
Abstract
Calsequestrin (CASQ) is the most abundant Ca2+ binding protein localized in the sarcoplasmic reticulum (SR) of skeletal and cardiac muscle. The genome of vertebrates contains two genes, CASQ1 and CASQ2. CASQ1 and CASQ2 have a high level of homology, but show specific patterns of expression. Fast-twitch skeletal muscle fibers express only CASQ1, both CASQ1 and CASQ2 are present in slow-twitch skeletal muscle fibers, while CASQ2 is the only protein present in cardiomyocytes. Depending on the intraluminal SR Ca2+ levels, CASQ monomers assemble to form large polymers, which increase their Ca2+ binding ability. CASQ interacts with triadin and junctin, two additional SR proteins which contribute to localize CASQ to the junctional region of the SR (j-SR) and also modulate CASQ ability to polymerize into large macromolecular complexes. In addition to its ability to bind Ca2+ in the SR, CASQ appears also to be able to contribute to regulation of Ca2+ homeostasis in muscle cells. Both CASQ1 and CASQ2 are able to either activate and inhibit the ryanodine receptors (RyRs) calcium release channels, likely through their interactions with junctin and triadin. Additional evidence indicates that CASQ1 contributes to regulate the mechanism of store operated calcium entry in skeletal muscle via a direct interaction with the Stromal Interaction Molecule 1 (STIM1). Mutations in CASQ2 and CASQ1 have been identified, respectively, in patients with catecholamine-induced polymorphic ventricular tachycardia and in patients with some forms of myopathy. This review will highlight recent developments in understanding CASQ1 and CASQ2 in health and diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Calsequestrin structure and expression
Identification and tissue distribution
It is been about 50 years since David MacLennan published his first article on the discovery of an acidic protein, unique of the sarcoplasmic reticulum (SR), able to bind up to 970 nmol of Ca2+ per mg, which he elegantly named calsequestrin (CASQ) (MacLennan and Wong 1971). From that time on, a number of studies agreed that CASQ represents the major Ca2+ binding protein of muscle cells. It specifically concentrates in the junctional cisternae of the SR facing the T-tubules (Franzini-Armstrong et al. 1987), although with some difference between skeletal and cardiac muscles. Actually, purification and sedimentation experiments showed that approximately only 30% of CASQ2 remained associated with cardiac SR junctional membranes, compared to approximately 70% of CASQ1 in skeletal muscle (Wei et al. 2009a). In the latter, it reaches a concentration of about 6 mM, with a total content estimated to reach 0.5 mg of calsequestrin per gram of wet weight, as measured in frog skeletal muscles (Volpe and Simon 1991).
CASQ is present in invertebrates and in all vertebrates, and a calsequestrin-like protein was identified even in plants (Furlan et al. 2016; Krause et al. 1989; Henson et al. 1989). Two CASQ genes are present in mammalian genomes: CASQ1 and CASQ2. These two isoforms are co-expressed in neonatal skeletal muscles; however, in rodents fast-twitch fibers, the CASQ2 isoform disappears between 2 and 4 weeks postnatally, while CASQ1 continues to be expressed (Sacchetto et al. 1993). On the contrary, slow twitch fibers maintain the expression of both isoforms even in the adult (Damiani et al. 1990). In cardiac muscle, only the CASQ2 isoform is expressed. The two isoforms differ in their amino acid composition and biochemical properties with CASQ1 being apparently more able to bind Ca2+ than CASQ2.
CASQ polymerization and Ca2+ binding
CASQ is characterized by its high capacity and low affinity for Ca2+. It can coordinately bind and release 40–50 Ca2+ ions per molecule per cycle with a Kd of about 1.1 mM under physiological conditions (Volpe and Simon 1991). These features make CASQ unequivocally suitable for its function, that is to manage the large amounts of Ca2+ necessary for muscle contraction. Hence it has been estimated that, at the beginning of each cycle of muscle contraction, the amount of Ca2+ released by calsequestrin is about twenty times higher than the pool of free Ca2+ in the SR (Pape et al. 2007).
