
Abstract
Angiotensin II (Ang II) dysregulation has been determined in many diseases. The CX3CL1/CX3CR1 axis, which has a key role in cardiovascular diseases, is involved in the proliferation and inflammatory cytokine production of vascular smooth muscle cells (VSMCs). In this study, we aim to explore whether Ang II has a role in the expression of CX3CL1/CX3CR1, thus contributing to the proliferation and pro-inflammatory status of VSMCs. Cultured mouse aortic VSMCs were stimulated with 100 nmol/L of Ang II, and the expression of CX3CR1 was assessed by western blot. The results demonstrated that Ang II significantly up-regulated CX3CR1 expression in VSMCs and induced the production of reactive oxygen species (ROS) and the phosphorylation of p38 MAPK. Inhibitors of NADPH oxidase, ROS, and AT1 receptor significantly reduced Ang II-induced CX3CR1 expression. Targeted disruption of CX3CR1 by transfection with siRNA significantly attenuated Ang II-induced VSMC proliferation as well as down-regulated the expression of proliferating cell nuclear antigen (PCNA). Furthermore, CX3CR1-siRNA suppressed the effect of Ang II on stimulating Akt phosphorylation. Besides, the use of CX3CR1-siRNA decreased inflammatory cytokine production induced by Ang II treatment. Our results indicate that Ang II up-regulates CX3CR1 expression in VSMCs via NADPH oxidase/ROS/p38 MAPK pathway and that CX3CL1/CX3CR1 axis contributes to the proliferative and pro-inflammatory effects of Ang II in VSMCs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Angiotensin II (Ang II) dysregulation has been determined in various chronic diseases, such as nephritis, hypertension, and chronic kidney disease [1]. As the key effector peptide of the renin-angiotensin system, Ang II plays an important role in cardiovascular diseases through mechanisms including endothelial dysfunction, vascular remodeling, cell oxidative stress, and pro-inflammatory cytokine secretion in the vascular wall [2]. Ang II exerts multiple effects through initiating downstream signaling molecules, such as extracellular signal-regulated kinase (ERK) 1/2, p38 MAPK, c-Jun N-terminal kinase, and protein kinase B (Akt) [3]. Prior studies have indicated that Ang II activates signaling response through NADPH oxidase-derived reactive oxygen species (ROS), leading to proliferation and inflammatory responses in vascular smooth muscle cells (VSMCs) [4]. VSMCs are the main component of vascular walls and are vital in maintaining the normal physiological function of blood vessels. Dysfunction of VSMCs is a pivotal pathogenesis of cardiovascular diseases. Cytokines secreted from VSMCs could induce cell proliferation, promote endothelial cell dysfunction, and activate leukocytes [5]. The proliferation and inflammation of VSMCs are closely linked with cardiovascular diseases including hypertension and atherosclerosis [6, 7].
As an important pro-inflammatory peptide, Ang II has been demonstrated to increase CX3CL1 expression in VSMCs [8]. CX3CL1 is a special chemokine existing in both soluble and membrane-bound form. Unlike most chemokine ligands that demonstrate affinity for multiple receptors, CX3CL1 exerts its effect through interacting with a unique receptor known as C-X3-C motif receptor 1 (CX3CR1). CX3CR1, a seven-transmembrane domain G-protein-coupled receptor, is expressed on not only leukocytes including monocytes, T cells, and natural killer (NK) cells but also nonmyeloid cells such as endothelial cells and aortic smooth muscle cells. Although CX3CL1 and CX3CR1 are not expressed in normal arteries, they are abundant in unstable atheromatous plaques [9]. Besides mediating leukocyte recruitment and adhesion, CX3CL1/CX3CR1 axis has been demonstrated to exert proliferative effect on VSMCs through an autocrine pathway [10]. Additionally, CX3CL1/CX3CR1 axis is shown to be implicated in inflammatory processes in various organs including the cardiovascular system [11]. Plaques with high expression of CX3CR1 exhibit more intensive inflammatory infiltrations and are more prone to rupture [12]. Butoi et al. [13] reported that the cross-talk between monocytes and VSMCs amplifies the inflammatory response in both cell types via the CX3CL1/CX3CR1 axis. Growing evidences from in vivo studies have revealed the crucial role of CX3CL1/CX3CR1 axis in cardiovascular diseases [14, 15]. However, the involvement of CX3CL1/CX3CR1 axis in Ang II-induced proliferation and inflammation in VSMCs remains unclear.
