
Abstract
The present study evaluates the effect of the naturally occurring dipeptide carnosine on primary cell cultures established from patients with glioblastoma multiforme. Surgically removed tumors were used to establish primary cell cultures that were incubated for 96 h with medium supplemented with carnosine at concentrations of 20, 40 and 50 mM. Following incubation, dehydrogenase activity, cellular adenosine triphosphate concentration (ATP), caspase activity, lactate dehydrogenase (LDH) release and the rate of DNA synthesis were determined. After 96 h of carnosine treatment a significant reduction in cellular ATP and dehydrogenase activity was detected already at a concentration of 20 mM carnosine. Carnosine (50 mM) reduced ATP concentration to 42.7 ± 13.5% (n = 6) and dehydrogenase activity to 41.0 ± 19.3% (n = 6) compared to untreated cells. Additional experiments revealed no sign of enhanced apoptosis or necrosis in the presence of carnosine. However, a quantitative bromo-desoxy-uridine-based proliferation assay demonstrated a clear effect of carnosine on DNA synthesis reducing its rate down to 50% (2 cultures) and 10% (4 cultures). Therefore, it can be concluded that carnosine is obviously able to inhibit proliferation of cells derived from glioblastoma. Since it is a naturally occurring substance that appears to be non-toxic to normal tissue and is able to penetrate the blood–brain barrier it may be a candidate for a therapeutic agent that may reduce proliferation of neoplastic cells even in vivo and especially in cases of glioblastoma multiforme.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Carnosine (β-alanyl-l-histidine) was the first peptide ever discovered (Gulewitsch and Amiradzibi 1900) and its structure was determined in the very beginning of the last century. Its name was derived from meat, the material it was isolated from. The dipeptide is usually present in different organs with the highest concentrations (up to 20 mM) in skeletal muscles of many vertebrates including humans (Boldyrev and Severin 1990; Scriver et al. 1983). Carnosine and the related dipeptide homocarnosine (γ-amino-butyryl-l-histidine) are in fact the most common dipeptides in humans (Bauer 2005). Early experiments indicated that carnosine is also found in the brain (Pisano et al. 1961). Later it was shown to be present in concentrations between 0.7 and 2.0 mM, depending on the brain region (Margolis 1980). In fact, it is likely that carnosine can directly be synthesized by glial cells (Bauer et al. 1982). The enzyme responsible for its synthesis is carnosine synthetase (Kalyankar and Meister 1959). Despite its natural occurrence and the long period of time carnosine has been studied, its physiological role is still a subject of controversial discussion (for detailed reviews see (Boldyrev 2000; Guiotto et al. 2005). Several studies suggest that carnosine is multifunctional (Hipkiss 1998; Quinn et al. 1992). Briefly, it was considered to have pH-buffering functions (Smith 1938), to be a metal chelator (Baran 2000) or may even function as a neurotransmitter in the olfactory bulb (Nadi et al. 1980). In addition, it was found to protect against excitotoxic cell death (Boldyrev et al. 1999). The most intensively discussed aspect is carnosine’s role as a scavenger of radical oxygen species (Kohen et al. 1988) and its role as a potential anti-senescence drug (Gallant et al. 2000). In addition to carnosine’s protection of proteins against reactive oxygen species, protection against reactive nitrogen species (Fontana et al. 2002) and lipid peroxidation products as well as against hypochlorid anions was demonstrated (Hipkiss et al. 1997, 1998b). Since carnosine also protects proteins against glycating sugars (Hipkiss et al. 1995), methylglyoxal (Hipkiss and Chana 1998), advanced glycation endproducts (Hipkiss et al. 1998a) and amyloid-β peptides (Preston et al. 1998) it was suggested that carnosine might be beneficial with regard to the therapy of Alzheimer (Hipkiss 2007; Preston et al. 1998; Reddy et al. 2005).
A further interesting observation with regard to a possible therapeutic value of carnosine was made by Holliday and McFarland. These authors demonstrated that carnosine has an inhibitory effect on cultured neoplastic cells. (Holliday and McFarland 1996). In their experiments seven human cell lines and two rodent cell lines were used. Two of the human lines were derived from SV40-transformed fibroblasts and the others from cervical carcinoma, lung carcinoma, osteogenic sarcoma, bladder carcinoma and prostate carcinoma. Encouraged by these data, we asked whether carnosine may be a candidate for the treatment of glioblastoma multiforme, the most malignant primary tumor of the central nervous system in adults. In order to study this interesting aspect of carnosine function we investigated the effect of carnosine on cell cultures derived from surgically removed brain tumors.
