
Abstract
Producing a functional anti-IL-2Rα antibody in Leishmania tarentolae, a trypanosomatid protozoan non-pathogenic to human, is a cost-effective and safe alternative for the production of therapeutic recombinant proteins. Expression cassettes encoding heavy and light chains of the anti-IL-2Rα antibody were cloned into two separate pLEXSY vectors. The plasmids were then used to transfect L. tarentolae laboratory strain p10 and stable expression of a recombinant humanized anti-IL-2Rα antibody was obtained. The heavy and light chains successfully assembled to produce a tetrameric 150 kDa antibody. The antibody was glycosylated and functional as assessed by enzymatic deglycosylation and cell proliferation assays respectively. L. tarentolae can be used as an efficient, cost and labor-effective expression system for the production of therapeutic recombinant proteins.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Biopharmaceuticals constitute a considerable proportion of marketed drugs. Monoclonal antibodies (mAbs) and related products are the main biopharmaceuticals. MAbs are used as biomedical (Chon and Zarbis-Papastoitsis 2011) research molecules for diagnostic, and therapeutic purposes (Stockwin and Holmes 2003; Majidi et al. 2009). The required therapeutic doses of mAbs however are much higher than that of other biological products (Jones et al. 2007; Iberg and Hawiger 2019). Prokaryotic hosts constitute the most widely used protein expression systems. Albeit limitations to prokaryotic expression of antibodies, i.e., improper folding and assembly of the antibody fragments, improper disulfide bond formation, and lack of glycosylation have led to the employment of mammalian expression systems (De Marco 2009; Ke and Berkmen 2014; Dhara et al. 2018).
Glycosylation is a eukaryotic post-translational modification which is critical to antibody function (Jefferis et al. 1998; Jefferis 2005). Leishmania tarentolae, a trypanosomatid protozoan not pathogenic to humans (Breitling et al. 2002; Niimi 2012), has been developed as an alternative eukaryotic protein expression system for the production of recombinant proteins with mammalian-like posttranslational modifications. The advantages of this system include easy handling, faster growth rate, and cost- effectivness (Niimi 2012; Fritsche et al. 2007). These features render it a promising system for the production of biopharmaceutical proteins.
Anti-IL-2Rα antibodies prevent IL-2–induced clonal expansion of activated lymphocytes and decrease their survival through binding to the α chain of IL-2 receptor (Church 2003; Kandus et al 2010). This can be effective in treatment of leukemias and autoimmune diseases, such as rheumatoid arthritis (Ben-Ari 2004). The IgG1 antibody, has a single glycosylation site (Hossler et al. 2009; Del Val et al. 2016), Asp296 on the heavy chain. In the current study, L. tarentolae was used as a new host for the production of this antibody.
Materials and Methods
Several culture media, including LB, BHI, and RPMI1640, were used to prepare a compound LBR medium (Table 1). RPMI1640 was supplemented with 10% (v/v) FBS. To avoid bacterial contamination, penicillin, 50 IU and treptomycin 50 μg/ml were added.
Strains and Culture Conditions
Leishmania tarentolae laboratory strain p10 (Jena Bioscience, Germany) was cultured in RPMI1640 supplemented with 10% FBS at 26 °C while shaking at 70 rpm. New medium was added every 2 to 3 days. TOP10 Escherichia coli cells (CinnaGene, Iran) were used to propagate recombinant plasmids.
Plasmid Construction
The amino acid sequences of the two chains of humanized anti-IL-2Rα antibody were obtained from NCBI, PDB, and the drug bank (DB00111) databases.