Since CX3CL1/CX3CR1 axis is implicated in cell proliferation and inflammatory responses, we attempted to explore whether Ang II has a role in the expression of CX3CR1 in VSMCs and then, to investigate the roles of CX3CL1/CX3CR1 axis in Ang II-induced VSMC proliferation and inflammatory cytokine production.
MATERIALS AND METHODS
Materials
Anti-CX3CR1 antibody was purchased from Sigma Chemical (St. Louis, MO, USA). Anti-phospho-p38 MAPK, anti-p38 MAPK, anti-phospho-Akt, anti-Akt, anti-PCNA, anti-TNF-α, anti-IL-6, anti-TGF-β1, and anti-GAPDH antibodies were bought from Wanlei Bio (Shenyang, China). SYBR® Premix Ex Taq™ II, PrimeScript™ RT reagent kit, and TRIzol reagent were from Takara Bio Inc. (Otsu, Japan). Diphenyleneiodonium chloride (DPI), N-acetyl-l-cysteine (NAC), and Ang II were from Sigma Chemical (St. Louis, MO, USA). Dihydroethidium (DHE), Lipo6000™ transfection reagent, Cell Counting Kit-8 (CCK-8) kit, and other chemicals were obtained from Beyotime Institute of Biotechnology (Shanghai, China) unless otherwise indicated.
Cell Culture
A mouse aorta origin vascular smooth muscle cell (MOVAS) line was purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). MOVAS cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (both from HyClone, Logan, UT, USA). The cells were incubated at 37 °C with 5% CO2 in a humidified incubator and were passaged at a ratio of 1:3. The 5th to 12th passages of cells were used for the following experiments. In all the experiments, cells at 80 to 90% confluence were starved in serum-free DMEM for 24 h before stimulation. All the pretreatments were administered 60 min before Ang II (100 nmol/L) treatment.
Transfection
We purchased the mouse small interfering RNA (siRNA) of CX3CR1 from Ribo Bio (Guangzhou, China). Transient transfection with CX3CR1-siRNA (50 nmol/L) was performed using the Lipo6000™ transfection reagent in accordance with the manufacturer’s protocols. A nonsense sequence served as the negative control (NC).
Cell Proliferation Assay
CCK-8 kit was used to evaluate the proliferation of MOVAS cells [16]. MOVAS cells were seeded in 96-well cell culture plates in 100 μL of medium and incubated at 37 °C in a humidified incubator containing 5% CO2. The cells were transfected with CX3CR1-siRNA or NC-siRNA 48 h prior to exposure to Ang II and then, incubated with Ang II (100 nmol/L) for 24 h. Cells without Ang II treatment were designated as control cells. Finally, 10 μL of WST-8 solution was added into each well and incubated for 1 h at 37 °C. The absorbance of these wells was measured at 450 nm using a Multiskan Spectrum spectrophotometer (Thermo Electron Corporation, MA, USA).
Western Blotting
After incubation with Ang II, the cells were rinsed twice with ice-cold phosphate buffer saline. The cells were lysed with radio immunoprecipitation assay (RIPA) lysis buffer for 30 min at 4 °C, and the soluble lysates were centrifuged at 12,000 rpm at 4 °C for 15 min. The concentrations of protein were determined using bicinchoninic acid protein kit. All of the samples were diluted in SDS loading buffer, followed by a boiling water bath for 10 min. The equal amounts of protein samples were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) by electrophoresis and then transferred onto polyvinylidene fluoride (PVDF) membranes. The membranes were blocked with 5% skimmed milk in Tris-buffered saline with Tween-20 (TBST) for 60 min and then hybridized overnight with primary antibodies against CX3CR1 (1:1000), PCNA (1:500), phospho-p38 (1:500), total p38 (1:500), phospho-Akt (1:500), total Akt (1:500), anti-TNF-α (1:500), anti-IL 6 (1:500), and anti-TGF-β1 (1:500) at 4 °C. After washing three times with TBST, the membranes were incubated with indicated secondary antibodies for 1 h at 37 °C. After the membranes were subjected to three 10-min washes in TBST, the signals of target proteins were visualized using the enhanced chemiluminescent reagent (Merck Millipore, Billerica, MA, USA) and the immunoreactive bands were quantified by using VILBER Fusion FX7 imaging system (Fusion FX7, Vilber Lourmat, France). The intensity of the detected bands was analyzed with FusionCapt Advance software (Vilber Lourmat, France) and normalized against the housekeeping protein GAPDH, except for the intensity of the phosphorylated p38 and Akt bands, which were normalized to intensity of the total p38 and Akt bands, respectively.