Methods
Enzymes and Reagents
If not stated otherwise, all chemicals were obtained from Sigma-Aldrich and Merck (Germany) and were of analytical grade.
Patients and Tumors
Tumor samples were obtained from freshly resected tumors. All patients provided written informed consent according to the German laws as confirmed by the local committee. Surgery was performed between 2003 and 2006 at the ‘Clinic for Neurosurgery’ at the Medical Faculty in Leipzig (Germany). All samples were histologically diagnosed as glioblastoma multiforme. In the experiments presented, low passage cultures were used (<passage 10) and since the experiments were performed over a longer period of time not every investigation could have been done with each of the cultures. In total, 13 cultures derived from patients of both genders were used for the experiments (Table 1).
Cell Lines and Primary Cells
The human glioblastoma cell line T98G was obtained from the ATCC and cultivated in DMEM (Gibco, BRL, Eggenstein) supplemented with 10% fetal calf serum (FCS gold, PAA Cölbe, Germany), 2 mM Glutamax (PAA), 50 μg/ml streptomycin and 30 μg/ml penicillin at 37°C, 5% CO2 in humidified air.
Primary cell cultures were established as follows: glioblastoma cells were isolated from tumor tissue of patients with histopathologically confirmed glioblastoma multiforme. Freshly removed tumor tissue was suspended in sterile culture tubes and immediately transferred to the laboratory. The tissue was washed with PBS (phosphate buffered saline) to be cleaned from blood and cauterized tissue. The purified tissue was minced by a scalpel blade. After mincing, small tissue pieces were transferred to a 25 cm2 culture flask (TPP, Trasadingen, Switzerland) sprinkled with AmnioMax complete medium (Gibco). Cells were incubated for 30 min at room temperature and finally, 1 ml AmnioMax complete medium was added. Incubation was now performed at 37°C, 5% CO2 and humidified air in an incubator. Medium was changed after 72 h. As soon as a confluent layer was obtained, cells were removed from confluent culture flasks by use of accutase (PAA) and transferred to 75 cm2 culture flasks (TPP). AmnioMax Medium with AmnioMax Supplement was used for the first 3 weeks of cultivation. Thereafter, and in the experiments described, DMEM Medium supplemented with 10% fetal calf serum, 2 mM Glutamax, 50-μg/ml streptomycin and 30-μg/ml penicillin was used for cultivation.
Cell Based Assays
Generally, 2,500 cells per well of a 96 well plate (black, clear bottom; μClear, Greiner Bio One, Frickenhausen, Germany) were suspended in 200 μl of medium. After 4 h, medium was exchanged and fresh medium containing carnosine at the specified concentrations or normal medium was added and cells were incubated for different times as indicated in the individual experiments. All cell-based assays were performed with five independently treated wells for each condition used. If not stated otherwise, all experiments were performed in DMEM medium containing 1 mM pyruvate.
Intracellular adenosine triphosphate (ATP) levels were measured by the CellTiter-Glo Assay (Promega, Mannheim, Germany) as described (Gaunitz and Heise 2003). Briefly, after incubation in the absence or presence of carnosine medium was removed and exchanged for 50 μl of fresh medium without carnosine. Then, 50 μl of CellTiter-Glo reagent were added. Ten minutes after addition of reagent and incubation at room temperature luminescence was determined using a SpectraMax M5 Multilabel Reader (Molecular Devices, Ismaning, Germany) in luminescence mode with an integration time of 500 ms.
For the determination of dehydrogenase activities the CellTiter-Blue assay (Promega) was employed. Briefly, before the start of the assay cells received 100 μl of fresh medium without carnosine and 20 μl of CellTiter-Blue reagent. Then, cells were incubated for 90 min at 37°C, 5% CO2 and in humidified air in an incubator. Finally, fluorescence was determined using an M5 Multilabel Reader with an excitation wave length of 560 nm and an emission wave length of 590 nm.
Lactate dehydrogenase (LDH) release into the medium was measured in order to assess loss of membrane integrity which is in turn an indicator of necrosis. The measurements were done using the CytoTox-ONE assay (Promega). Briefly, 50 μl of cell culture medium from the cells was transferred to new 96 well plates and after 30 min at room temperature 50 μl of CytoTox-ONE assay reagent were added. After 10 min incubation at room temperature 25 μl of stop solution (supplied with the CytoTox-ONE reagent) were added and fluorescence was determined using an M5 Multilabel Reader with an excitation wave length of 560 nm and an emission wave length of 590 nm.