Humanized Anti-CD25 Heavy Chain 1
QVQLVQSGAEVKKPGSSVKVSCKASGYTFTSYRMHWVRQAPGQGLEWIGYINPSTGYTEY
NQKFKDKATITADESTNTAYMELSSLRSEDTAVYYCARGGGVFDYWGQGTTLTVSSGPSV
FPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSV
VTVPSSSLGTQTYICNVNHKPSNTKVDKKAEPKSCDKTHTCPPCPAPELLGGPSVFLFPP
KPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSV
LTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSRDELTKNQVSL
TCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSC
SVMHEALHNHYTQKSLSLSPGK
> Humanized Anti-CD25 Light Chain 1
DIQMTQSPSTLSASVGDRVTITCSASSSISYMHWYQQKPGKAPKLLIYTTSNLASGVPAR
FSGSGSGTEFTLTISSLQPDDFATYYCHQRSTYPLTFGSGTKVEVKRTVAAPSVFIFPPS
DEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTL
SKADYEKHKVYACEVTHQGLSSPVTKSFNR
Their molecular characteristics were adjusted based on Zenapax monograph (CAS Number: 152923-56-3: https://www.clearsynth.com/en/CSO16306.html). Protein sequences were reverse-translated to DNA using an online codon optimization tool (Integrated DNA Technologies. https://www.euidtdnacom/CodonOpt). Restriction enzymes sites for BglII and NheI were inserted at the 5′ and 3′ ends respectively. Vector pLEXSY (Jena Bioscience, Germany) was used as the backbone for designing the expression construct. The expression construct was designed as follows: a signal peptide sequence optimized for high-level protein expression in CHO cell line (SEQ ID No 9: https://patents.google.com/patent/US20100240097A1/en. Sequence: MMRPIVLVLLFATSALAQV (Young and Rance 2012) was placed upstream of the sequences encoding the humanized heavy or light chain of anti-IL-2Rα antibody; ACC sequence (threonine amino acid) was placed between BglII restriction site and the signal peptide, to increase expression levels, according to the pLEXSY manufacturer guidelines.
Using signalp 4.1 server, in silico prediction of the signal peptide cleavage site in the constructs confirmed the expected cleavage site. The expression constructs were commercially synthesized and provided as inserts in recombinant pGH vectors (GenRay, China). These vectors were then digested by BglII and NheI restriction enzymes to either produce a 1407 bp fragment encoding the heavy chain or a 708 bp fragment encoding the light chain. The longer and shorter fragments were cloned between BglII and NheI restriction sites of pLEXSY-hyg2 and pLEXSY-neo2 (Jena Bioscience, Germany) vectors, respectively, generating pLEXSY-hyg2-H chain and pLEXSY- neo2-L chain plasmids. The vectors were then used to transform TOP10 Escherichia coli cells; the recombinant clones were selected and identified by colony PCR using P1442 (CCGACTGCAACAAGGTGTAG) and A264 (CATCTATAGAGAAGTACACGTAAAAG) primers flanking the multiple cloning site of pLEXSY plasmid. The selected clones were confirmed by sequencing of the PCR products (Bioneer, Korea).
Transfection of L. tarentolae with the Expression Constructs
The recombinant vectors were extracted from E. coli using a plasmid extraction kit (Bioneer, Korea) and digested with SwaI restriction enzyme (Fermentas, Lithuania), yielding the following fragments: 2864 bp (the same fragment length for both vectors), and 5679 bp (pLEXSY-neo2-L chain) or 6600 bp (pLEXSY-hyg2-H chain). After electrophoresis, the respective expression cassettes (5679 bp and 6600 bp) were eluted from the gel (AccuPrep® Gel Purification kit- Bioneer, Korea) for subsequent transfections of L. tarentolae. L. tarentolae P10 was cultured in RPMI1640 medium supplemented with 10% (v/v) FBS for 48 h and the cell suspension was adjusted to 1 × 108 parasites/ml in electroporation buffer (Eppendorf, Germany). Electroporation (Eppendorf, Germany) was performed using 5–10 μg of DNA (1300 V/5 ms). About 24 h after culturing in RPMI1640 supplemented with 10% (v/v) FBS, hygromycin B (25 μg/ml) and neomycin G480 (25 μg/ml) were added. Stringent selection of colonies were performed by increasing the concentration of hygromycin up to 200 μg/ml and neomycin up to 100 μg/ml, over 10 days. To evaluate the insertion of the expression cassettes into the ssu locus of L. tarentolae, following DNA extraction, the cassettes were amplified using F3001 and A264 primers (Table 2).
