
Abstract
Motivated by a genome-wide association study to discover risk variants for the progression of Age-related Macular Degeneration (AMD), we develop a computationally efficient copula-based score test, in which the dependence between bivariate progression times is taken into account. Specifically, a two-step estimation approach with numerical derivatives to approximate the score function and observed information matrix is proposed. Both parametric and weakly parametric marginal distributions under the proportional hazards assumption are considered. Extensive simulation studies are conducted to evaluate the Type I error control and power performance of the proposed method. Finally, we apply our method to a large randomized trial data, the Age-related Eye Disease Study, to identify susceptible risk variants for AMD progression. The top variants identified on Chromosome 10 show significantly differential progression profiles for different genetic groups, which are critical in characterizing and predicting the risk of progression-to-late-AMD for patients with mild to moderate AMD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Our research is motivated by a genome-wide association study (GWAS) on identifying risk variants for the progression of a bilateral eye disease—Age-related Macular Degeneration (AMD). AMD is a common, polygenic, and progressive neurodegenerative disease, which is a leading cause of blindness in the developed world (Swaroop et al. 2009; The Eye Diseases Prevalence Research Group 2004). The overall prevalence of AMD in the US population of adults who are 40 years and older is estimated to be 1.47%, with more than 1.75 million citizens having AMD. Both common and rare variants associated with AMD risk (i.e., whether or not the disease will develop) have been identified in multiple large-scale case-control association studies (Fritsche et al. 2013, 2016). However, the genetic causes for AMD progression have not been well-studied. Several studies evaluated the effects of a few known AMD risk variants on its disease progression (Seddon et al. 2009, 2014). These studies analyzed only one eye per subject (e.g., the faster progressed eye). Recently, Sardell et al. (2016) and Ding et al. (2017) evaluated a set of known AMD risk variants on progression using both eyes with a robust marginal Cox model, where the between-eye correlation was taken into account. All these aforementioned studies on AMD progression only analyzed a small set of known AMD risk variants. Very recently, Yan et al. (2018) performed a GWAS on AMD progression using the robust Cox regression approach.
The Age-related eye disease study (AREDS) was a multi-center, controlled, randomized clinical trial of AMD and age-related cataract, sponsored by the National Eye Institute (AREDS Group 1999). It was designed to assess the clinical course and risk factors for the development and progression of AMD and cataract. The study collected DNA samples of consenting participants and performed genome-wide genotyping. With progression times available for both eyes and a large collection of SNPs to be tested, our endeavor was to develop a stable, robust and computationally efficient test procedure for bivariate time-to-event data.
For multivariate survival analysis, Hougaard (2000) and Joe (1997) provided thorough reviews and examples. One of the earliest distribution families for correlated bivariate measurements is the Copula family (Clayton 1978), originated from the Sklar’s Theorem (Sklar 1959), of which the joint distribution is modeled as a function of each marginal distribution together with an dependence parameter. Another popular approach for analyzing multivariate survival data is the frailty model, which was originally proposed by Oakes (1982). In this approach, a common latent frailty variable, as a random effect, introduces the correlation between survival times. The frailty model is typically suitable for a situation where the number of clusters is not large and the parameter of interest is at a cluster level. The third approach is a marginal method, which was developed under the Generalized Estimation Equation framework (Wei et al. 1989; Lee et al. 1992). A robust sandwich estimator from the estimating equation is used to obtain the variance-covariance matrix of the regression parameter. Although the within-cluster correlation is taken into account in this approach, the strength of such correlation cannot be estimated and the joint survival probability cannot be obtained. Given that the objective of our study is to discover risk variants for the progression of this bilateral disease, we propose to develop a test procedure based on copula models, so that we can (1) assess the genetic effect on a marginal (population) level, and (2) estimate the joint progression-free profiles for different genetic groups. In the meantime, we can model the strength of the dependence between the two margins by the dependence parameter in the copula.
In the GWAS setting, the score test is usually preferred to other likelihood-based tests, such as the Wald test or the likelihood ratio test (Cantor et al. 2010; Sha et al. 2011). This is because the score test needs to fit the model only once, under the null of no SNP effect, rather than fitting millions of (alternative) models for each SNP. This can save computational time significantly. We develop a computationally efficient copula-based score test procedure for analyzing bivariate time-to-event data, and apply it on AREDS to identify significant variants associated with AMD progression.
The paper is organized as follows. Section 2 introduces the proposed test procedure. Section 3 presents simulation studies for evaluating type-I error control and power performance under various settings. Section 4 presents the real data analysis on AREDS using the proposed method. Finally in Sect. 5, we discuss the practical challenges and possible extensions of the proposed method.
2 Methods
2.1 Copula model for bivariate time-to-event data
First, we introduce the notation for bivariate time-to-event data. Assume that there are n subjects. Let \((T_{1i},T_{2i})\) and \((C_{1i},C_{2i}), i= 1,\ldots ,n\), denote the bivariate failure times and censoring times for the ith subject, respectively. Denote by \(\varDelta _i=(\varDelta _{1i},\varDelta _{2i})\) the censoring indicator and \(X_i=(X_{1i}, X_{2i})\) the risk factors for the ith subject. We consider right censoring and assume that given covariates X, \((T_1,T_2)\) and \((C_1,C_2)\) are independent. Then for each subject, we observe
Let \(S(t_{1},t_{2})=P(T_{1}\ge t_{1}, T_{2} \ge t_{2})\) denote the joint survival function for \((T_1,T_2)\) and let \(f(t_1,t_2)=\partial ^2S(t_1,t_2)/\partial t_1\partial t_2\) denote its corresponding density function. Denote by \(\theta \) all the parameters in \(S(t_{1},t_{2})\), then the joint likelihood for the observed data \(\{D_i\}_{i=1}^n\) can be written as
where \((\delta _{1i},\delta _{2i}) \in \{(0,0),(0,1),(1,0),(1,1)\}\).
