II type 2 receptor (AT2R) antagonist PD123319 in Ang II-stimulated adventitial fibroblasts, the results showed that the addition of losartan, but not PD123319, abolished the decrease in CAT protein expression induced by Ang II (Fig. 2a). Then, we constructed specific AT1R siRNA to block AT1R expression in adventitial fibroblasts. As shown in our previous study [18], the AT1R protein expression in the cells transfected with AT1R siRNA was significantly lower than that in the cells transfected with NC siRNA (P < 0.01) (Fig. 2b). After inhibiting AT1R expression by AT1R siRNA, the CAT downregulation induced by Ang II was reversed markedly (P < 0.01) (Fig. 2c). These results suggest that CAT expression induced by Ang II is mediated through AT1R.

AT1R was involved in Ang II-induced CAT downregulation in adventitial fibroblasts. a Effects of losartan and PD123319 on Ang II-induced decrease in CAT protein expression. Cells were preincubated with losartan or PD123319 (10−5 M each) for 30 min before the addition of Ang II (10−7 M) for 24 h. b Transfection efficacy of AT1R siRNA. Cells were transfected with NC siRNA or AT1R siRNA (100 nM each) for 48 h. c Effect of AT1R siRNA transfection on Ang II-induced decrease in CAT protein expression. Cells were transfected with NC siRNA or AT1R siRNA (100 nM each) for 48 h before exposure to Ang II (10−7 M) for an additional 24 h. The whole cell lysates and total RNA were analyzed by Western blotting and RT–PCR. Data are means±SD from three independent experiments. *P < 0.01 versus control, # P < 0.01 versus Ang II
Modulation of Ang II-induced ROS formation and fibroblast phenotype differentiation by PEG-CAT
Adventitial fibroblasts in the control state showed a slight basal ROS production. Ang II significantly increased ROS production in fibroblast homogenates as compared with controls (P < 0.01). The cell-permeant H2O2-scavenging enzyme, PEG-CAT, reduced the increase in ROS production induced by Ang II (P < 0.01 vs. Ang II) (Fig. 3a).

PEG-CAT modulated ROS generation and α-SMA expression induced by Ang II. a Effect of PEG-CAT on Ang II-induced ROS production. Cells were preincubated with PEG-CAT (1,000 U/ml) for 30 min before exposure to Ang II (10−7 M) for 4 h, and then stained with 10 μM 2′,7′-H2DCFH-DA. b Effect of PEG-CAT on Ang II-induced α-SMA expression. Cells were preincubated with PEG-CAT (1000 U/ml) for 30 min before exposure to Ang II (10−7 M) for 24 h. Oxidant levels were determined by fluorescence-activated cell sorting analysis of 2′,7′-DCF fluorescence, and the whole cell lysates were analyzed by Western blotting. Data are means±SD from three independent experiments. *P < 0.01 versus control, # P < 0.01 versus Ang II
To determine the specific role of ROS in Ang II-induced fibroblast phenotype differentiation, we examined the effect of PEG-CAT on Ang II-induced α-SMA expression, an indicator of fibroblasts differentiation to myofibroblasts. Preincubation with this compound significantly suppressed α-SMA expression induced by Ang II (Fig. 3b). These findings suggest that Ang II-mediated downregulation of CAT is capable of inducing oxidative stress and phenotypic differentiation of adventitial fibroblasts.
Effects of ERK1/2 and PI3-kinase inhibitors on CAT expression induced by Ang II in adventitial fibroblasts
In order to determine the signaling molecules involving in Ang II-stimulated CAT downregulation, the activation of ERK1/2 pathway was first confirmed. As shown in Fig. 4a, the phosphorylation of ERK1/2 was elevated in Ang II-stimulated adventitial fibroblasts, which was significantly lowered by losartan preincubation and AT1R transfection (P < 0.01), demonstrating that Ang II induces ERK1/2 phosphorylation through AT1R. We further tested the expression of CAT and α-SMA in Ang II-induced adventitial fibroblasts after blocking ERK1/2 pathway with ERK1/2 inhibitor PD98059. The results showed that the addition of PD98059 significantly diminished Ang II-induced decrease in CAT protein and mRNA expression (P < 0.01 vs Ang II) (Fig. 4b). Similarly, the α-SMA production by Ang II-stimulated fibroblast was also abolished by PD98059 (Fig. 4c).