Expression of Recombinant Anti-IL-2Rα Antibody
Ice-cold supernatant of the selected positive transfected cells grown for 48 h in agitated cultures in LBR medium was precipitated with ice-cold trichloroacetic acid (TCA, final concentration 10%, v/v) for 30 min and centrifuged at 10,000×g and 4 °C for 5 min. The pellet was washed with ethanol, dried, and resuspended in SDS-PAGE loading buffer. Reduced and non-reduced samples were prepared and run on a 12% SDS PAGE gel. Western blotting analysis was performed using mouse polyclonal anti-human IgG conjugated with horseradish peroxidase (HRP) (Dako, Denmark). Densitometric measurement of the protein yield was performed using ImageJ.1.50a software, and the yield calculated based on a standard curve.
Purification of the Humanized Anti-IL-2Rα Antibody
The cultured transfected L.taranolae promastigotes were centrifuged at 1000×g for 5 min, and the pellet resuspended in fresh medium. The supernatant was collected and the centrifugation repeated, at 8000×g for 5 min, to obtain a second supernatant. The second supernatant was used for IgG1 mAb purification using Protein G affinity chromatography (Amersham, UK) (Sugino and Niimi 2012). The concentration of the purified antibody was measured using a biophotometer (Eppendorf, Germany).
In-Cell ELISA Assay (ICE Assay)
Human peripheral blood lymphocytes (PBLs) were isolated from blood samples obtained from healthy volunteers who provided informed consent, according to the guidelines of the local ethics committee. PBLs were isolated on Ficoll/Hypaque gradients (Lymphodex, Germany) and were cultured at a concentration of 1 × 106 cells/ml in the presence of T cell mitogen phytohemagglutinin (PHA, 5 μg/ml), in a 96-well microplate. After 72 h, ICE assay (Mitoscience company, https://www.ispybio.com/search/protocols/elisa_protocol_26.pdf) was performed using PHA-activated T cells expressing the IL-2α receptor and non-activated T cells as a control group in duplicate.. The anti-IL-2Rα antibody was used as the primary antibody (5 μg/ml) and HRP-conjugated mouse anti-human antibody (1:1000 dilution) as the secondary antibody. The blank wells (duplicate) did not include the primary antibody. After incubation with the substrate TMB (3,3′,5,5′- tetramethylbenzidine), the colorimetric signal was measured within 20 min using a microplate reader, at 650 nm.
Immunocytochemistry (ICC Assay)
PHA-activated and non-activated T cells were stained using an ICC procedure similar to the ICE method, except that the water-insoluble substrate DAB (3,3′-diaminobenzidine) was used for visualizing antibody binding to the target.
Biological Activity Assay (BrdU Assay)
Inhibition of PHA-activated T cell proliferation by the anti-IL-2Rα antibody was assessed using Roche BrdU assay kit (cat. no. 11647229001). Isolated PBLs were diluted to 1 × 106 cells/ml in RPMI1640 supplemented with 10% (v/v) FBS, and cultured in 96-well microtiter plates (100 μl of the cell suspension/well; the blank wells were empty; all measurements were performed in triplicate). Wells containing untreated cells were designated as control group 1 (cell proliferation control) and those treated with PHA only as control group 2. The experimental group was treated with both PHA(5 μg/ml) and anti-IL2Rα. All groups were incubated at 37 °C and 5% CO2. 500 to 700 μg of the purified anti-IL-2Rα antibody was added to the experimental group after 2 h of incubation. BrdU was added to the cell cultures after 48 h of incubation and the cells were incubated for an additional 24 h. Harvested cells were stained with antibodies included in the kit, followed by absorbance measurements (370 nm/492 nm) according to the manufacturer’s instructions (https://www.sigmaaldrich.com/content/dam/sigma-aldrich/docs/Roche/Bulletin/1/11647229001bul.pdf).