Copula functions provide a parametric assumption about the dependence between two correlated margins. A bivariate copula is a function defined as \(\{C_\eta : [0,1]^2 \rightarrow [0,1] : (u,v)\rightarrow C_\eta (u,v), \eta \in R\}\) (Nelsen 2006). Assume that U, V are both uniformly distributed random variables. The parameter \(\eta \) in copula function describes the dependence between U and V. By Sklar’s theorem (Sklar 1959), one can model the joint distribution by modeling the copula function and the marginal distributions separately. The theorem states that, if marginal survival functions \(S_{1}(t_1)=P(T_1 > t_1) \) and \(S_{2}(t_2)=P(T_2 > t_2)\) are continuous, then there exists a unique copula function \(C_\eta \) such that for all \(t_1 \ge 0, t_2 \ge 0\),
Define the density function for \(C_\eta \) to be \(c_\eta = \partial ^2 C_\eta (u,v)/\partial \, u \partial \, v\), then the joint density function of \(T_1\) and \(T_2\) can be expressed as
Copula functions allow robust modeling of dependence structures and have nice properties. For example, the rank-based dependence measurement Kendall’s \(\tau \) can be directly obtained as a function of \(\eta \) in some copula models.
In this work, we focus on Archimedean copula family, which is one of the most popular copula families because of its flexibility and simplicity. Two most frequently used Archimedean copulas in survival analysis are:
Clayton copula (Clayton 1978)
and
Gumbel-Hougaard copula (Gumbel 1960)
The Clayton copula models lower tail dependence in survival functions, while the Gumbel copula models upper tail dependence in survival functions. For the Clayton copula, the dependence parameter \(\eta \) corresponds to the Kendall’s \(\tau \) as \(\tau ={\eta } / (\eta +2)\). Thus, \(T_1\) and \(T_2\) are positively associated when \(\eta >0\) and are independent when \(\eta \rightarrow 0\). While for the Gumbel copula, \(\tau =(\eta -1) / {\eta }\), meaning \(T_1\) and \(T_2\) are positively associated when \(\eta >1\) and are independent when \(\eta =1\).
Under the copula model, the joint likelihood function (2.1) can be rewritten as
2.2 Copula-based generalized score test
We consider testing each single SNP in a GWAS setting. Specifically, we are interested in testing whether a given SNP is associated with disease progression, after adjusting for other risk factors. We consider the marginal distributions under the Cox proportional hazards (PH) assumption. We then further denote by \(S_0=(S_{01}, S_{02})\) the baseline survival functions for \(T_1\) and \(T_2\), and \(\beta = (\beta _{ng}, \beta _g)\) the regression coefficients, where \(\beta _{ng}\) are the coefficients of non-genetic risk factors and \(\beta _g\) is the coefficient of the SNP. We assume that the regression coefficients \(\beta \) are the same for \(T_1\) and \(T_2\), which is scientifically plausible for the bilateral eye disease we consider here. However, the method can be easily generalized to the situation where each \(T_k\) has its own regression coefficients.
Denote by \(\theta =(\beta =(\beta _{ng},\beta _g),\eta ,S_{0}=(S_{01},S_{02}))\) the full parameter set for the copula model. We are interested in testing whether or not \(\beta _g=0\). Thus we further separate \(\theta \) into two parts: \(\theta _1 =\beta _g\), which is the parameter of interest (to be tested), and \(\theta _2 = (\beta _{ng},\eta ,S_{0})\), which is the nuisance parameter. Then the null hypothesis can be expressed as \(H_0: \theta _1=\beta _g=0\) and \(\theta _2\) is arbitrary.
The biggest advantage of score test in a GWAS setting is, one only needs to estimate the unknown parameters once under the null model without any SNP effect (i.e., \(\theta _1 = \beta _g = 0\)), since the non-genetic covariates are the same no matter which SNP is being tested. The score test is much less computationally intensive as compared to the likelihood ratio or the Wald test. In addition, when the testing SNP has a low minor allele frequency (MAF), maximizing the complex log-likelihood under a copula model (to obtain the parameter estimates) may produce an unstable result. Therefore, we propose to use the score test for our problem.
Assume that \({\hat{\theta }}_0=(\theta _1 = 0, \theta _2={\hat{\theta }}_{20})\) is the restricted maximum likelihood estimate (MLE) of \(\theta \) from (2.2) under the restriction \(\theta _1=0\), then the corresponding score function and Fisher’s information can be written as
where \(U_j(\cdot ) ={\partial \log L}/{\partial \theta _j}, \ j=1, 2\), and
where \( I_{11}, I_{12}, I_{21}\) and \(I_{22}\) are partitions of the information matrix \({\mathcal {I}}\) by \(\theta _1\) and \(\theta _2\). By Cox and Hinkley (1979), we can obtain the generalized score test statistics as
where \({\mathcal {I}}^{11}=({\mathcal {I}}^{-1})_{11}=(I_{11}-I_{12}I_{22}^{-1}I_{21})^{-1}\).
In practice, the observed information matrix \({\mathcal {J}}({\hat{\theta }}_0)\) is often used in the score test. With bivariate copula models, the first and second order derivatives of \(\log L(D|\theta )\) usually have very complex forms and the forms depend on the specific copula model as well as the marginal distributions. Thus, we propose to use numerical differentiation through Richardson’s extrapolation (Lindfield and Penny 1989) to approximate the score function and the observed information matrix, denoted by \({\tilde{U}}\) and \(\tilde{{\mathcal {J}}}\). This numerical approximation only requires a close-formed log-likelihood function. Therefore, the generalized score test statistic we propose is
which asymptotically follows a \(\chi ^2\) distribution with degrees of freedom being the dimension of \(\theta _1\) under the null.
2.3 Choice of marginal distributions
We assume that the marginal distributions are from the PH family, which can be written as
where \(\lambda _{0k}(\cdot )\) is the baseline hazard function for the kth margin, \(x_{ki}\) are the covariates for the ith subject with kth margin. In general, the covariates can be either subject-specific or margin-specific. For example, one can consider a fully parametric distribution such as the Weibull distribution,
or the Gompertz distribution,
where \(\lambda _k\) is the scale parameter and \(\gamma _k\) is the shape parameter. In this case, the full parameter set \(\theta \) is \((\beta =(\beta _{ng},\beta _g),\eta , \gamma _k, \lambda _k)\).