Induction of apoptosis was assessed by measuring the activity of caspase 3/7 using the Apo-ONE Homogeneous Caspase-3/7 Assay (Promega). Briefly, caspase substrate was diluted 100× in the supplied assay buffer and 100 μl of this reagent was directly added to cells that had freshly received 100 μl of fresh medium without carnosine. Then, cells were incubated for 90 min at 37°C, 5% CO2 and in humidified air in an incubator. Finally, fluorescence was determined using an M5 Multilabel Reader with an excitation wave length of 485 nm and an emission wave length of 530 nm.
Proliferation Assay
Proliferation was assessed by the determination of DNA synthesis using an enzyme-linked immunosorbent assay (ELISA) measuring 5-bromo-2′-desoxyuridine (BrdU) incorporation into newly synthesized DNA (Cell Proliferation ELISA, BrdU (chemiluminescent); Roche, Mannheim, Germany). The assay was performed according to manufacturers recommendations. Briefly, cells were cultivated for 96 h in cell culture medium containing different concentrations of carnosine. Then, BrdU was added and cultivation was continued for 2 h. Afterwards, culture medium was removed and cells were fixed for 30 min at room temperature by the supplied ‘FixDenat’ solution that partially denatures DNA. Finally, Anti-BrdU antibody peroxidase conjugate was added and incubated at room temperature for 90 min. After three washes with the supplied washing buffer (each 5 min at room temperature) the substrate was added and incubation proceeded for 3 min at room temperature. The resulting chemiluminescence was measured using a Spectra Max M5 Multilabel Reader in luminescence mode.
Statistics
All data are expressed as Mean ± standard deviation. Analysis of significance was performed using the two sample t-test implemented in the Origin 7.5 Software (OriginLab Corporation, Northampton, USA).
Results
Effect of Carnosine on ATP Concentration in Cells from the Line T98G
In an initial experiment it was asked whether carnosine is able to influence cell metabolism in a cell line derived from a human glioblastoma. Therefore, cells from the line T98G were incubated at a density of 2,500 cells per well in 96 well plates in the presence of 50 mM carnosine. This concentration was chosen since Holliday and McFarland demonstrated that HFF-1 cells derived from human foreskin were well growing in medium with this concentration of carnosine (Holliday and McFarland 1996, 2000; McFarland and Holliday 1994). In contrast to the experiments by Holliday and McFarland our experiments were performed in medium with pyruvate in order to duplicate an environment more consistent with an in vivo situation. Cells were harvested at different times after the start of the culture. A bioluminescence assay (CellTiter-Glo) determining the concentration of ATP was used for the assessment of viability. In fact, this bioluminescence approach is highly sensitive with regard to the assessment of cytotoxic effects (Crouch et al. 1993; Kangas et al. 1984). In detail, the ATP concentration of cultures treated with carnosin (50 mM) was compared to the concentration measured in cultures harvested at the same time but cultured in medium without carnosine. The result of the experiment is shown in Fig. 1. Already 24 h after incubation the ATP concentration in the treated cultures was reduced to 85.3 ± 1.4% compared to control. Ninety-six hours later the concentration was reduced to 25.6 ± 1.5%. Thus, exposure to carnosine (50 mM) significantly decreased intracellular ATP concentrations in a time-dependent manner.

ATP concentration in cells from the line T98G treated with carnosine. Cells from the human glioblastoma line T98G were exposed to 50 mM carnosine for different periods of time (4, 24, 48, 72 and 96 h). ATP concentration was determined using the CellTiter-Glo assay. Cells not treated with carnosine but incubated for the same time were used as control and set to 100%. Numbers indicate the level of significance (P-values)
Carnosine Reduces ATP Concentration and Dehydrogenase Activity in Cells Isolated from Human Glioblastoma Multiforme
Encouraged by the inhibitory effect of carnosine on cells from the line T98G even in the presence of pyruvate similar experiments were carried out using a primary culture of cells from a human glioblastoma. In a first experiment, cells isolated from a human tumor were cultivated in the presence or absence of pyruvate (1 mM) and at different concentrations of carnosine. Twenty millimole was chosen as the lowest concentration since this is the concentration of carnosine in skeletal muscle (Boldyrev and Severin 1990) and should therefore be a concentration not disturbing physiological functions. Cells were exposed to carnosine for 96 h and then ATP concentration and dehydrogenase activity in the cultures were determined. The result is presented in Fig. 2. As can be seen, increasing concentrations of carnosine lead to a continuous decline in ATP concentration and dehydrogenase activity. In the absence of pyruvate these effects were more pronounced, although at a concentration of 50 mM carnosine, dehydrogenase activity was only slightly higher in the presence of pyruvate.