Inhibitory effect of Ang II on CAT expression in adventitial fibroblasts was mediated by ERK1/2 pathway but independent of PI3-kinase. a Effects of losartan and AT1R siRNA transfection on Ang II-induced ERK1/2 activation. Cells were preincubated with losartan (10−5 M) for 30 min or transfected with NC siRNA or AT1R siRNA (100 nM each) for 48 h before exposure to Ang II (10−7 M) for 10 min. b Effect of PD98059 on Ang II-induced decrease in CAT protein and mRNA expressions. Cells were preincubated with PD98059 (10−5 M) for 30 min before the addition of Ang II (10−7 M) for 24 h. c Effect of PD98059 on Ang II-induced α-SMA expression. Cells were preincubated with PD98059 (10−5 M) for 30 min before the addition of Ang II (10−7 M) for 24 h. d Effect of LY294002 on Ang II-induced ERK1/2 activation. Cells were exposed to LY294002 (10−5 M) for 30 min and then stimulated with Ang II (10−7 M) for 10 min. e Effect of LY294002 on Ang II-induced CAT downregulation. Cells were preincubated with LY294002 (10−5 M) for 30 min before the addition of Ang II (10−7 M) for 24 h. The whole cell lysates were analyzed by Western blotting. Data are means±SD from three independent experiments. *P < 0.01 versus control, # P < 0.01 versus Ang II
It has been described that ERK1/2 is a downstream target of PI3-kinase, and the activation of the ERK1/2 pathway is regulated by PI3-kinase [13]. Thus, we examined the effect of PI3-kinase inhibitor on Ang II-induced ERK1/2 phosphorylation and CAT downregulation. Stimulation with the PI3-kinase inhibitor LY294002 did not modify the effect of Ang II on ERK1/2 phosphorylation (Fig. 4d) as well as CAT downregulation (Fig. 4e). These results suggest that the PI3-kinase pathway is not involved in Ang II-induced ERK1/2 activation and downregulation of CAT.
Discussion
The main findings of the present study demonstrate that Ang II decreases the protein and mRNA expressions as well as activity level of CAT in vascular adventitial fibroblasts from adult rats, but without influencing the expressions and activities of Mn-SOD, Cu/Zn-SOD, and GPx. Moreover, the inhibition of CAT mediated by Ang II led to oxidative stress and fibroblasts phenotypic differentiation. The mechanisms of these potentially harmful vascular effects are partially clarified in the present study. Our results suggest that ERK1/2 pathway is involved in the decrease of CAT through a mechanism that is dependent of AT1R activation.