Glycosylation Analysis
Deglycosylation of asparagine-linked glycans was done with N-glycosidase F (PNGase F, Sigma) and the SDS-PAGE electrophoretic mobility of the protein before and after the treatment was compared.
Statistical Analysis
Data were analyzed using SPSS.17 software by two-sided unpaired Student’s t-test. The level of statistical significance was set at p < 0.05.
Results
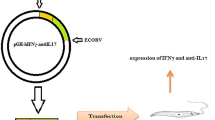
The heavy and light chain genes of anti-IL2Rα antibody were synthesized and cloned into pLEXSY vectors. Figure 1 shows the schematic design of gene construction for expression in L. tarentolae.

Schematic design of gene construction
Recombinant pLEXYS vectors were digested by Saw1 restriction enzyme. The linear fragment transfected into L. tarentolae contained the genes for antibody, antibiotics and the homologous arms. The integration of expression cassettes into the L. tarentolae ssu gene was verified by PCR (Fig. 2).

Confirmation of genomic integration of the anti-IL-2Rα antibody heavy and light chain genes by PCR. Diagnostic PCR was performed using F3001 forward primer hybridizing to the ssu gene of L. tarentolae and A264 reverse primer hybridizing within the construct. Lane 1, PCR products from transfected cells, 1900 bp and 2600 bp fragments, confirming integration of light and heavy chains, respectively; lane 2, 10,000 bp DNA ladder
Confirmation of Protein Expression in Leishmania Using SDS-PAGE and Western Blot
The expression of the recombinant 150 kDa anti-IL-2Rα antibody was confirmed by SDS-PAGE and western blot analysis under non-reducing conditions (Fig. 3).

Analysis of the expressed recombinant 150 kDa anti-IL-2Rα antibody by SDS-PAGE (a) and western blotting (b). a Lane 1, purified recombinant anti-IL-2Rα antibody; lane2, protein size marker (kDa). b Lane 1, human IgG as control; lane 2, purified recombinant anti-IL-2Rα antibody; lane 3, protein size marker (kDa)
Fourty eight hours after culture of cells in a medium without FBS the cultures were centrifuged and the proteins in the supernatant were precipitated with TCA and run on 12% SDS-PAGE gel and stained with Coomassie Brilliant Blue G250. The total amount of protein produced was estimated using the analysis of bands by ImageJ.1.50a software and compared with the standard sample.
The Specificity of Anti-IL-2Rα Antibody
The specificity of anti-IL-2Rα antibody was examined by ICE. Student’s t-test analysis of the mean absorbance values indicated significant differences between the experimental (activated T cells) and control group (untreated T cells) (p = 0.005). The results revealed that the recombinant anti-IL-2Rα antibody specifically binds to the IL-2α receptor expressed on the activated T cells but not to untreated T cells (Fig. 4).

ICE assay with the expressed recombinant anti-IL-2Rα antibody. The experiment was performed using T cells activated by PHA, with T cells not activated by PHA serving as a negative control. Mean absorbance values: negative control, 0.11 ± 0.008; experimental group: 0.497 ± 0.013 (mean ± SD, n = 2). Student’s t-test analysis of the mean absorbance values indicated significant differences between the experimental and control groups (p = 0.005)
ICC Evaluation and Activity of Anti-IL-2Rα Antibody
ICC analysis verified binding of the recombinant anti-IL-2Rα antibody to the surface of proliferating T cells expressing IL-2α receptor but not to non-expressing T cells (Fig. 5).

ICC assay with PHA-activated lymphocytes (a) and non-activated lymphocytes as a control (b), using the expressed recombinant anti-IL-2Rα antibody. The recombinant anti-IL-2Rα antibody was bound to the IL-2Rα expressed on T lymphocytes activated by PHA, and identified by a secondary antibody in a colorimetric reaction. The control group, which did not express IL- 2Rα, was not recognized by the anti-IL-2Rα antibody and no colorimetric reaction was observed
To assess the overall function of the antibody we used inhibition of PHA-activated T cell proliferation by the anti-IL-2Rα antibody test using Roche BrdU assay kit (Fig. 6).