Real-Time Quantitative PCR
Total RNA was extracted from MOVAS cells using TRIzol reagent according to the manufacturer’s instructions. Approximately 1000 ng of total RNA was reverse transcribed using the PrimeScript™ RT reagent kit. PCR was performed using a CFX96™ Real-Time PCR detection system (Bio-Rad, USA) and SYBR® Premix Ex Taq™ II under the following cycling conditions: 40 cycles of 95 °C for 15 s and 58 °C for 60 s. The data were normalized to GAPDH levels, and the abundance of mRNA was calculated using the 2-ΔΔCT method. The primer sequences were listed in Table 1.
Fluorescence Microscopy Analysis for Intracellular ROS
The ROS generated in the cells was detected by DHE probe, which is capable of staining superoxide anion (O2−). The cells were pretreated with 1 μmol/L of losartan, 10 μmol/L of DPI (an NADPH oxidase inhibitor), and 1 mmol/L of NAC (an ROS scavenger), respectively, and then stimulated by Ang II (100 nmol/L) for 1 h. After incubation, the cells were washed twice with Hanks’ balanced salt solution (HBSS) and incubated with DHE (10 μmol/L) for 30 min at 37 °C in a light-protected humidified chamber. Then, the cells were washed three times with HBSS. A fluorescence microscope (Axio Observer Z1, Carl Zeiss, Oberkochen, Germany) was applied at set excitation and emission wavelengths in these experiments. Images were taken at same condition and mean fluorescence intensity quantification was performed for comparison with each group by using Image-Pro Plus 6.0 software (Media Cybernetics, MD, USA).
Statistical Analysis
All of the data were shown as the mean ± SEM. GraphPad Prism 5 software (GraphPad Software, Inc., San Diego, CA) and SPSS 19.0 software (IBM Corporation, NY) were applied to perform the statistical analyses. One-way ANOVA followed by post hoc Bonferroni test was performed when multiple comparisons were made. All differences considered statistically different when the analysis satisfied the two-sided statistical significance of p < 0.05.
RESULTS
Ang II Increased the Expression of CX3CL1/CX3CR1 in MOVAS Cells
To examine the expression level of CX3CR1 stimulated by Ang II, western blot and quantitative PCR (qPCR) were performed to detect protein expression and mRNA level, respectively. Figure 1a shows the protein expression of CX3CR1 in cells after treatment with Ang II (100 nmol/L) for 0, 12, 24, or 36 h. Figure 1b quantifies the protein expression of CX3CR1 after incubation with different concentrations of Ang II on 10, 100, and 1000 nmol/L. The data showed that Ang II markedly increased CX3CR1 protein expression in a time- and dose-dependent manner. The peak protein expression of CX3CR1 occurred after treatment for 24 h. When the cells were treated with Ang II for 12 h, the mRNA levels of CX3CR1 as well as CX3CL1 were increased compared with the levels observed in the control cells. Pretreating the cells with losartan, an inhibitor of Ang II type 1 receptor (AT1R), blocked the observed increases in both CX3CL1 and CX3CR1 mRNA levels (Fig. 1c, d).

Ang II increases CX3CL1/CX3CR1 expression in MOVAS cells. a MOVAS cells were treated with Ang II for the indicated time intervals. b MOVAS cells were treated with Ang II at different doses for 24 h. The protein expression of CX3CR1 was determined by western blot. c, d MOVAS cells were treated with Ang II for 12 h. The mRNA levels of CX3CL1 and CX3CR1 were determined by qPCR. All of the relative protein expression levels were normalized to the GAPDH expression. All of the data are shown as the mean ± SEM. n = 4 independent experiments. *P < 0.05 vs. control; #P < 0.05 vs. Ang II.
Ang II Activated p38 MAPK Signaling Pathway Through NADPH Oxidase-Derived ROS in MOVAS Cells
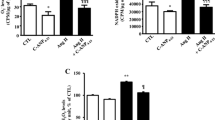
To define the association between ROS levels and p38 MAPK activation induced by Ang II, DPI (an NADPH oxidase inhibitor) and NAC (an ROS scavenger) were applied. DHE probe was used to measure the ROS levels produced in MOVAS cells after treatment with Ang II. As shown in Fig. 2a, Ang II enhanced ROS production by approximately three folds, and pretreating the cells with losartan, DPI, and NAC effectively blocked the observed increase in ROS production. The level of phosphorylated p38 was detected by western blot after the cells were treated with Ang II for 30 min. In agreement with previous reports, the phosphorylated p38 level was more than two folds higher relative to the level observed in the control cells. The Ang II-induced p38 MAPK activation was largely suppressed by the pretreatment with DPI and NAC (Fig. 2b). Thus, these data indicated that ROS production via NADPH oxidase mediated Ang II-induced activation of p38 MAPK signaling pathway in MOVAS cells.