In some circumstances, a specific parametric marginal distribution may not fit the data properly. Kim et al. (2007) has shown that the dependence parameter estimation in copula models is not robust to misspecification of the marginal distributions. Thus, a relaxed assumption may be more desired for marginal distributions. For example, the piecewise constant hazards assumption given by
where \(0=a_{0k}<a_{1k}<\cdots <a_{rk}=\max {y_{ik}}\) are pre-specified cutoff points, can be considered. The full parameter set \(\theta \) in this case will be \((\beta =(\beta _{ng},\beta _g),\eta , \rho _{jk})\). More generally, one could also consider nonparametric marginal distributions and may use the Breslow estimator (Breslow 1972) for baseline hazards, which essentially treats \(\lambda _{0k}(\cdot )\) as piecewise constants between all uncensored failure times. We focus on parametric and weakly parametric marginal distributions. We also explore the non-parametric margin case in terms of parameter estimation, without fully establishing its asymptotic properties.
Under the PH family, the marginal survival function for \(T_{ki}\) given covariate \(X_{ki}\) can be expressed as
where \(S_{0k}(t_{ki}) = \exp \left\{ -\int _0^{t_{ki}} \lambda _{0k}(s) \ ds\right\} \).
2.4 Two-step estimation procedure for \({\hat{\theta }}_0\)
In order to derive the above score test statistic in (2.3), we need to estimate \(\theta \) under \(H_0\). Motivated by the two-stage estimation approach from Shih and Louis (1995), we propose a two-step maximum likelihood estimation procedure to obtain the restricted MLE \({\hat{\theta }}_0=(0,{\hat{\theta }}_{20})\). In step 1, we first obtain initial estimates of the parameters in marginal distributions (i.e., \(\beta _{ng}\) and \(S_{0}\)) based on marginal likelihood functions. Then we maximize the pseudo joint likelihood (with the initial estimates of \(\beta _{ng}\) and \(S_{0}\) plugged in) to get an initial estimate of the dependence parameter \(\eta \). Then in step 2, we maximize the joint likelihood with estimates from step 1 being initial values to obtain the final estimate \({\hat{\theta }}_0\). Detailed steps are provided below:
-
(1)
Obtain initial estimates of \(\theta _0\):
-
(a)
\(({\hat{\beta }}_{ng}^{(1)},{\hat{S}}_{0}^{(1)})={\mathop {{{\,\mathrm{\arg \max }\,}}}\limits _{(\beta _{ng},S_{0})}} \log L_0(\beta _{ng},S_{0})\), where \(L_0\) denotes the marginal likelihood function under the null (\(\beta _g=0\));
-
(b)
\({\hat{\eta }}^{(1)}={\mathop {{{\,\mathrm{\arg \max }\,}}}\limits _\eta } \log L({\hat{\beta }}_{ng}^{(1)},\eta , {\hat{S}}_{0}^{(1)})\), where \(L({\hat{\beta }}_{ng}^{(1)},\eta , {\hat{S}}_{0}^{(1)})\) is the pseudo joint likelihood function with \(\beta _{ng}\) and \(S_0\) replaced by their initial estimates from (a).
-
(a)
-
(2)
Maximize the joint likelihood function with initial value \(({\hat{\beta }}_{ng}^{(1)},{\hat{\eta }}^{(1)},{\hat{S}}_{0}^{(1)})\) to get final estimates \({\hat{\theta }}_{20}=({\hat{\beta }}_{ng},{\hat{\eta }},{\hat{S}}_{0})={\mathop {{{\,\mathrm{\arg \max }\,}}}\limits _{(\beta _{ng},\eta , S_{0})}} \log L(\beta _{ng}, \eta , S_{0}).\)
The standard two-step estimation procedure for copula models stops after the step 1(b), since the dependence parameter \(\eta \) is of the primary interest. Note that, the initial estimates from the step 1 \(({\hat{\beta }}_{ng}^{(1)},{\hat{\eta }}^{(1)},{\hat{S}}_{0}^{(1)})\) are already consistent and asymptotically normal (Shih and Louis 1995). However one cannot directly use Hessian matrices from the step 1(a) to obtain variance estimates for \({\hat{\beta }}_{ng}\). The second step produces correct variance covariance estimates for all the parameters by using the joint likelihood. In theory, the model parameters can be estimated by a one-step MLE procedure (i.e., step 2). The purpose of the first stage (step 1a and 1b) is to provide good initial values of all unknown parameters \(({\beta }_{ng},{\eta },{S}_{0})\) for the MLE procedure in the second step, which could save computing time and reduce algorithm failure rate due to suboptimal initial values. We demonstrate this in our simulation studies.
For nonparametric marginal baseline hazard case, such as using the Breslow estimator, a pseudo-maximum likelihood (PML) estimation can be used in the step 2 by fixing the cumulative baseline hazard \(\varLambda _{0k}(t)\) with its estimate from the marginal model in step 1(a) and only updating \((\beta _{ng},\eta )\). In this way, the estimates for the regression coefficients and the dependence parameter are still consistent and asymptotically normal. However, the Hessian matrix from the PML in the step 2 cannot be directly used for estimating the variance of \({\hat{\beta }}_{ng}\) and \({\hat{\eta }}\). One solution is to use bootstrapped variance estimates, for example, see Lawless and Yilmaz (2011).