There is a growing evidence that oxidative stress plays a major role in many vascular diseases. Ang II has been demonstrated to regulate ROS generation in cardiac, vascular smooth muscle, endothelial, adventitial, and mesangial cells [19]. Infusion of Ang II increases NADPH oxidase activity and superoxide anion production in rat aorta [20]. While in cultured rat cardiac fibroblasts, Ang II not only increases superoxide anion production and intracellular ROS formation but also decreases the activity levels of Cu/Zn-SOD and Mn-SOD [21, 22]. It is well established that adventitia is a major source of ROS in the rabbit and rat aorta [23], and vascular adventitial fibroblasts NADPH oxidase-derived ROS is the sensor and messenger for the early development of vascular disease [24]. Pro-inflammatory Ang II induces oxidative stress by increasing superoxide production in adventitial fibroblasts within hours [4] as well as regulating the expression of antioxidant enzymes. Contradictory findings have been reported regarding the effect of Ang II on the activities and expressions of CAT, Mn-SOD, Cu/Zn-SOD, and GPx in various cells and tissues [9, 25], implying that the effect of Ang II on them are highly cell-specific. In this report, although Ang II decreased CAT expression and activity in a time-dependent and dose-dependent manner after stimulation for 6 h, there was a transient increase in CAT protein and mRNA levels. Recent studies have highlighted the importance of CAT in the clearance of high concentrations of H2O2, showing an adaptive increase of CAT activity after H2O2 infusion [26, 27]. These changes in CAT are likely to be compensatory responses to oxidative stress. Many transcription factors, including NF-E2-related factor (Nrf2) and peroxisome proliferator-activated receptor γ (PPAR γ), have been reported to regulate CAT expression and activity [18, 28]. In adventitial fibroblasts taken from adult rats, we have observed that Ang II decreases PPAR γ expression but increases Nrf2 protein levels in a time-dependent manner. This mechanism could account for the alteration of CAT expression in the early and later phases of Ang II stimulation. Moreover, Ang II did not affect CAT mRNA stability, and CAT protein synthesis induced by Ang II was not suppressed by cycloheximide. It was suggested that CAT expression suppression was linked to a downregulation of its mRNA levels, thereby indicating that the alterations in CAT expression resulted from an altered gene expression rather than from alterations in post-transcriptional regulation. In addition, we clearly document a novel role for AT1R in mediating Ang II-induced downregulation of CAT, as indicated by the finding that the Ang II effect was blocked by losartan and AT1R siRNA but not by PD123319 or NC siRNA. This is in agreement with the previous results obtained in other cell types [9, 12].
ROS generation and phenotypic differentiation of fibroblasts into myofibroblasts have been speculated to be of special importance in the initiation and progression of many vascular diseases. Ang II induces ROS formation via NADPH oxidase stimulation, resulting in fibroblast phenotypic differentiation [29]. Consistent with this observation, we found that ROS generation and α-SMA expression were significantly increased in Ang II-stimulated fibroblasts. Since the intracellular redox status is also governed by antioxidant enzymes, the cell-permeant specific scavenger of H2O2, PEG-CAT, was employed to incubate adventitial fibroblasts before stimulation with Ang II. Treatment with this agent has been reported to be effective in reducing H2O2 levels and oxidant-mediated injury [30]. As referred previously, PEG-CAT abolished Ang II-induced ROS and α-SMA productions. Thus, it is possible that oxidative stress and fibroblasts phenotypic differentiation observed after stimulation with Ang II may be induced by inhibition of CAT expression and activity, indicating that modulation of CAT expression and activity seems to be a promising strategy in the prevention or treatment of pathological vascular structural changes.
Mitogen-activated protein kinases (MAPKs) participate in signal transduction classically associated with cell differentiation, cell growth, and cell death [31]. Of the three major MAPKs, ERK1/2 is a key growth signaling kinase. Once activated, ERK1/2 could phosphorylate a wide array of intracellular targets and result in the reprogramming of gene expression. Several reports indicated that Ang II stimulated activation of ERK1/2 in different cell types [12, 32]. In the present study, we observed that losartan and AT1R siRNA blocked Ang II-induced ERK1/2 activation, suggesting that, in vascular adventitial fibroblasts, this activation is AT1R dependent. Notably, we proved that inhibiting ERK1/2 activity with PD98059 attenuated the Ang II-induced decrease in CAT expression and increase in α-SMA expression in adventitial fibroblasts. These data provide strong support to the idea that Ang II-mediated activation of ERK1/2 is capable of inducing the downregulation of CAT, contributing to oxidative stress and phenotypic differentiation of adventitial fibroblasts.