Ang II increases CX3CR1 expression via NADPH oxidase/ROS/p38 MAPK pathway. a MOVAS cells were pretreated with losartan (1 μmol/L), DPI (10 μmol/L), or NAC (1 mmol/L), and then treated with Ang II for 1 h. The generation of ROS was determined by DHE (10 μmol/L) probe. All of the mean fluorescence intensities were standardized to control samples. b MOVAS cells were pretreated with DPI or NAC, and then treated with Ang II for 30 min. The protein expression of phosphorylated p38 MAPK was determined by western blot. c MOVAS cells were pretreated with losartan, DPI, NAC, or SB203580 (25 μmol/L), then treated with Ang II for 24 h. The protein expression of CX3CR1 was determined by western blot. The relative level of phosphorylated p38 expression was normalized to total p38 expression; the relative level of CX3CR1 expression was normalized to GAPDH expression. All of the data are shown as the mean ± SEM. n = 4 independent experiments. *P < 0.05 vs. control; #P < 0.05 vs. Ang II.
Ang II Increased CX3CR1 Expression via AT1R/NADPH Oxidase/ROS-Dependent p38 MAPK Signaling Pathway in MOVAS Cells
It has been reported that ROS-dependent activation of MAPKs is required for inflammatory responses [1, 17]. We next investigated the signaling pathways involved in Ang II-induced CX3CR1 expression, initially focusing on p38 MAPK: one of the major MAPK signaling pathways. MOVAS cells were pretreated with losartan, DPI, NAC, or SB203580 (a specific inhibitor of p38 MAPK) separately, and then, CX3CR1 protein expression was analyzed by western blot after exposure to Ang II for 24 h. As shown in Fig. 2c, the pretreatment with losartan, DPI, NAC, and SB203580 strongly blocked Ang II-induced CX3CR1 protein expression in MOVAS cells. The results displayed that Ang II induced ROS generation via NADPH oxidase, leading to activation of p38 MAPK signaling pathway, which consequently up-regulated the protein expression of CX3CR1.
Targeted Disruption of CX3CR1 Attenuated Ang II-Induced Proliferation of MOVAS Cells
To clarify whether the CX3CL1/CX3CR1 axis is involved in mediating the proliferative effect of Ang II, we disrupted CX3CR1 expression by RNA interference. The efficiencies of siRNA were determined via qPCR and western blot, respectively. MOVAS cells showed an approximately 70% decrease in CX3CR1 mRNA level and a 60% reduction in the protein expression after 48-h transfection with CX3CR1-specific siRNA, when compared with those of the control cells (Fig. 3a, b). After the transient transfection for 48 h, the cells were treated with Ang II for 24 h. The proliferation of MOVAS cells was evaluated using the CCK-8 kit. Ang II exerted an approximately threefold increase in MOVAS cell proliferation as compared with that of the control cells. Transfection with CX3CR1-siRNA significantly attenuated the effect of Ang II on MOVAS cell proliferation (Fig. 3c). In addition, western blot assay showed that Ang II augmented proliferating cell nuclear antigen (PCNA) protein expression in MOVAS cells, which was also obviously suppressed by transfection with CX3CR1-siRNA (Fig. 3d). The NC-siRNA did not influence the effect of Ang II on MOVAS cell proliferation. We further investigated the signaling pathways involved in CX3CR1-mediated proliferation, focusing on Akt signaling pathway: a key regulator of proliferation and survival in VSMCs. We found that the phosphorylation of Akt was significantly increased in respond to Ang II treatment and this effect was ameliorated by disruption of CX3CR1 (Fig. 3e). The results indicated that CX3CL1/CX3CR1 axis mediated the Ang II-induced proliferation of MOVAS cells via Akt signaling pathway.

Targeted disruption of CX3CR1 attenuates Ang II-induced proliferation of MOVAS cells. a, b CX3CR1 expression was disrupted by siRNA, and the mRNA level and protein expression of CX3CR1 were detected by qPCR and western blot, respectively. c CX3CR1 expression was disrupted by siRNA, and then, the cells were treated with Ang II for 24 h. The cell proliferation was determined by CCK-8 assay. d MOVAS cells were treated with Ang II for 24 h. The protein expression of PCNA was determined by western blot. e MOVAS cells were treated with Ang II for 30 min. The Akt phosphorylation was determined by western blot. The relative levels of CX3CR1 and PCNA expressions were normalized to GAPDH expression; the relative level of phosphorylated Akt expression was normalized to total Akt expression. All of the data are shown as the mean ± SEM. n = 4 independent experiments. *P < 0.05 vs. control; #P < 0.05 vs. Ang II.