2.5 Model selection and diagnostics
Several model selection procedures have been proposed for copula-based time-to-event models. The Akaike’s Information Criteria (AIC) (Akaike 1998) and Bayesian Information Criteria (BIC) (Schwarz 1978) have been widely used for model selection purpose in copula models. Wang and Wells (2000) proposed a model selection procedure based on nonparametric estimation of the bivariate joint survival function within the class of Archimedean copulas. For model diagnostics, Chen et al. (2010) proposed a penalized pseudo-likelihood ratio test for copula models in non-censored data. Recently, Zhang et al. (2016) proposed a goodness-of-fit test for copula models using the pseudo in-and-out-of sample (PIOS) method. Then Mei (2016) extended this PIOS method to censored survival data without covariates. For simplicity, we use AIC for selecting a proper model in our real data analysis.
3 Simulation study
In this section, we evaluate the finite sample performance of the proposed test procedure through various simulation studies and compare it to the Wald test under the Cox PH model with robust variance estimate (Lee et al. 1992). The Wald test from the Cox model under independence assumption is also included for type-I error control simulations.
3.1 Data generation
Recall that the bivariate joint survival function under a copula model is \(S(t_1,t_2)=C_\eta (S_1(t_1),S_2(t_2))\), where \(U=S_1(T_1)\), \(V=S_2(T_2)\) each follows a uniform distribution U[0, 1]. Define \(W_v(u) = h(u,v) = P(U \le u |V=v)\), which equals to \(\partial C_{\eta }(u,v)/\partial v\). To generate bivariate survival data \((t_{1i}, t_{2i})\), \(i=1,..,n\), we first generate \(v_i\) and \(w_i\) from two independent U[0, 1] distributions. Then let \(w_i=h(u_i,v_i)(=C_{\eta }(u_i,v_i)/\partial v_i)\) and solve for \(u_i\) from the inverse of h function \(h^{-1}\). Finally, we obtain \(t_{1i}\) and \(t_{2i}\) from \(S^{-1}_1(u_i)\) and \(S^{-1}_2(v_i)\) respectively. We generate censoring times \(c_{1i}\) and \(c_{2i}\) from uniform distribution U(0, C) with C chosen to yield desired censoring rate.
The value for the dependence parameter \(\eta \) is chosen to introduce weak or strong dependence, represented by Kendall’s \(\tau \) = 0.2 and 0.6, respectively. We generate SNP data from a multinomial distribution with values \(\{0, 1, 2\}\) and probabilities \(\{(1-p)^2,2p(1-p),p^2\}\), where p is the MAF, chosen to be \(40\%\) or \(5\%\). We also include a continuous non-genetic risk factor \(X_{ng,k}\) (\(k=1, 2\)), generated from a normal distribution \(N(6,2^2)\), where the mean and standard deviation are decided based upon our AREDS data.
In all simulations, the sample size is \(N=500\) and we choose the same baseline marginal distribution for the two survival times (i.e., \(S_{01}(t)=S_{02}(t)\)). For type-I error control simulations, the SNP effect \(\beta _g\) is set to be 0. We replicate 100,000 runs and evaluate the type-I error at various \(\alpha \) levels: 0.05, \(10^{-2}\), \(10^{-3}\) and \(10^{-4}\), respectively. These small tail levels (such as \(\alpha =10^{-3}\) or \(10^{-4}\)) are evaluated since our application involves a large number of SNPs to be tested, the test performance at these smaller tail levels is more critical. For power evaluation, we replicate 1000 runs under each SNP effect size, where a range of \(\beta _g\) values are picked to represent weak to strong SNP effects.
3.2 Simulation I: parameter estimation
We first examined the parameter estimation and computing performance between our proposed two-step estimation procedure and the one-step MLE procedure (i.e., step 2). Table 1 reports the results from the situation where data were simulated from the Clayton copula with Weibull margins. The baseline hazard parameters were set to be \(\lambda =0.01\) and \(\gamma =2\) and the censoring rate was set to be 50%. Both procedures achieve accurate parameter estimates with appropriate coverage probabilities. However, on average, the two-step procedure saves about 36% computing time compared to the one-step procedure. Moreover, the one-step procedure causes about 23% failures due to non-convergence of the optimization while the two-step procedure only causes 0.4% failures.
Then we examined the parameter estimation from various copula models in Table 2. Data were simulated from the Clayton copula with Weibull margins. We fitted four models: marginal Cox with robust variance estimates (Cox-R), Clayton copula with Weibull (Clayton-WB), piecewise constant with \(r=4\) (Clayton-PW) and Breslow (Clayton-BS) margins, respectively. Note that for the Clayton-BS model, the variance estimates were obtained through bootstraps, since the analytic form of the asymptotic variance (of the parametric parameters) has not been established under this case.
As we can see from the table, all four models produce virtually unbiased parameter estimates with satisfactory coverage probabilities for the regression parameters. The variances of \({\hat{\beta }}_{ng}\) and \({\hat{\beta }}_g\) from Clayton-WB and Clayton-PW are smaller than those from Cox-R and Clayton-BS, since the latter two models use non-parametric baseline cumulative hazards estimates which are more variable than the parametric models. For the dependence parameter \(\eta \), the biases under Clayton-WB and Clayton-BS models are minimal and the coverage probabilities are close to the nominal level. However, Clayton-PW produces non-negligible bias for \(\eta \) under the scenario with \(\tau =0.6\). These simulations provide reassurance that the proposed copula model with the two-step estimation procedure performs well in finite samples. When the purpose is to test the regression parameters, the specification of marginal distributions seem to be less critical.
3.3 Simulation II: score test under correctly specified models
In this section, we evaluated the score test performance under correctly specified models. The true models are from Clayton copula with Weibull or Gompertz marginal distributions. With Weibull margin, we chose \(\lambda =0.01\) and \(\gamma =2\), and with Gompertz margin, we chose \(\lambda =0.2\) and \(\gamma =0.05\). In both scenarios, we also fitted copula models with piecewise constant hazards margins. We evaluated three censoring rates, 25%, 50% and 75% and only present the results from 50% censoring here. The other two censoring rates yield very similar results in terms of type-I error control and power performance, and thus are omitted.