The results of the BrdU assay. The mean reading for non-activated lymphocytes (control 1) was 0.505 ± 0.1. The mean readings for PHA-activated lymphocytes that were either treated with the expressed recombinant anti-IL-2Rα antibody or untreated with the antibody (control 2) were 0.594 ± 0.07 and 2.153 ± 0.19, respectively. The difference in the rates of cellular proliferation between the experimental group and control group 1 was not significant (p = 0.29; two-sided unpaired Student’s t-test). The proliferation rate of control group 2 was significantly greater than that of the experimental group (p = 0.0002). The data are presented as the mean ± SD (n = 3)
The proliferation of activated T cells was clearly inhibited by the anti-IL-2Rα antibody. Cell proliferation of the antibody-treated group (experimental; + PHA, + antibody) was almost the same as in control group 1 (no PHA or antibody activation; p = 0.29). The proliferation of PHA- activated control group (control group 2; +PHA, no antibody) was significantly higher than those of the experimental and control groups (p = 0.0002). As anticipated, N-glycan removal by PNGase F resulted in an SDS-PAGE mobility shift of the heavy chain of the antibody (Fig. 7).

Enzymatic deglycosylation of the heavy chain of the recombinant anti-IL-2Rα antibody under reducing conditions. N-glycosylation of the expressed recombinant antibody was evaluated by PNGase F treatment. Electrophoretic mobility of the purified heavy chain of the recombinant antibody treated with the enzyme (lane 1) and without enzymatic treatment (lane 2) is shown. Lane 3, protein size marker (kDa)
Discussion
MAbs and related products comprise a major proportion of the biopharmaceutical market (Chon and Zarbis-Papastoitsis 2011) but their high therapeutic doses necessitate a large-scale and cost- effective manufacturing process (Jones et al. 2007). Although recombinant protein production in prokaryotic expression systems is cost-effective, the production of full-length monoclonal antibodies in these systems presents a number of challenges due to the size and complexity of the expressed molecules. Consequently, the mammalian cells are the primary choice for the production of mAbs, with the CHO cell lines the most commonly used expression system. Proper folding and human-like post-translational modifications of the recombinant proteins comprise the main advantages of these systems (Khan 2013). However, recombinant protein production in the mammalian cell lines requires more expensive equipment and materials than other expression systems, e.g., E. coli. Moreover, these expression systems are at a risk of contamination with animal viruses (Berting et al. 2010). Considering the above, a novel protein expression system based on L. tarentolae, was developed (Fritsche et al. 2007; Niimi 2012).
Here, we produced a recombinant humanized anti-IL-2Rα antibody in L. tarentolae cultured in a low-cost medium (LBR). Identification of a full-length 150 kDa antibody in the culture medium by SDS-PAGE and western blotting under non-reducing conditions indicated correct assembly of the four chains, suggesting proper formation of disulfide bridges. This was in accordance with the findings of Sugino and Niimi (Sugino and Niimi 2012) who produced functional human laminin (LM)-332, a large heterotrimeric glycoprotein, in L. tarentolae. It was also consistent with the observations of Jørgensen et al. (2014) who expressed scFv-Fc in this system. No accumulation of antibody fragments was detected in cell lysates (data not shown), which implies that the CHO-specific signal peptide incorporated in the expression constructs was processed by L. tarentolae. A similar successful secretion of truncated human TPA protein with its native signal sequence in this expression system was reported by Nazari and Davoudi (2011). ICE and ICC assays revealed that the produced recombinant antibody specifically binds to its endogenous target (i.e., the alpha chain of IL-2 receptor). Glycosylation of the heavy chain, an important eukaryotic post- translational modification, was also verified. The average dose of the expressed anti-IL-2Rα antibody required for the inhibition of IL-2-dependent lymphocyte proliferation was 5–7 mg/ml, which is comparable with the required amount of commercial daclizumab (5–10 mg/ml). The recombinant protein yield in the L. tarentolae (Lai et al. 2019) expression system was 2 mg/l after 48 h culture in LBR medium, which was comparable with similar studies that reported 0.1 to 6 mg of secreted protein per ml (Basile and Peticca 2009). The expression levels of mAbs in mammalian cell systems were initially on the order of 100–500 mg/l (Jones et al. 2007). However, recent advances in mammalian cell line development, and media and bioreactor optimizations reportedly resulted in up to 10 g protein/l yields and cell densities of over 20 million cells/ml in fed-batch processes (Hunter et al 2018). To date, no genetic manipulations of L. tarentolae LEXSY host P10 to adapt this expression system to large-scale production of recombinant proteins have been reported. In summary, we expressed, purified, and evaluated human anti-IL-2Rα antibody in L. tarentolae. L. This protozoan can thus be used as a host for the production of therapeutic monoclonal antibodies. The low-cost growth medium and equipment, easy handling, low risk of contamination by viruses or prions, and mammalian-like posttranslational modifications constitute main advantages of this expression system.