Dehydrogenase activity and ATP concentration in a primary culture of glioblastoma cells under the influence of carnosine and pyruvate. Cells from a human glioblastoma (#2) were exposed to carnosine (20, 40, 50 mM) for 96 h in the presence or absence of pyruvate (1 mM). Dehydrogenase activity (A) and ATP concentration (B) were determined and compared to cells not treated with carnosine. Numbers indicate the level of significance (P-values) determined by Student’s t-test
Since both experiments indicated that carnosine might be a functional inhibitor of glioblastoma growth it was asked whether the effects seen can also be detected in primary cultures of glioblastoma cells from other patients. Therefore, in a first series of experiments cells from different patients were exposed to 50 mM carnosine for 18, 42, 68 and 90 h. ATP concentrations (left panels in Fig. 3) and dehydrogenase activities (right panels in Fig. 3) were compared to untreated cells set as 100%. As can be seen in Fig. 3, the cells from different patients responded differently, although ATP concentration was in most cases already reduced after an 18-h incubation period. Incubation of cells for 42, 68 and 90 h with carnosine (50 mM) caused a time-dependent reduction of ATP concentration with the exception of cells from patient “20/05” (#7 in the left upper panel of Fig. 3). Surprisingly, these cells seem to recover from the treatment as also demonstrated by an increase of dehydrogenase activity (right upper panel in Fig. 3). However, ATP concentration in cells from patient #7 were still significantly below control at 42 and 90 h under the influence of carnosine and dehydrogenase activity was significantly below control at all harvest times. Another deviation was seen in cells from patient “36/05” (#8 in the lower right panel of Fig. 3), since 18 h after addition of carnosine dehydrogenase activity of these cells was determined to be higher than control. However, this deviation was statistically not significant. More importantly, ATP concentration and dehydrogenase activity in cells of patient #8 were significantly affected after 68 h of exposure to carnosine.

ATP concentration and dehydrogenase activities in primary cultures of glioblastoma cells in the presence of carnosine at different incubation times. Cells from six patients were incubated in the presence of carnosine (50 mM). The left panels indicate the ATP concentration determined and the right panels the corresponding dehydrogenase activities with respect to control. The numbers in the graph indicate the level of significance (P-values) determined by Student’s t-test. If no numbers are indicated the level of significance was not below 0.05 (P < 0.05). The dashed lines indicate the 100% control level
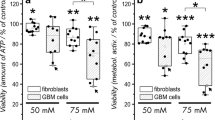
In order to explore whether lower concentrations of carnosine may also be effective, a second set of experiments was performed. In Fig. 4 the effect of carnosine on cells isolated from glioblastomas of six different patients is presented. As can be seen, all cells responded to increasing concentrations of carnosine with a decline in ATP concentration (left panels in Fig. 4) and a decline of dehydrogenase activity (right panels in Fig. 4). ATP concentration in all glioblastoma derived cells was reduced to 78.0 ± 10.7% (20 mM), 63.7 ± 9.3% (40 mM) and 42.7 ± 13.5% (50 mM). Correspondingly, dehydrogenase activity was reduced to 77.1 ± 16.4% (20 mM), 71.2 ± 15.0% (40 mM) and 41.0 ± 19.3% (50 mM). Therefore, incubation of cells with carnosine (20–50 mM) caused a significant concentration-dependent decrease in ATP concentration and dehydrogenase activity. Combining the data from Figs. 3 and 4 for 4 days incubation at a concentration of 50 mM carnosine (12 separate experiments), the dehydrogenase activity was determined to be reduced to 39.83 ± 14.74% and ATP concentration to 48.28 ± 16.90%. Changes induced by carnosine exposure were usually highly significant as determined by student’s t-tests (see information given in Figs. 3 and 4).