Table 3 presents type-I error rates under different \(\alpha \) levels for four models, namely, (1) the Cox model under independence assumption (Cox-I), (2) Cox-R, (3) the copula model with parametric marginal distributions (Cop-PM, either Weibull or Gompertz), and (4) the copula model with piecewise constant marginal distributions (Cop-PW). The test performance under the copula model with nonparametric margins (i.e., Breslow) was not examined due to its large computational time (for bootstrapped variance estimates), especially under low \(\alpha \)’s.
It is clearly seen that when \(\hbox {MAF}=40\%\), all models, except Cox-I, control the type-I error well. However, when \(\hbox {MAF}=5\%\), Cox-R yields inflated type-I error rates at all \(\alpha \) levels, especially with lower \(\alpha \) levels. For example, with data generated from Clayton–Weibull, the type-I error from Cox-R is 0.003 and 0.0007 for \(\alpha =0.001\) and 0.0001, respectively, which is 3 or 7 times of the expected value. The two copula models control type-I error very well under both common and rare allele frequency scenarios, with Cop-PW showing slightly more conservative type-I error comparing to Cop-PM. The Cox-I model always inflates the type-I error, which is not surprising.

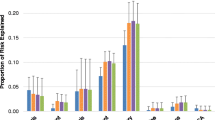
Simulation results for power comparison between Cox-R, Clayton-WB and Clayton-PW models over different genetic effect sizes. Number of replicates = 1000, sample size = 500
Figure 1 presents the power curves over different genetic effect sizes for the three models that can control type-I error: Cox-R, Clayton-WB, and Clayton-PW. When the dependence between margins is strong, both copula models yield better power as compared to Cox-R. The parametric copula method is slightly more powerful than the weakly parametric copula model, which is as expected. When the dependence is weak, all three models produce similar power.
We also fitted the robust Weibull method for the case where the marginal distributions are Weibull. The results (in terms of both type I error control and power) are very close to the results from Cox-R (not shown). Therefore, the inflated type-I error issue when MAF is small exists in the robust parametric marginal model as well.
3.4 Simulation-III, score test under misspecified models
In this section, we evaluated the method performance in situations where either the copula function or the marginal distributions are misspecified. In the case of copula function being misspecified, data were generated from the Gumbel copula with Weibull margins. For misspecification on marginal distributions, data were generated from the Clayton copula with Gompertz margins. In both scenarios, data were fitted by the Clayton copula with Weibull margins or piecewise constant hazards margins.
Table 4 presents type-I errors under different \(\alpha \) levels for the two misspecified scenarios. The same four models as in Sect. 3.3 were compared. Under both scenarios, two Cox model approaches do not depend on copula model specifications (so long as the marginal distributions are still from the PH family), and thus yield similar performance as those in Table 3. When the copula function is misspecified, the parametric copula model (Cop-PM) shows an obvious inflation on type-I errors, especially when the dependence is strong. The copula model with piecewise constant margins (Cop-PW) shows a smaller degree of inflation on type-I error rates. When the marginal distributions were misspecified, Cop-PM shows a conservative type-I error control while Cop-PW produces type-I errors closer to the nominal levels. Overall, Cop-PW is more robust against incorrectly specified models.
4 Real data analysis
We implemented our proposed method on AREDS data to identify genetic variants associated with the progression of late-AMD. All the phenotype and genotype data of AREDS are located from the public available website dbGap (accession: phs000001.v3.p1, and phs001039.v1.p1, respectively) and have been reported by our previous studies (Ding et al. 2017; Yan et al. 2018). In this longitudinal study, each subject was followed every 6 months (in the first 6 years) or 1 year (after year 6) for about 12 years. A severity score, scaled from 1 to 12 (with larger value indicating more severe disease), was recorded for each eye of each participant at every visit. We analyzed 629 Caucasian participants who had at least one eye in moderate AMD stage at baseline, defined by severity scores between 4 to 8. The time-to-late AMD was calculated for each eye of these participants, defined as the time from the baseline visit to the first visit when the severity score reached 9 or above. The overall censoring rate was 54% for our analysis sample. In this work, we specifically tested the common variants (i.e. SNPs with MAF \(\ge 5\%\)) from chromosome 10, since one of the most significant regions associated with AMD risk (i.e., the ARMS2 gene region) is on chromosome 10. In total, we analyzed around 350,000 SNPs. To decide which non-genetic risk factors to include in the model, we considered the same variables as in Ding et al. (2017) and performed univariable analysis using the Clayton copula with Weibull margins (Table 5). Variables with a \(p<0.05\) were included in the final copula model, which are baseline age and baseline severity score.
To decide which copula function and marginal distribution to select for this dataset, we considered two copula functions, Clayton and Gumbel, and three marginal distributions, Weibull, Gompertz and piecewise constant with \(r=4\). Table 6 presents the AIC values for each model under the null hypothesis (\(H_0: \beta _g=0\)). The Weibull margins under both copula models produce similar AIC values, which are smaller than other AICs. We performed analyses using both Gumbel and Clayton copulas with Weibull margins and their results are very similar. We also analyzed the data using Cox-R and copula with piecewise constant margins (Clayton-PW) models, as they are more robust to model misspecification based on our simulations.
Table 7 presents five most significant variants discovered from our analysis. In addition to the p values from the Clayton-WB model, we also report p values from Cox-R and Clayton-PW models. As we can see, the p values from Clayton-WB are all smaller than those from the other two models. One top variant rs2672599 is a known common variant from the ARMS2 gene region with MAF \(=35\%\). The estimated hazard ratio for this SNP is 1.42, with a 95% CI \(=[1.23, 1.65]\) (from Clayton-WB). Figure 2a is the marginal (eye-level) Kaplan-Meier (K-M) plot, which shows this variant can separate AMD progression curves quite well. Two of these five variants (rs2672599 and rs2284665) have also been reported in Yan et al. (2018) to be associated with AMD progression.