Targeted Disruption of CX3CR1 Decreased Ang II-Induced Production of TNF-α, IL-6, and TGF-β1 in MOVAS Cells
To investigate the involvement of CX3CL1/CX3CR1 axis in Ang II-induced VSMC inflammatory cytokine production, the cells were transfected with CX3CR1-siRNA or NC-siRNA 48 h prior to their exposure to Ang II. The mRNA levels of TNF-α, IL-6, and TGF-β1 were measured by qPCR after a 12-h incubation of Ang II. The protein expressions of TNF-α, IL-6, and TGF-β1 were measured by western blot after a 24-h incubation of Ang II. Ang II stimulation led to significant increases in both the mRNA levels and protein expressions of TNF-α, IL-6, and TGF-β1 in MOVAS cells. The use of CX3CR1-siRNA decreased the mRNA levels of TNF-α, IL-6, and TGF-β1 by 44, 45, and 38% respectively as compared with the Ang II group (Fig. 4a–c). Besides, the use of CX3CR1-siRNA decreased the protein expressions of TNF-α, IL-6, and TGF-β1 by 47, 27, and 23% respectively as compared with the Ang II group (Fig. 4d–f). In contrast, NC-siRNA showed no effect on suppressing the inflammatory cytokine production induced by Ang II.

Targeted disruption of CX3CR1 decreases Ang II-induced production of TNF-α, IL-6, and TGF-β1. a–c CX3CR1 expression was disrupted by siRNA, and then, the cells were treated with Ang II for 12 h. The mRNA levels of TNF-α, IL-6, and TGF-β1 were determined by qPCR. d–f CX3CR1 expression was disrupted by siRNA, and then, the cells were treated with Ang II for 24 h. The protein expressions of TNF-α, IL-6, and TGF-β1 were determined by western blot. All of the relative protein expression levels were normalized to the GAPDH expression. All of the data are shown as the mean ± SEM. n = 4 independent experiments. *P < 0.05 vs. control; #P < 0.05 vs. Ang II.
DISCUSSION
Ang II is an important vasoactive peptide which performs a vital part in the pathogenesis, such as cell growth, inflammation, and migration of VSMCs [1, 18]. The results of this study demonstrated that Ang II induced CX3CR1 expression in VSMCs via p38 MAPK signaling pathway, which was transactivated by an NADPH oxidase/ROS cascade. Furthermore, CX3CL1/CX3CR1 axis contributed to the proliferation of VSMCs through Akt signaling pathway and was involved in Ang II-induced production of TNF-α, IL-6, and TGF-β1.
Unbalanced production of ROS has been determined in various cardiovascular diseases [19]. Oxidative stress, induced by generation of ROS, can directly exert vascular damage and intensify vascular wall dysfunction. The relationship between Ang II and ROS has raised great concern for years. The NADPH oxidase family members are proteins that transfer electrons across biological membranes. In brief, the electron acceptor is oxygen and the product of the electron-transfer reaction is a superoxide. Therefore, NADPH oxidase enzymes can serve as major sources of the superoxide anions in the vascular system [20]. Consistently, our data showed that NADPH oxidase inhibitor significantly decreased Ang II-induced superoxide anion production in VSMCs. Pretreatment with the inhibitor of NADPH oxidase or an ROS scavenger resulted in decreased expression of CX3CR1 stimulated with Ang II in VSMCs, suggesting that NADPH oxidase/ROS cascade was involved in Ang II-induced CX3CR1 expression. These results indicated that ROS probably played a key role in Ang II-induced CX3CR1 expression.
MAPK signaling pathways regulate various cellular programs by relaying extracellular signals to intracellular responses. In mammals, MAPK enzymes coordinately regulate cell proliferation, differentiation, motility, and survival. The best known MAPK enzymes include the extracellular signal-regulated kinases 1 and 2 (ERK1/2), c-Jun amino terminal kinases 1 to 3, p38, and ERK5 families. MAPKs were previously shown to regulate the expression of CX3CR1 in VSMCs [21]. However, the role of p38 MAPK in the Ang II-induced CX3CR1 expression was rarely investigated. Our results indicated that Ang II markedly activated the phosphorylation of p38, which further up-regulated CX3CR1 expression. Moreover, it was demonstrated that Ang II-stimulated p38 MAPK was mediated through NADPH oxidase/ROS cascade in VSMCs. Therefore, p38 MAPK signaling pathway might play a key role in Ang II-induced NADPH oxidase/ROS-dependent CX3CR1 expression.