ATP concentration and dehydrogenase activities in primary cultures of glioblastoma cells in the presence of different concentrations of carnosine. Cells from six patients were exposed to 20, 40 and 50 mM carnosine for 96 h. ATP concentration (left panels) and dehydrogenase activity (right panels) were determined and compared to cells not treated with carnosine. The numbers in the graph indicate the level of significance (P-values) determined by Student’s t-test. If no numbers are indicated the level of significance was not below 0.05 (P < 0.05)
Influence of Carnosine on Necrosis, Apoptosis and Cell Proliferation of Cells Derived from Human Glioblastoma
The experiments presented above suggest an influence of carnosine on the vitality of cells derived from human glioblastoma as indicated by reduced ATP production and dehydrogenase activity. In fact, the experiments were accompanied by the determination of LDH release to the medium. LDH release indicates loss of membrane integrity and is therefore commonly used as an indicator of necrosis. However, the experiments did not indicate any necrosis. In addition, there was no indication of enhanced caspase activity ruling out apoptosis (data not shown). Therefore, it was asked whether the different activity of dehydrogenases and concentrations of ATP in the presence of carnosine may be caused by a reduced proliferation rate. In order to answer this question a quantitative BrdU assay was employed to determine DNA-synthesis as an indicator of cell proliferation. Figure 5 shows the result of experiments with cells from different glioblastoma multiforme patients. Again, cells were cultivated for 96 h in the presence of different concentrations of carnosine and the level of proliferation was compared to untreated control. All cells responded to the presence of carnosine with reduced proliferation. However, at the highest concentration of carnosine (50 mM) two cultures exhibited a 50% reduction of proliferation whereas all other cultures had a level of proliferation of 10% of control. Although we do not have an answer to this difference of response one has to be aware that 50% is already a strong effect that needs to be investigated in future experiments using animal models. In addition, at 40 and 50 mM carnosine reduction of proliferation was highly significant in all cultures (see data given in Fig. 5).

BrdU incorporation in primary cultures of glioblastomas cells in the presence of different concentrations of carnosine. Cells from six patients were exposed to 20, 40 and 50 mM carnosine and cell proliferation was compared to untreated cells. The numbers in the graph indicate the level of significance (P-values) determined by Student’s t-test. If no numbers are indicated the level of significance was not below 0.05 (P < 0.05)
Discussion
Glioblastoma multiforme (GBM) is the most malignant primary tumor of the central nervous system in adults. Until few years ago the standard therapy regimen consisted in surgical resection followed by radiotherapy and an optional chemotherapy with nitrosourea-based agents. Recently, the alkylating agent temozolomide was shown to improve the overall survival rate of GBM patients by about 30% (Norden and Wen 2006). In fact, the phase 3 trial of the European Organisation for the Research and Treatment of Cancer (EORTC) and the National Cancer Institute of Canada (NCIC) demonstrated, that radiotherapy combined with concomitant temozolomide administration prolonged the median survival rate as well as the 2 year survival rate of patients with newly diagnosed glioblastoma (Stupp et al. 2005). However, although the prolonged survival of 2.5 months may appear to be promising, the currently observed overall survival is still very poor and even in the most favourable situations most patients die within 2 years after first diagnosis. Active research on GBM is performed since several decades as indicated by roughly 10,000 citations, indexed by the National Library of Medicine. A number of new therapeutic concepts is currently studied, such as the aforementioned alkylator temozolomide (Brandes 2003). In addition, targeted molecular therapies against key elements of signalling pathways appear to be promising (Jagannathan et al. 2006). It is beyond the scope of this manuscript to discuss the different approaches currently performed in order to treat GBM. However, it has to be realized that there is currently only slow progress in the treatment of GBM. Therefore, there is a huge demand to investigate substances which might be useful for therapy, at least in combination with other therapies.