In addition to the score test result for each variant, we can obtain both estimated joint and conditional survival functions from copula models, which can be used to establish a predictive model for progression-free probabilities. We demonstrate these using fitted results from Clayton-WB model. For example, Figure 2b plots the joint 5-year progression-free probability contours (i.e., neither eye is progressed by year 5) for subjects having the same baseline severity score (\(=5.8\)) and age (\(=69.6\)) but different genotypes of the variant rs2672599. Figure 2c plots the conditional 5-year progression-free probability of the remaining years for one eye, given that the other eye has progressed at year 5. It is clearly seen that in both plots, the three genotype groups are well separated, with the AA group having the largest progression-free probabilities.
We further picked two variants, rs72798393 from the gene LOC101928913 and rs2672599 from the gene ARMS2, and plotted the predicted 5-year joint progression-free probabilities by genotype, varying the eye-level baseline severity score values (Fig. 3). We can see that carrying more T allele of rs72798393 leads to larger progression-free probabilities, indicated by the overall lighter color of the plot. On the other hand, carrying more C allele of rs2672599 leads to smaller progression-free probabilities, indicated by the overall darker color of the plot. Within each genotype group, having larger value of the baseline severity scores leads to smaller progression-free probabilities.
Moreover, in Fig. 4, we plotted the predicted joint progression-free probability function \(P(t_{1,i-1}<t_1<t_{1,i},t_{2,i-1}<t_2<t_{2,i})\) within a bivariate time interval varying the interval values of \((t_{1,i-1}, t_{1,i}, t_{2,i-1}, t_{2,i})\) for subjects in different genotype groups of rs2672599. It is clearly seen that the joint progression-free probabilities decrease as the years increase, with smaller probabilities in subjects carrying more C alleles. We can also see that the two eyes are more likely to progress within the similarly years, observed by the darker color cloud around the diagonal lines, indicating that the two eyes are correlated in terms of progression. The estimated \({\hat{\eta }}\) from the Clayton-WB model with SNP rs2672599 (and other two non-genetic risk factors) included is 1.12, corresponding to Kendall’s \({\hat{\tau }}=0.36\), which also indicates a moderate dependence between the two eyes.

The estimated AMD progression profiles separated by a top SNP rs2672599 (from ARMS2 gene region): a the eye-level K-M plot, with the total number of eyes and the percentage of progressed eyes in each genetic group given in parenthesis. b The (Clayton-WB) estimated joint progression-free probability contours (the baseline severity score and age are fixed at their mean values: 5.8 and 69.6, respectively). c The (Clayton-WB) estimated conditional progression-free probabilities of remaining years (since year 5) for one eye, given the other eye has been progressed by 5 year (the baseline severity score and age are also fixed at their mean values: 5.8 and 69.6, respectively)

Predicted joint 5-year progression-free probabilities \(P(T_1>5,T_2>5)\) for subjects with mean age 69.6 and various baseline severity scores (between 4 and 8), separated by genetic groups defined by rs72798393 (from gene LOC101928913) (top panel) or rs2672599 (from gene ARMS2) (bottom panel)

Predicted joint progression-free probability \(P(t_{1,i-1}<t_1<t_{1,i},t_{2,i-1}<t_2<t_{2,i})\) for subjects in different genotype groups of rs2672599. The baseline severity score and age are fixed at their mean values: 5.8 and 69.6, respectively
5 Discussion and conclusion
In this work, we developed a computationally efficient copula-based score test procedure for bivariate time-to-event data. The copula model provides flexibility in modeling the dependence and marginal distributions separately. The two-step estimation approach with numerical derivatives to approximate the score function and the observed information matrix works well and is computationally feasible for the GWAS setting that we consider here. The proposed method has been demonstrated to produce correct type-I error control and satisfactory power performance when model assumptions are met. The proposed method has been implemented in R with key functions can be found in GitHub (https://github.com/yingding99/CopulaRC).
Compared to the robust Cox model, which is frequently used in analyzing multivariate survival data, our copula-based method is more powerful when the model is correctly specified. Moreover, our method appears to be more robust against low MAF in controlling type-I errors.
Our approach uses copula to model the dependence of two margins. Certain equivalence between Archimedean copulas and shared frailty models has been claimed in the literature. For example, the joint distribution functions of the Clayton copula model and the Gamma frailty model have the same mathematical expression. However, as shown in Goethals et al. (2008), the two joint distributions are essentially different due to the difference in their corresponding marginal functions. The two joint distribution functions are identical only when the two margins are independent. Therefore, the two types of approaches are fundamentally different.
Several directions may be pursued to extend the current proposed method. First, instead of using one-parameter copula functions as we consider here, one may consider using a two-parameter copula function, which is more flexible to characterize the dependence structure of the bivariate data. For example, Chen (2012) has introduced a framework for estimating two-parameter copula models. In that setting, the dependence is described jointly by two parameters in the copula function. Both Clayton and Gumbel copulas are special or limiting scenarios of the two-parameter copula family.
Secondly, for modeling marginal distributions, in addition to fully parametric or nonparametric approaches, a semiparametric sieve-based smoothing technique may be used to estimate baseline hazards (He and Lawless 2003; Ding and Nan 2011). In that case, the semiparametric M-estimation theory applies and the variance estimates for \({\hat{\beta }}\) and \({\hat{\eta }}\) can be obtained from the joint sieved log-likelihood in step 2.
Lastly, in our AREDS data, the actual time-to-late-AMD are interval censored due to intermittent assessment times. We currently treat them as right censored data given that the interval lengths are fairly small and similar for all subjects. However, it is worthwhile to extend this test procedure to handle bivariate interval-censored data. All these directions are currently under investigation.
Application of the proposed method on AREDS data jointly model the progression profiles in both eyes, which, to the best of our knowledge, has not been done in any previous studies on AMD progression. The findings provide new insights about genetic causes on AMD progression, which is critical to establish novel and reliable predictive models of AMD progression to accurately identify high-risk patients at an early stage. Our proposed methods are applicable to general bilateral diseases and are particularly powerful for performing tests on a large number of markers.