In addition, we explored the upstream signaling involved in Ang II-induced ERK1/2 activation. It has been reported that PI3-kinase activates the ERK1/2 pathway by several stimuli [23]. We demonstrated that the activation of ERK1/2 by Ang II was not suppressed by PI3-kinase inhibitor LY294002, indicating that this process is independent on PI3-kinase activation. Moreover, preincubation with LY294002 did not block Ang II-induced decrease in CAT expression. These results suggest that PI3-kinase pathway does not play a dominant role in the Ang II-induced ERK1/2 activation and the downregulation of CAT in adventitial fibroblasts.
In conclusion, the present results provide the first evidence that Ang II decreases CAT expression and activity in aortic fibroblasts, at least in part, through mechanisms that include ERK1/2 signaling and dependent of AT1R. The decreased CAT levels induced by Ang II led initially to ROS generation and adventitial fibroblasts activation followed by a phenotypic transformation to myofibroblasts. The dependence of these changes on CAT downregulation is supported by the observation that overexpression of CAT suppressed these responses to Ang II. Hence, it is tempting to speculate that in vivo rise in Ang II levels may play a key role in mediating oxidative stress and then adventitial remodeling. Blockade of CAT downregulation might be a potential therapeutic target in the treatment of Ang II-induced vessel wall damage.
Abbreviations
- Ang II:
-
Angiotensin II
- AT1R:
-
Angiotensin II type 1 receptor
- α-SMA:
-
α-Smooth muscle actin
- CAT:
-
Catalase
- ERK1/2:
-
Extracellular signal-regulated kinase 1/2
- GPx:
-
Glutathione peroxidase
- H2O2 :
-
Hydrogen peroxide
- MAPKs:
-
Mitogen-activated protein kinases
- PI3-kinase:
-
Phosphoinositide-3-kinase
- ROS:
-
Reactive oxygen species
- SOD:
-
Superoxide dismutase
References
Touyz RM (2003) The role of angiotensin II in regulating vascular structural and functional changes in hypertension. Curr Hypertens Rep 5:155–164
Touyz RM (2004) Reactive oxygen species, vascular oxidative stress, and redox signaling in hypertension: what is the clinical significance? Hypertension 44:248–252. doi:10.1161/01.HYP.0000138070.47616.9d
Lassègue B, Griendling KK (2004) Reactive oxygen species in hypertension; An update. Am J Hypertens 17:852–860. doi:10.1016/j.amjhyper.2004.02.004
Shen WL, Gao PJ, Che ZQ, Ji KD, Yin M, Yan C, Berk BC, Zhu DL (2006) NADPH oxidase-derived reactive oxygen species regulate angiotensin II-induced adventitial fibroblast phenotypic differentiation. Biochem Biophys Res Commun 339:337–343. doi:10.1016/j.bbrc.2005.10.207
Brezniceanu ML, Liu F, Wei CC, Chénier I, Godin N, Zhang SL, Filep JG, Ingelfinger JR, Chan JS (2008) Attenuation of interstitial fibrosis and tubular apoptosis in db/db transgenic mice overexpressing catalase in renal proximal tubular cells. Diabetes 57:451–459. doi:10.2337/db07-0013
Godin N, Liu F, Lau GJ, Brezniceanu ML, Chénier I, Filep JG, Ingelfinger JR, Zhang SL, Chan JS (2010) Catalase overexpression prevents hypertension and tubular apoptosis in angiotensinogen transgenic mice. Kidney Int 77:1086–1097. doi:10.1038/ki.2010.63
Choi SI, Kim TI, Kim KS, Kim BY, Ahn SY, Cho HJ, Lee HK, Cho HS, Kim ER (2009) Decreased catalase expression and increased susceptibility to oxidative stress in primary cultured corneal fibroblasts from patients with granular corneal dystrophy type II. Am J Pathol 175:248–261. doi:10.2353/ajpath.2009.081001