VSMCs can initiate excessive proliferation in response to vascular injury [22], which is involved in the development of cardiovascular diseases, such as atherosclerosis, hypertension, and vessel stenosis. There is little CX3CL1 expression in VSMCs under normal condition. However, overexpression of CX3CL1 in VSMCs can be observed under inflammatory condition [23]. In addition to chemotaxis, increasing evidences have shown that CX3CL1/CX3CR1 axis plays an important role in the proliferation of VSMCs through an autocrine pathway [10]. The activation of CX3CR1 directly leads to a cascade of cellular events, such as the generation of inositol triphosphate, the release of calcium, and the activation of Akt [24], NF-κB [10], and Jak/Stat 5 [25] signaling pathways. CX3CL1/CX3CR1 axis provides the survival signals for VSMCs and has both anti-apoptosis and proliferative effects on human VSMCs via activation of ERK and Akt signaling pathways [26]. However, the role of this chemokine axis in Ang II-treated VSMCs remains uncertain. Our results demonstrated that transfection with CX3CR1-siRNA not only effectively attenuated the proliferative effect of Ang II but also down-regulated the expression of PCNA, a cell cycle-related protein which is known as a molecular marker of proliferation. PI3K/Akt is an essential intracellular signaling pathway in regulating the migration and proliferation of VSMCs [27]. Ang II stimulates a variety of intracellular signaling pathways, including the Akt and the MAPK signaling pathways, contributing to the proliferation, migration, and inflammation of VSMCs [28, 29]. It was reported that inhibition of Akt signaling pathway can suppress Ang II-induced proliferation of VSMCs [30]. In the present study, we found that silencing of CX3CR1 suppressed Ang II-induced activation of Akt signaling pathway, suggesting that CX3CL1/CX3CR1 axis probably mediated Ang II-induced VSMCs proliferation through activation of PI3K/Akt signaling pathway. These results indicated that the activation of CX3CL1/CX3CR1 axis could be at least one of the mechanisms contributing to the proliferation of VSMCs induced by Ang II.
CX3CL1/CX3CR1 axis can work as a signal transduction pathway to coordinate functions of CX3CL1 and other cytokines. Although most of in vivo studies have established the pro-inflammatory role of CX3CL1/CX3CR1 axis [31, 32], the underlie mechanism by which CX3CL1/CX3CR1 axis modulates other cytokines is still unclear. Additionally, the related data from in vitro experiments are often limited and controversial. For instance, Mizutani et al. [33] reported that pretreatment with 0.03 nM of CX3CL1 significantly reduced LPS-induced TNF-α secretion from macrophages, suggesting that CX3CL1 might play a protective role in inflammatory response. We therefore investigated the impact of CX3CL1/CX3CR1 axis on Ang II-induced inflammatory cytokine production in VSMCs. Interestingly, silencing of CX3CR1 decreased the expressions of TNF-α, IL-6, and TGF-β1 in MOVAS cells stimulated with Ang II. Therefore, it was tempting to speculate that CX3CL1/CX3CR1 axis also plays a key role in regulating Ang II-induced secretion of TNF-α, IL-6, and TGF-β1 in VSMCs, which may in turn exert autocrine/paracrine effects on VSMCs and likely on other vascular cells. However, the specific mechanism by which CX3CL1/CX3CR1 axis modulates other cytokines is still unclear. Further studies are needed to elucidate the complex role of CX3CL1/CX3CR1 axis in the whole cytokine network.
Taken together, this study indicated that Ang II can up-regulate the expression of CX3CR1 in VSMCs in vitro via NADPH oxidase/ROS/p38 MAPK pathway, and CX3CL1/CX3CR1 axis plays an important role in modulating Ang II-induced cell proliferation and inflammatory cytokine production in VSMCs in vitro. Accordingly, CX3CL1/CX3CR1 axis may serve as a potential therapeutic target for cardiovascular diseases, including atherosclerosis, hypertension, and vessel stenosis. To specifically address the contribution of the CX3CL1/CX3CR1 axis in those diseases, further studies involving modulation of the axis in VSMC-specific deficient mice need to be performed.
References
Cat, A.N.D., A.C. Montezano, D. Burger, et al. 2013. Angiotensin II, NADPH oxidase, and redox signaling in the vasculature. Antioxidants & Redox Signaling 19 (10): 1110–1120.