References
Akaike H (1998) Information theory and an extension of the maximum likelihood principle. In: Parzen E, Tanabe K, Kitagawa G (eds) Selected Papers of Hirotugu Akaike. Springer, New York, pp 477–485
AREDS Group (1999) The age-related eye disease study (AREDS): design implications. Control Clin Trials 20(6):573–600
Breslow NE (1972) Discussion of the paper by D. R. Cox. J R Stat Soc Ser B 34:216–217
Cantor RM, Lange K, Sinsheimer JS (2010) Prioritizing GWAS results: a review of statistical methods and recommendations for their application. Am J Hum Genet 86(1):6–22
Chen X, Fan Y, Pouzo D, Ying Z (2010) Estimation and model selection of semiparametric multivariate survival functions under general censorship. J Econom 157(2):129–142
Chen Z (2012) A flexible copula model for bivariate survival data. PhD thesis, University of Rochester
Clayton DG (1978) A model for association in bivariate life tables and its application in epidemiological studies of familial tendency in chronic disease incidence. Biometrika 65(1):141–151
Cox DR, Hinkley DV (1979) Theor Stat. Chapman & Hall/CRC, London
Ding Y, Nan B (2011) A sieve m-theorem for bundled parameters in semiparametric models, with application to the efficient estimation in a linear model for censored data. Ann Stat 39(1):2795–3443
Ding Y, Liu Y, Yan Q, Fritsche LG, Cook RJ, Clemons T, Ratnapriya R, Klein ML, Abecasis GR, Swaroop A, Chew EY, Weeks DE, Chen W, The AREDS2 Research Group (2017) Bivariate analysis of age-related macular degeneration progression using genetic risk scores. Genetics 206(1):119–133
Fritsche LG, Chen W, Schu M, Yaspan BL, Yu Y, Thorleifsson G, Zack DJ, Arakawa S, Cipriani V, Ripke S, Igo RP Jr, Buitendijk GHS, Sim X, Weeks DE, Guymer RH, Merriam JE, Francis PJ, Hannum G, Agarwal A, Armbrecht AM, Audo I, Aung T, Barile GR, Benchaboune M, Bird AC, Bishop PN, Branham KE, Brooks M, Brucker AJ, Cade WH, Cain MS, Campochiaro PA, Chan CC, Cheng CY, Chew EY, Chin KA, Chowers I, Clayton DG, Cojocaru R, Conley YP, Cornes BK, Daly MJ, Dhillon B, Edwards AO, Evangelou E, Fagerness J, Ferreyra HA, Friedman JS, Geirsdottir A, George RJ, Gieger C, Gupta N, Hagstrom SA, Harding SP, Haritoglou C, Heckenlively JR, Holz FG, Hughes G, Ioannidis JPA, Ishibashi T, Joseph P, Jun G, Kamatani Y, Katsanis N, Keilhauer C, Khan JC, Kim IK, Kiyohara Y, Klein BEK, Klein R, Kovach JL, Kozak I, Lee CJ, Lee KE, Lichtner P, Lotery AJ, Meitinger T, Mitchell P, Mohand-Sad S, Moore AT, Morgan DJ, Morrison MA, Myers CE, Naj AC, Nakamura Y, Okada Y, Orlin A, Ortube MC, Othman MI, Pappas C, Park KH, Pauer GJT, Peachey NS, Poch O, Priya RR, Reynolds R, Richardson AJ, Ripp R, Rudolph G, Ryu E, Sahel JA, Schaumberg DA, Scholl HPN, Schwartz SG, Scott WK, Shahid H, Sigurdsson H, Silvestri G, Sivakumaran TA, Smith RT, Sobrin L, Souied EH, Stambolian DE, Stefansson H, Sturgill-Short GM, Takahashi A, Tosakulwong N, Truitt BJ, Tsironi EE, Uitterlinden A, van Duijn CM, Vijaya L, Vingerling JR, Vithana EN, Webster AR, Wichmann HE, Winkler TW, Wong TY, Wright AF, Zelenika D, Zhang M, Zhao L, Zhang K, Klein ML, Hageman GS, Lathrop GM, Stefansson K, Allikmets R, Baird PN, Gorin MB, Wang JJ, Klaver CCW, Seddon JM, Pericak-Vance MA, Iyengar SK, Yates JRW, Swaroop A, Weber BHF, Kubo M, DeAngelis MM, Lveillard T, Thorsteinsdottir U, Haines JL, Farrer LA, Heid IM, Abecasis GR, AMD Gene Consortium (2013) Seven new loci associated with age-related macular degeneration. Nat Genet 45(4):433–439
Fritsche LG, Igl W, Bailey JNC, Grassmann F, Sengupta S, Bragg-Gresham JL, Burdon KP, Hebbring SJ, Wen C, Gorski M, Kim IK, Cho D, Zack D, Souied E, Scholl HPN, Bala E, Lee KE, Hunter DJ, Sardell RJ, Mitchell P, Merriam JE, Cipriani V, Hoffman JD, Schick T, Lechanteur YTE, Guymer RH, Johnson MP, Jiang Y, Stanton CM, Buitendijk GHS, Zhan X, Kwong AM, Boleda A, Brooks M, Gieser L, Ratnapriya R, Branham KE, Foerster JR, Heckenlively JR, Othman MI, Vote BJ, Liang HH, Souzeau E, McAllister IL, Isaacs T, Hall J, Lake S, Mackey DA, Constable IJ, Craig JE, Kitchner TE, Yang Z, Su Z, Luo H, Chen D, Ouyang H, Flagg K, Lin D, Mao G, Ferreyra H, Stark K, von Strachwitz CN, Wolf A, Brandl C, Rudolph G, Olden M, Morrison MA, Morgan DJ, Schu M, Ahn J, Silvestri G, Tsironi EE, Park KH, Farrer LA, Orlin A, Brucker A, Li M, Curcio CA, Mohand-Sad S, Sahel JA, Audo I, Benchaboune M, Cree AJ, Rennie CA, Goverdhan SV, Grunin M, Hagbi-Levi S, Campochiaro P, Katsanis N, Holz FG, Blond F, Blanch H, Deleuze JF, Igo RP Jr, Truitt B, Peachey NS, Meuer SM, Myers CE, Moore EL, Klein R, Hauser MA, Postel EA, Courtenay MD, Schwartz SG, Kovach JL, Scott WK, Liew G, Tan AG, Gopinath B, Merriam JC, Smith RT, Khan JC, Shahid H, Moore AT, McGrath JA, Laux R, Brantley MA Jr, Agarwal A, Ersoy L, Caramoy A, Langmann T, Saksens NTM, de Jong EK, Hoyng CB, Cain MS, Richardson AJ, Martin TM, Blangero J, Weeks DE, Dhillon B, van Duijn CM, Doheny KF, Romm J, Klaver CCW, Hayward C, Gorin MB, Klein ML, Baird PN, den Hollander AI, Fauser S, Yates JRW, Allikmets R, Wang JJ, Schaumberg DA, Klein BEK, Hagstrom SA, Chowers I, Lotery AJ, Lveillard T, Zhang K, Brilliant MH, Hewitt AW, Swaroop A, Chew EY, Pericak-Vance MA, DeAngelis M, Stambolian D, Haines JL, Iyengar SK, Weber BHF, Abecasis GR, Heid IM (2016) A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat Genet 48(2):134–143