The work presented demonstrates that the growth of cells isolated from human glioblastoma multiforme is affected by carnosine also at physiological concentrations. We could show that carnosine acts on proliferation as deduced from reduced incorporation of BrdU and results in a reduced production of ATP and diminished dehydrogenase activities. Although carnosine does not completely abolish the growth of cells it has to be considered that with regard to GBM overall survival of patients diagnosed for the disease is currently very poor as already outlined above. As Holliday and McFarland have shown in there experiments with transformed neoplastic cell lines, carnosine was only active in the absence of pyruvate (Holliday and McFarland 1996; McFarland and Holliday 1994). In contrast to these experiments, we observed that primary cells from human GBM are inhibited in growth also in the presence of pyruvate. Therefore, the dipeptide may be useful for reducing tumor growth and prolonging survival rates even under physiological conditions. With regard to a possible clinical application for the treatment of brain tumors the question remains which concentrations of carnosine in the central nervous system can be achieved. Unfortunately, it is difficult to say whether transporters known to transport carnosine like PEPT1 or PEPT2 may transport carnosine across the blood brain barrier from blood to brain at elevated carnosine blood concentrations (Teuscher et al. 2000). With regard to this question it is interesting, that carnosine can actually influence brain function as already demonstrated by the successful treatment of children with autistic spectrum disorder by oral carnosine supplementation (Chez et al. 2002). Actually, many years ago carnosine was shown to serve as a model peptide for investigating peptide uptake by brain cells (Schulz et al. 1987).
Among the demanding experiments that need to be performed next is to study the effect of carnosine on tumor growth in an animal model. The second question of course is to figure out how carnosine exhibits its function on growth at the molecular level.
Concluding Remarks
Although known since more than a decade the naturally occurring dipeptide carnosine is still an enigmatic substance. Since it is able to inhibit proliferation of cells derived from human glioblastoma multiforme it may be a candidate for adjuvant therapy of this disease. In addition, a detailed analysis on how the substance affects tumor growth at the molecular level may hopefully guide a way to the development of substances with an even higher potential.
References
Baran EJ (2000) Metal complexes of carnosine. Biochemistry (Mosc) 65:789–797
Bauer K (2005) Carnosine and homocarnosine, the forgotten, enigmatic peptides of the brain. Neurochem Res 30:1339–1345
Bauer K, Hallermayer K, Salnikow J, Kleinkauf H, Hamprecht B (1982) Biosynthesis of carnosine and related peptides by glial cells in primary culture. J Biol Chem 257:3593–3597
Boldyrev A, Song R, Lawrence D, Carpenter DO (1999) Carnosine protects against excitotoxic cell death independently of effects on reactive oxygen species. Neuroscience 94:571–577
Boldyrev AA (2000) Problems and perspectives in studying the biological role of carnosine. Biochemistry (Mosc) 65:751–756
Boldyrev AA, Severin SE (1990) The histidine-containing dipeptides, carnosine and anserine: distribution, properties and biological significance. Adv Enzyme Regul 30:175–194
Brandes AA (2003) State-of-the-art treatment of high-grade brain tumors. Semin Oncol 30:4–9
Chez MG, Buchanan CP, Aimonovitch MC, Becker M, Schaefer K, Black C, Komen J (2002) Double-blind, placebo-controlled study of l-carnosine supplementation in children with autistic spectrum disorders. J Child Neurol 17:833–837
Crouch SP, Kozlowski R, Slater KJ, Fletcher J (1993) The use of ATP bioluminescence as a measure of cell proliferation and cytotoxicity. J Immunol Methods 160:81–88
Fontana M, Pinnen F, Lucente G, Pecci L (2002) Prevention of peroxynitrite-dependent damage by carnosine and related sulphonamido pseudodipeptides. Cell Mol Life Sci 59:546–551
Gallant S, Semyonova M, Yuneva M (2000) Carnosine as a potential anti-senescence drug. Biochemistry (Mosc) 65:866–868
Gaunitz F, Heise K (2003) HTS compatible assay for antioxidative agents using primary cultured hepatocytes. Assay Drug Dev Technol 1:469–477
Guiotto A, Calderan A, Ruzza P, Borin G (2005) Carnosine and carnosine-related antioxidants: a review. Curr Med Chem 12:2293–2315