Kasal, D.A., and E.L. Schiffrin. 2012. Angiotensin II, aldosterone, and anti-inflammatory lymphocytes: Interplay and therapeutic opportunities. International Journal of Hypertension 2012: 829786.
Touyz, R.M., and E.L. Schiffrin. 2000. Signal transduction mechanisms mediating the physiological and pathophysiological actions of angiotensin II in vascular smooth muscle cells. Pharmacological Reviews 52: 639–672.
Montezano, A.C., D.C.A. Nguyen, F.J. Rois, et al. 2014. Angiotensin II and vascular injury. Current Hypertension Reports 16 (6): 431.
Doran, A.C., N. Meller, and C.A. McNamara. 2008. Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology 28: 812–819.
Ragolia, L., T. Palaia, E. Paric, and J.K. Maesaka. 2003. Prostaglandin D2 synthase inhibits the exaggerated growth phenotype of spontaneously hypertensive rat vascular smooth muscle cells. The Journal of Biological Chemistry 278: 22175–22181.
Rudijanto, A. 2007. The role of vascular smooth muscle cells on the pathogenesis of atherosclerosis. Acta Medica Indonesiana 39: 86–93.
Zheng, Lulu, Yongwen Cao, Shao Liu, et al. 2014. Neferine inhibits angiotensin II-induced rat aortic smooth muscle cell proliferation predominantly by downregulating fractalkine gene expression. Experimental and Therapeutic Medicine 8: 1545–1550.
Lucas, A.D., C. Bursill, T.J. Guzik, J. Sadowski, K.M. Channon, and D.R. Greaves. 2003. Smooth muscle cells in human atherosclerotic plaques express the fractalkine receptor CX3CR1 and undergo chemotaxis to the CX3C chemokine fractalkine (CX3CL1). Circulation 108: 2498–2504.
Chandrasekar, B., S. Mummidi, R.P. Perla, et al. 2003. Fractalkine (CX3CL1) stimulated by nuclear factor kappaB (NF-kappaB)-dependent inflammatory signals induces aortic smooth muscle cell proliferation through an autocrine pathway. The Biochemical Journal 373: 547–558.
Xuan, W., Y. Liao, B. Chen, et al. 2011. Detrimental effect of fractalkine on myocardial ischaemia and heart failure. Cardiovascular Research 92: 385–393.
Masztalewicz, Marta, Przemysław Nowacki, Łukasz Szydłowski, et al. 2017. High expression of CX3C chemokine receptor 1 (CX3CR1) in human carotid plaques is associated with vulnerability of the lesions. Folia Neuropathologica 55 (2): 174–181.
Butoi, E.D., A.M. Gan, I. Manduteanu, D. Stan, et al. 2011. Cross talk between smooth muscle cells and monocytes/activated monocytes via CX3CL1/CX3CR1 axis augments expression of pro-atherogenic molecules. Biochimica et Biophysica Acta 1813: 2026–2035.
Moatti, D., S. Faure, F. Fumeron, et al. 2001. Polymorphism in the fractalkine receptor CX3CR1 as a genetic risk factor for coronary artery disease. Blood 97: 1925–1928.
Ikejima, H., T. Imanishi, H. Tsujioka, et al. 2010. Upregulation of fractalkine and its receptor, CX3CR1, is associated with coronary plaque rupture in patients with unstable angina pectoris. Circulation Journal 74: 337–345.
Sun, H.J., et al. 2015. Salusin-beta contributes to vascular remodeling associated with hypertension via promoting vascular smooth muscle cell proliferation and vascular fibrosis. Biochimica et Biophysica Acta 1852: 1709–1718.
Yu, M., Y. Zheng, H.X. Sun, and D.J. Yu. 2012. Inhibitory effects of enalaprilat on rat cardiac fibroblast proliferation via ROS/P38MAPK/TGF-β1 signaling pathway. Molecules 17: 2738–2751.
Savoia, C., D. Burger, N. Nishigaki, et al. 2011. Angiotensin II and the vascular phenotype in hypertension. Expert Reviews in Molecular Medicine. https://doi.org/10.1017/S:1462399411001815.
Khan, M., F. Hassan, S. Roy, et al. 2014. Measurement of reactive oxygen species in cardiovascular disease. Hypertension 49 (4): 359–370.
Cai, H., K.K. Griendling, and D.G. Harrison. 2003. The vascular NAD(P)H oxidases as therapeutic targets in cardiovascular diseases. Trends in Pharmacological Sciences 24: 471–478.