References
Basile G, Peticca M (2009) Recombinant protein expression in Leishmania tarentolae. Mol Biotechnol 43:273–278. https://doi.org/10.1007/s12033-009-9213-5
Ben-Ari ET (2004) Dual purpose: some cancer therapies used to treat autoimmune diseases. J Natl Cancer Inst 96:577–579. https://doi.org/10.1093/jnci/96.8.577
Berting A, Farcet MR, Kreil TR (2010) Virus susceptibility of Chinese hamster ovary (CHO) cells and detection of viral contaminations by adventitious agent testing. Biotechnol Bioeng 106:598–607. https://doi.org/10.1002/bit.22723
Breitling R et al (2002) Non-pathogenic trypanosomatid protozoa as a platform for protein research and production. Protein Expr Purif 25:209–218. https://doi.org/10.1016/S1046-5928(02)00001-3
Church A (2003) Clinical advances in therapies targeting the interleukin-2 receptor. QJM 96:91–102. https://doi.org/10.1093/qjmed/hcg014
Chon JH, Zarbis-Papastoitsis G (2011) Advances in the production and downstream processing of antibodies. New Biotechnol 28:458–463. https://doi.org/10.1016/j.nbt.2011.03.015
Del Val IJ, Polizzi KM, Kontoravdi C (2016) A theoretical estimate for nucleotide sugar demand towards Chinese Hamster Ovary cellular glycosylation. Sci Rep 6:28547. https://doi.org/10.1038/srep28547
De Marco A (2009) Strategies for successful recombinant expression of disulfide bond-dependent proteins in Escherichia coli. Microb Cell Fact 8:26. https://doi.org/10.1186/1475-2859-8-26
Dhara VG, Naik HM, Majewska NI, Betenbaugh MJ (2018) Recombinant antibody production in CHO and NS0 cells: differences and similarities. Biodrugs 32:571–584. https://doi.org/10.1007/s40259-018-0319-9
Fritsche C, Sitz M, Weiland N, Breitling R, Pohl HD (2007) Characterization of the growth behavior of Leishmania tarentolae–a new expression system for recombinant proteins. J Basic Microbiol 47:384–393. https://doi.org/10.1002/jobm.200710111
Hossler P, Khattak SF, Li ZJ (2009) Optimal and consistent protein glycosylation in mammalian cell culture. Glycobiology 19:936–949. https://doi.org/10.1093/glycob/cwp079
Hunter M, Yuan P, Vavilala D, Fox M (2018) Optimization of protein expression in mammalian cells. Curr Protoc Protein Sci 95(1):e77. https://doi.org/10.1002/cpps.77
Iberg CA, Hawiger D (2019) Advancing immunomodulation by in vivo antigen delivery to DEC-205 and other cell surface molecules using recombinant chimeric antibodies. Int Immunopharmacol 73:575–580. https://doi.org/10.1016/j.intimp.2019.05.037
Jefferis R (2005) Glycosylation of recombinant antibody therapeutics. Biotechnol Prog 21:11–16. https://doi.org/10.1021/bp040016jCCC
Jefferis R, Lund J, Pound JD (1998) IgG-Fc-mediated effector functions: molecular definition of interaction sites for effector ligands and the role of glycosylation. Immunol Rev 163:59–76. https://doi.org/10.1111/j.1600-065X.1998.tb01188
Jones SD, Castillo FJ, Levine HL (2007) Advances in the development of therapeutic monoclonal antibodies. BioPharm Int 20:96–114
Jørgensen ML, Friis NA, Just J, Madsen P, Petersen SV, Kristensen P (2014) Expression of single-chain variable fragments fused with the Fc-region of rabbit IgG in Leishmania tarentolae. Microb Cell Fact 13:9. https://doi.org/10.1186/1475-2859-13-9