Gulewitsch W, Amiradzibi S (1900) Ueber das Carnosin, eine neue organische Base des Fleischextraktes. Ber Deut Chem Ges 33:1902–1903
Hipkiss AR (1998) Carnosine, a protective, anti-ageing peptide? Int J Biochem Cell Biol 30:863–868
Hipkiss AR (2007) Could carnosine or related structures suppress Alzheimer’s disease? J Alzheimers Dis 11:229–240
Hipkiss AR, Chana H (1998) Carnosine protects proteins against methylglyoxal-mediated modifications. Biochem Biophys Res Commun 248:28–32
Hipkiss AR, Michaelis J, Syrris P (1995) Non-enzymatic glycosylation of the dipeptide l-carnosine, a potential anti-protein-cross-linking agent. FEBS Lett 371:81–85
Hipkiss AR, Preston JE, Himsworth DT, Worthington VC, Keown M, Michaelis J, Lawrence J, Mateen A, Allende L, Eagles PA, Abbott NJ (1998a) Pluripotent protective effects of carnosine, a naturally occurring dipeptide. Ann N Y Acad Sci 854:37–53
Hipkiss AR, Preston JE, Himswoth DT, Worthington VC, Abbot NJ (1997) Protective effects of carnosine against malondialdehyde-induced toxicity towards cultured rat brain endothelial cells. Neurosci Lett 238:135–138
Hipkiss AR, Worthington VC, Himsworth DT, Herwig W (1998b) Protective effects of carnosine against protein modification mediated by malondialdehyde and hypochlorite. Biochim Biophys Acta 1380:46–54
Holliday R, McFarland GA (1996) Inhibition of the growth of transformed and neoplastic cells by the dipeptide carnosine. Br J Cancer 73:966–971
Holliday R, McFarland GA (2000) A role for carnosine in cellular maintenance. Biochemistry (Mosc) 65:843–848
Jagannathan J, Prevedello DM, Dumont AS, Laws ER (2006) Cellular signaling molecules as therapeutic targets in glioblastoma multiforme. Neurosurg Focus 20:E8
Kalyankar GD, Meister A (1959) Enzymatic synthesis of carnosine and related beta-alanyl and gamma-aminobutyryl peptides. J Biol Chem 234:3210–3218
Kangas L, Gronroos M, Nieminen AL (1984) Bioluminescence of cellular ATP: a new method for evaluating cytotoxic agents in vitro. Med Biol 62:338–343
Kohen R, Yamamoto Y, Cundy KC, Ames BN (1988) Antioxidant activity of carnosine, homocarnosine, and anserine present in muscle and brain. Proc Natl Acad Sci USA 85:3175–3179
Margolis FL (1980) Carnosine: an olfactory neuropeptide. In: Barker JL, Smith TG Jr (eds) The role of peptides in neuronal function. Marcel Decker, New York, pp 545–572
McFarland GA, Holliday R (1994) Retardation of the senescence of cultured human diploid fibroblasts by carnosine. Exp Cell Res 212:167–175
Nadi NS, Hirsch JD, Margolis FL (1980) Laminar distribution of putative neurotransmitter amino acids and ligand binding sites in the dog olfactory bulb. J Neurochem 34:138–146
Norden AD, Wen PY (2006) Glioma therapy in adults. Neurologist 12:279–292
Pisano JJ, Wilson JD, Cohen L, Braham D, Udenfried S (1961) Isolation of gamma-aminobutyrylhistidine (homocarnosine) from brain. J Biol Chem 236:499–502
Preston JE, Hipkiss AR, Himsworth DT, Romero IA, Abbott JN (1998) Toxic effects of beta-amyloid(25–35) on immortalised rat brain endothelial cell: protection by carnosine, homocarnosine and beta-alanine. Neurosci Lett 242:105–108
Quinn PJ, Boldyrev AA, Formazuyk VE (1992) Carnosine: its properties, functions and potential therapeutic applications. Mol Aspects Med 13:379–444
Reddy VP, Garrett MR, Perry G, Smith MA (2005) Carnosine: a versatile antioxidant and antiglycating agent. Sci Aging Knowledge Environ 18:e12
Schulz M, Hamprecht B, Kleinkauf H, Bauer K (1987) Peptide uptake by astroglia-rich brain cultures. J Neurochem 49:748–755
Scriver CR, Perry TL, Nutzenadel W (1983), In: Stanbury JB et al. (eds) The metabolic basis of inherited disease, 5th edn. McGraw-Hill, New York, pp 570–585
Smith EC (1938) The buffering of muscle in rigor; protein, phosphate and carnosine. J Physiol 92:336–343
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352:987–996
Teuscher NS, Novotny A, Keep RF, Smith DE (2000) Functional evidence for presence of PEPT2 in rat choroid plexus: studies with glycylsarcosine. J Pharmacol Exp Ther 294:494–499
Acknowledgement
We would like to thank Mr. Baran-Schmidt for technical assistance in preparing the primary cultures of glioblastomas.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Renner, C., Seyffarth, A., de Arriba, S.G. et al. Carnosine Inhibits Growth of Cells Isolated from Human Glioblastoma Multiforme. Int J Pept Res Ther 14, 127–135 (2008). https://doi.org/10.1007/s10989-007-9121-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10989-007-9121-0