Gan, A.M., E.D. Butoi, A. Manea, et al. 2013. Inflammatory effects of resistin on human smooth muscle cells: Up-regulation of fractalkine and its receptor, CX3CR1 expression by TLR4 and Gi-protein pathways. Cell and Tissue Research 351: 161–174.
Panchatcharam, M., et al. 2010. Enhanced proliferation and migration of vascular smooth muscle cells in response to vascular injury under hyperglycemic conditions is controlled by beta3 integrin signaling. The International Journal of Biochemistry & Cell Biology 42: 965–974.
Ludwig, A., T. Berkhout, K. Moores, et al. 2002. Fractalkine is expressed by smooth muscle cells in response to IFN-gamma and TNF-alpha and is modulated by metalloproteinase activity. Journal of Immunology 168: 604–612.
Kansra, V., C. Groves, J.C. Gutierrez Ramos, and R.D. Polakiewicz. 2001. Phosphatidylinositol 3-kinase-dependent extracellular calcium influx is essential for CX(3)CR1-mediated activation of the mitogen-activated protein kinase cascade. The Journal of Biological Chemistry 276 (34): 31831–31838.
Yang, X.P., R.A. Jones, and J.H. Rivers. 2007. Fractalkine upregulates intercellular adhesion molecule-1 in endothelial cells through CX3CR1 and the Jak-Stat5 pathway. Circulation Research 101 (10): 1001–1008.
White, Gemma E., Thomas C.C. Tan, Alison E. John, et al. 2010. Fractalkine has anti-apoptotic and proliferative effects on human vascular smooth muscle cells via epidermal growth factor receptor signaling. Cardiovascular Research 85: 825–835.
Zhang, Feng, Xingsheng Ren, Mingxia Zhao, et al. 2016. Angiotensin-(1–7) abrogates angiotensin II-induced proliferation, migration and inflammation in VSMCs through inactivation of ROS-mediated PI3K/Akt and MAPK/ERK signaling pathways. Scientific Reports 6: 34621.
Xi, X.P., et al. 1999. Central role of the MAPK pathway in ang II-mediated DNA synthesis and migration in rat vascular smooth muscle cells. Arteriosclerosis, Thrombosis, and Vascular Biology 19: 73–82.
Shen, Y.J., et al. 2014. Cardamonin inhibits angiotensin II-induced vascular smooth muscle cell proliferation and migration by downregulating p38 MAPK, Akt, and ERK phosphorylation. Journal of Natural Medicines 68: 623–629.
Chiou, Wen Fei, Chien-Chih Chen, and Bai-Luh Wei. 2011. 3, 4-di-O-Caffeoylquinic acid inhibits angiotensin-II-induced vascular smooth muscle cell proliferation and migration by downregulating the JNK and PI3K/Akt signaling pathways. Evidence-based Complementary and Alternative Medicine 2011: 634502.
Niess, J.H., and G. Adler. 2010. Enteric flora expands gut lamina propria CX3CR1+ dendritic cells supporting inflammatory immune responses under normal and inflammatory conditions. Journal of Immunology 184 (4): 2026–2037.
Koziolek, M.J., G.A. Müller, A. Zapf, et al. 2010. Role of CX3C-chemokine CX3C-L/fractalkine expression in a model of slowly progressive renal failure. Nephrology, Dialysis, Transplantation 25: 684–698.
Mizutani, N., T. Sakurai, T. Shibata, et al. 2007. Dose-dependent differential regulation of cytokine secretion from macrophages by fractalkine. Journal of Immunology 179 (11): 7478–7487.
Acknowledgments
We sincerely thank Lixue Chen, Wenhui Yan, and Xiaojuan Deng at the central laboratory of the First Affiliated Hospital of Chongqing Medical University for their technical assistance while we conducted our experiments.
Funding
This study was funded by Wu Jieping Medical Foundation (No. 320.6750.16196) and the Medical Research and Developmental Fund of the First Affiliated Hospital of Chongqing Medical University (No. PYJJ2017-02).
Author information
Authors and Affiliations
Contributions
Chengsheng Li and Yunfeng Xia conceived the study and designed the experiments. Chengsheng Li, Jin He, and Xiaoyi Zhong performed all of the experiments. Chengsheng Li analyzed the data and wrote the manuscript. Hua Gan and Yunfeng Xia reviewed and revised the paper.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Li, C., He, J., Zhong, X. et al. CX3CL1/CX3CR1 Axis Contributes to Angiotensin II-Induced Vascular Smooth Muscle Cell Proliferation and Inflammatory Cytokine Production. Inflammation 41, 824–834 (2018). https://doi.org/10.1007/s10753-018-0736-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-018-0736-4