Okutan H, Ozcelik N, Yilmaz H, Uz E (2005) Effects of caffeic acid phenethyl ester on lipid peroxidation and antioxidant enzymes in diabetic rat heart. Clin Biochem 38:191–196. doi:10.1016/j.clinbiochem.2004.10.003
Lijnen PJ, van Pelt JF, Fagard RH (2010) Downregulation of manganese superoxide dismutase by angiotensin II in cardiac fibroblasts of rats: association with oxidative stress in myocardium. Am J Hypertens 23:1128–1135. doi:10.1038/ajh.2010.128
Leung PS, Chan YC (2009) Role of oxidative stress in pancreatic inflammation. Antioxid Redox Signal 11:135–165. doi:10.1089/ars.2008.2109
Viedt C, Soto U, Krieger-Brauer HI, Fei J, Elsing C, Kübler W, Kreuzer J (2000) Differential activation of mitogen-activated protein kinases in smooth muscle cells by angiotensin II: involvement of p22phox and reactive oxygen species. Arterioscler Thromb Vasc Biol 20:940–948
Beltrán AE, Briones AM, García-Redondo AB, Rodríguez C, Miguel M, Alvarez Y, Alonso MJ, Martínez-González J, Salaices M (2009) p38 MAPK contributes to angiotensin II-induced COX-2 expression in aortic fibroblasts from normotensive and hypertensive rats. J Hypertens 27:142–154. doi:10.1097/HJH.0b013e328317a730
Takeda K, Ichiki T, Tokunou T, Iino N, Takeshita A (2001) 15-Deoxy-∆12, 14-prostaglandin J2 and thiazolidinediones activate the MEK/ERK pathway through phosphatidylinositol 3-kinase in vascular smooth muscle cells. J Biol Chem 276:48950–48955. doi:10.1074/jbc.M108722200
Liu HW, Cheng B, Yu WL, Sun RX, Zeng D, Wang J, Liao YX, Fu XB (2006) Angiotensin II regulates phosphoinositide 3 kinase/Akt cascade via a negative crosstalk between AT1 and AT2 receptors in skin fibroblasts of human hypertrophic scars. Life Sci 79:475–483. doi:10.1016/j.lfs.2006.01.031
Zhu DL, Herembert T, Marche P (1991) Increased proliferation of adventitial fibroblasts from spontaneously hypertensive rat aorta. J Hypertens 9:1161–1168
Dobrian AD, Davies MJ, Schriver SD, Lauterio TJ, Prewitt RL (2001) Oxidative stress in a rat model of obesity-induced hypertension. Hypertension 37:554–560
Baas AS, Berk BC (1995) Differential activation of mitogen-activated protein kinases by H2O2 and O2 − in vascular smooth muscle cells. Circ Res 77:29–36
Yang WW, Zhang J, Wang HY, Shen WL, Gao PJ, Singh M, Fang NY (2011) Peroxisome proliferator-activated receptor γ regulates angiotensin II-induced catalase downregulation in adventitial fibroblasts of rats. FEBS Lett 585:761–766. doi:10.1016/j.febslet.2011.01.040
Griendling KK, Sorescu D, Lassègue B, Ushio-Fukai M (2000) Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arterioscler Thromb Vasc Biol 20:2175–2183
Rajagopalan S, Kurz S, Münzel T, Tarpey M, Freeman BA, Griendling KK, Harrison DG (1996) Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation Contribution to alterations of vasomotor tone. J Clin Invest 97:1916–1923. doi:10.1172/JCI118623
Lijnen P, Papparella I, Petrov V, Semplicini A, Fagard R (2006) Angiotensin II-stimulated collagen production in cardiac fibroblasts is mediated by reactive oxygen species. J Hypertens 24:757–766. doi:10.1097/01.hjh.0000217860.04994.54
Lijnen P, Petrov V, van Pelt J, Fagard R (2008) Inhibition of superoxide dismutase induces collagen production in cardiac fibroblasts. Am J Hypertens 21:1129–1136. doi:10.1038/ajh.2008.242
Pagano PJ, Clark JK, Cifuentes-Pagano ME, Clark SM, Callis GM, Quinn MT (1997) Localization of a constitutively active, phagocyte-like NADPH oxidase in rabbit aortic adventitia: enhancement by angiotensin II. Proc Natl Acad Sci USA 94:14483–14488