Goethals K, Janssen P, Duchateau L (2008) Frailty models and copulas: similarities and differences. J Appl Stat 35(9):1071–1079
Gumbel EJ (1960) Bivariate exponential distributions. J Am Stat Assoc 55(292):698–707
He W, Lawless JF (2003) Flexible maximum likelihood methods for bivariate proportional hazards models. Biometrics 59(4):837–848
Hougaard P (2000) Anal Multivar Surviv Data. Springer, New York
Joe H (1997) Multivariate models and dependence concepts. Chapman & Hall/CRC, London
Kim G, Silvapulle MJ, Silvapulle P (2007) Comparison of semiparametric and parametric methods for estimating copulas. Comput Stat Data Anal 51(6):2836–2850
Lawless JF, Yilmaz YE (2011) Semiparametric estimation in copula models for bivariate sequential survival times. Biom J 53(5):779–796
Lee EW, Wei LJ, Amato DA (1992) Cox-type regression analysis for large numbers of small groups of correlated failure time observations. In: Klein J, Goel P (eds) Surviv Anal State Art, vol 211. Springer, Dordrecht, pp 237–247
Lindfield GR, Penny JET (1989) Microcomputers in numerical analysis. Halsted Press, New York
Mei M (2016) A goodness-of-fit test for semi-parametric copula models of right-censored bivariate survival times. Master’s thesis, Simon Fraser University
Nelsen RB (2006) An introduction to Copulas. Springer, New York
Oakes D (1982) A model for association in bivariate survival data. J R Stat Soc Ser B 44(3):414–422
Sardell RJ, Persad PJ, Pan SS, Whitehead P, Adams LD, Laux R, Fortun JA, Brantley MA Jr, Kovach JL, Schwartz SG, Agarwal A, Haines JL, Scott WK, Pericak-Vance MA (2016) Progression rate from intermediate to advanced age-related macular degeneration is correlated with the number of risk alleles at the CFH locus. Investig Ophthalmol Visual Sci 57(14):6107–6115
Schwarz G (1978) Estimating the dimension of a model. Ann Stat 6(2):461–464
Seddon JM, Reynolds R, Maller J, Fagerness JA, Daly MJ, Rosner B (2009) Prediction model for prevalence and incidence of advanced age-related macular degeneration based on genetic, demographic, and environmental variables. Investig Ophthalmol Visual Sci 50(5):2044–2053
Seddon JM, Reynolds R, Yu Y, Rosner B (2014) Three new genetic loci are independently related to progression to advanced macular degeneration. PLoS ONE 9(1):1–11
Sha Q, Zhang Z, Zhang S (2011) An improved score test for genetic association studies. Genet Epidemiol 35(5):350–359
Shih JH, Louis TA (1995) Inferences on the association parameter in copula models for bivariate survival data. Biometrics 51(4):1384–1399
Sklar A (1959) Fonctions de répartition à n dimensions et leurs marges. Publications de L’Institut de Statistique de L’Université de Paris 8:229–231
Swaroop A, Chew EY, Rickman CB, Abecasis GR (2009) Unraveling a multifactorial late-onset disease: from genetic susceptibility to disease mechanisms for age-related macular degeneration. Annu Rev Genomics Human Genet 10:19–43
The Eye Diseases Prevalence Research Group (2004) Causes and prevalence of visual impairment among adults in the united states. Arch Ophthalmol 122(4):477–485
Wang W, Wells MT (2000) Model selection and semiparametric inference for bivariate failure-time data. J Am Stat Assoc 95(449):62–72
Wei LJ, Lin D, Weissfeld L (1989) Regression analysis of multivariate incomplete failure time data by modeling marginal distributions. J Am Stat Assoc 84(408):1065–1073
Yan Q, Ding Y, Liu Y, Sun T, Fritsche LG, Clemons T, Ratnapriya R, Klein ML, Cook RJ, Liu Y, Fan R, Wei L, Abecasis GR, Swaroop A, Chew EY, Group AR, Weeks DE, Chen W (2018) Genome-wide analysis of disease progression in age-related macular degeneration. Hum Mol Genet 27(5):929–940
Zhang S, Okhrin O, Zhou Q, Song P (2016) Goodness-of-fit test for specification of semiparametric copula dependence models. J Econom 193(1):215–233
Acknowledgements
This research is supported by the National Institute of Health (EY024226). We would like to thank the participants in the AREDS study, who made this research possible, and International AMD Genomics Consortium for generating the genetic data and performing quality check.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Sun, T., Liu, Y., Cook, R.J. et al. Copula-based score test for bivariate time-to-event data, with application to a genetic study of AMD progression. Lifetime Data Anal 25, 546–568 (2019). https://doi.org/10.1007/s10985-018-09459-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10985-018-09459-5