Kandus A, Arnol M, Omahen K, Oblak M, Vidan-Jeras B, Kmetec A, Bren AF (2010) Basiliximab versus daclizumab combined with triple immunosuppression in deceased donor renal transplantation: a prospective, randomized study. Transplantation 89:1022–1027. https://doi.org/10.1097/TP.0b013e3181d02496
Ke N, Berkmen M (2014) Production of disulfide-bonded proteins in Escherichia coli. Curr Protoc Mol Biol. https://doi.org/10.1002/0471142727.mb1601bs108
Khan KH (2013) Gene expression in mammalian cells and its applications. Adv Pharm Bull 3:257–263. https://doi.org/10.5681/apb.2013.042
Lai JY, Klatt S, Lim TS (2019) Potential application of Leishmania tarentolae as an alternative platform for antibody expression. Crit Rev Biotechnol 39:380–394. https://doi.org/10.1080/07388551.2019.1566206
Majidi J, Barar J, Baradaran B, Abdolalizadeh J, Omidi Y (2009) Target therapy of cancer: implementation of monoclonal antibodies and nanobodies. Hum Antib 18:81–100. https://doi.org/10.3233/HAB-2009-0204
Nazari R, Davoudi N (2011) Cloning and expression of truncated form of tissue plasminogen activator in Leishmania tarentolae. Biotechnol Lett 33:503–508. https://doi.org/10.1007/s10529-010-0470-y
Niimi T (2012) Recombinant protein production in the eukaryotic protozoan parasite Leishmania tarentolae: a review. Methods Mol Biol 824:307–315. https://doi.org/10.1007/978-1-61779-433-9_15
Stockwin L, Holmes S (2003) The role of therapeutic antibodies in drug discovery. Biochem Soc Trans 31:433–436. https://doi.org/10.1042/bst0310433
Sugino M, Niimi T (2012) Expression of multisubunit proteins in Leishmania tarentolae. Methods Mol Biol (Clifton, NJ) 824:317–325. https://doi.org/10.1007/978-1-61779-433-9_16
Young R, Rance J (2012) Mammalian expression vector with a highly efficient secretory signal sequence. Google Patents, https://patents.google.com/patent/US20100240097A1/en
Acknowledgements
This article was extracted from Akram Jalali`s PhD thesis and financially supported by the ‘School of Advanced Technologies in Medicine, Shahid Beheshti University of Medical Sciences (though Grant No. 5086), and was carried out at the Cellular and Molecular Biology Research Center, the directors of which we gratefully.
Funding
The study was funded by ‘School of Advanced Technologies in Medicine, Shahid Beheshti University of Medical Sciences, Tehran, Iran (though Grant No. 5086).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
There are no conflict of interests for authors.
Ethics Approval
The ethics process was approved by Shahid Beheshti University of Medical Sciences Ethical Committee with IR.SBMU.REC.1394.99 number.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Jalali, A., Bandehpour, M., Chegeni, R. et al. Expression, Purification, and Evaluation of Anti-IL-2Rα Antibody Secreted by Leishmania tarentolae. Int J Pept Res Ther 27, 301–307 (2021). https://doi.org/10.1007/s10989-020-10088-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10989-020-10088-6