Haurani MJ, Pagano PJ (2007) Adventitial fibroblast reactive oxygen species as autacrine and paracrine mediators of remodeling: Bellwether for vascular disease? Cardiovasc Res 75:679–689. doi:10.1016/j.cardiores.2007.06.016
Dieterich S, Bieligk U, Beulich K, Hasenfuss G, Prestle J (2000) Gene expression of antioxidative enzymes in the human heart: increased expression of catalase in the end-stage failing heart. Circulation 101:33–39
Scandalios JG (2005) Oxidative stress: molecular perception and transduction of signals triggering antioxidant gene defenses. Braz J Med Biol Res 38:995–1014. doi:10.1590/S0100-879X2005000700003
Wassmann S, Wassmann K, Nickenig G (2004) Modulation of oxidant and antioxidant enzyme expression and function in vascular cells. Hypertension 44:381–386. doi:10.1161/01.HYP.0000142232.29764.a7
Zhu H, Itoh K, Yamamoto M, Zweier JL, Li Y (2005) Role of Nrf2 signaling in regulation of antioxidants and phase 2 enzymes in cardiac fibroblasts: protection against reactive oxygen and nitrogen species-induced cell injury. FEBS Lett 579:3029–3036. doi:10.1016/j.febslet.2005.04.058
Zhang J, Fang NY, Gao PJ, Wu LY, Han WQ, Guo SJ, Shen WL, Zhu DL (2008) Peroxisome proliferator-activated receptor-γ agonists attenuate angiotensin II-induced collagen type I expression in adventitial fibroblasts. Clin Exp Pharmacol Physiol 35:72–77. doi:10.1111/j.1440-1681.2007.04748.x
Sousa T, Pinho D, Morato M, Marques-Lopes J, Fernandes E, Afonso J, Oliveira S, Carvalho F, Albino-Teixeira A (2008) Role of superoxide and hydrogen peroxide in hypertension induced by an antagonist of adenosine receptors. Eur J Pharmacol 588:267–276. doi:10.1016/j.ejphar.2008.04.044
Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH (2001) Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev 22:153–183
Chan YC, Leung PS (2009) Involvement of redox-sensitive extracellular-regulated kinases in angiotensin II-induced interleukin-6 expression in pancreatic acinar cells. J Pharmacol Exp Ther 329:450–458. doi:10.1124/jpet.108.148353
Acknowledgments
This research was supported by the Grants from Shanghai Municipal Health Bureau Youth Research Projects (No. 2010Y55) and the International Non-Governmental Cooperation Projects from the Shanghai Science and Technical Committee (No. 09410706900). The authors are grateful to Dr. Tianqing Peng, the University of Western Ontario, for his technical assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
Weiwei Yang and Jia Zhang equally contributed to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
11010_2011_915_MOESM1_ESM.tif
Supplementary Figures Effect of Ang II on activities and expressions of SOD and GPx in adventitial fibroblasts. Cells were stimulated with Ang II (10−7 M) for 0, 0.5, 2, 6, 12, 24, or 48 h, or with different concentrations (0, 10−8, 10−7, 10−6, 10−5 M) of Ang II for 24 h. The activities of SOD (S1 and S2) and GPx (S3 and S4) as well as protein and mRNA levels of Cu/Zn-SOD (S5, S6 and S7), Mn-SOD (S5, S8 and S9) and GPx (S5, S10 and S11) were analyzed by Western blotting and RT–PCR. Data are means±SD from three independent experiments
Rights and permissions
About this article
Cite this article
Yang, W., Zhang, J., Wang, H. et al. Angiotensin II downregulates catalase expression and activity in vascular adventitial fibroblasts through an AT1R/ERK1/2-dependent pathway. Mol Cell Biochem 358, 21–29 (2011). https://doi.org/10.1007/s11010-011-0915-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-011-0915-1