The ability of CASQ to bind Ca2+ is mainly due to the complex quaternary structure that this protein assumes in the SR lumen. Electron microscopy analysis of CASQ1 distribution in the SR of skeletal muscles showed that it is arranged in intricate gels, with repeated nodal points connecting short segments of linear polymers (Perni et al. 2013). Interestingly, Ca2+ dissociation experiments revealed that the presence of CASQ at high concentrations in a restricted space, such as when bound to the junctional membranes of the SR, accelerates Ca2+dissociation presumably as a result of cooperative molecular interactions (Beltrán et al. 2006). A number of reports based on biochemical data confirm that CASQ exists in a wide range of high-molecular-mass clusters (Sato et al. 1998; Maguire et al. 1997; Maguire et al. 1998). The first crystals of calsequestrin were obtained from canine cardiac muscle (Hayakawa et al. 1994) and rabbit skeletal muscle (Wang et al. 1998). These revealed the presence of three highly negative thioredoxin-like domains surrounding a hydrophilic center. Following Ca2+ binding, the three domains collapse and CASQ monomers start to polymerize. Accordingly, Park and co-workers proposed a model where Ca2+-induced formation of one front-to-front contact between two monomers leads to dimerization followed by Ca2+-induced formation of a second contact, this time following back-to-back interactions among dimers, to bring polymerization in a ribbon like structure (Park et al. 2003). The assembly in large polymers represents a pivotal factor in determining the Ca2+ buffering capacity of CASQ. Analysis of the primary sequence and of the crystal structure of CASQ1 revealed the presence of low and high affinity Ca2+ binding sites; in addition, 45Ca2+ overlay assays indicated that the asp-rich or CAS (consecutive aspartate stretch at the C-terminus) region, localized at the interface of back-to-back dimers, represents a major Ca2+ binding motif in CASQ. Further experiments showed that front-to-front and back-to-back polymerization can induce the formation of negatively charged cavities that may accommodate additional Ca2+ binding sites (Park et al. 2003; Sanchez et al. 2012a). This observation was further supported by the model described by Kumar and collaborators. They showed that, at low Ca2+ concentration, the CAS region can accommodate 6 to 8 Ca2+ ions and acquires a compact structure. Under these conditions, the negative charges of the CAS region get neutralized, thus allowing back-to-back stacking. The remaining regions of CASQ1 contain additional Ca2+-binding sites that can progressively bind more Ca2+ ions as the [Ca2+] increases, thanks to the formation of additional Ca2+-dependent Ca2+ binding sites (Kumar et al. 2013).
Structural modeling of CASQ1 and CASQ2 suggested that the differences in the Ca2+ binding capacity observed in the two calsequestrin isoforms may be explained by differences in their ability to form front-to-front and back-to-back interfaces (Park et al. 2003, 2004). In particular, the different composition of the CAS region in CASQ1 and CASQ2 may correlate with isoform-specific Ca2+-dependent polymerization properties (Bal et al. 2015). Finally, glycosylation and phosphorylation of CASQ were also proposed to correlate with polymer formation and/or Ca2+ binding capacity (Sanchez et al. 2011, 2012b; McFarland et al. 2011; Beard et al. 2008).
Ca2+ binding and polymerization have been suggested to also regulate the SR retention of CASQ (Gatti et al. 1997, 2001; Houle et al. 2006). At difference with many proteins of the ER, CASQ lacks the Golgi retrieval COOH-terminal signal KDEL; original studies proposed that it may travel from the Golgi to SR by clathrin-coated vesicles; however, the molecular mechanisms responsible for general ER/SR retention and trafficking of CASQ have not been completely defined yet (Thomas et al. 1989; Tijskens et al. 2003; Nori et al. 2004; Ram et al. 2004). On the other hand, it appears to be quite clear that neither polymerization, nor post-translational modifications are strictly involved in CASQ1 sorting to the junctional SR in skeletal muscle cells (Nori et al. 1999, 2000, 2001, 2006); this, in turn, has been proposed to depend on protein interactions with the ryanodine receptor, triadin and junctin (Cho et al. 2007; Boncompagni et al. 2012; Oddoux et al. 2009; Cacheux et al. 2020).
The quaternary Ca 2+ release complex and the regulation of the excitation–contraction coupling mechanism
CASQ interactions with RyR, triadin and junctin
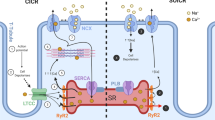
In striated muscles, CASQ is part of a quaternary complex composed of the ryanodine receptor calcium release channel and of two single transmembrane domain proteins of the junctional SR, triadin and junctin (Zhang et al. 1997; Mitchell et al. 1988; Glover et al. 2002; Jones et al. 1995). These four proteins were found to directly and indirectly interact one with each other and their reciprocal interactions were shown to be important for regulation of Ca2+ release during muscle contraction. Indeed, within this quaternary complex, CASQ plays a dual role, that is to keep the large amount of Ca2+ necessary for muscle contraction in close proximity of RyRs and to directly and/or indirectly regulate channel opening and activity.
In skeletal muscle, a direct interaction was described between CASQ1 and triadin. In particular, the CAS sequence in CASQ1 was found to bind a region containing a KEKE motif localized in the lumen of skeletal muscle triadin (Guo and Campbell 1995; Shin et al. 2000; Beard and Dulhunty 2015; Rossi et al. 2014a). Interaction of CASQ1 with triadin was proposed to be important to anchor the protein at the junctional region of the SR and to promote the junctional coupling between calsequestrin and the RyR (Guo and Campbell 1995; Kobayashi et al. 2000).
A region homologous to the KEKE motif observed in triadin is also present in the luminal domain of junctin, suggesting that interaction between CASQ1 and junctin can also occur via KEKE motifs (Dulhunty et al. 2009), while the junctin binding site in CASQ has been proposed to include the CAS region and a region encompassing amino acids 86–191 (Beard and Dulhunty 2015; Shin et al. 2003). It has been also proposed that CASQ phosphorylation may enhance binding to junctin at low Ca2+ concentration (Beard et al. 2008). Finally, a direct interaction was reported between CASQ1 and RyR1 (Herzog et al. 2000).
The same network of interactions between CASQ2, triadin, junctin and RyR2 was described in cardiac muscle, where it plays a key role in the regulation of the cardiac excitation–contraction coupling (Kobayashi et al. 2000; Rani et al. 2016; Handhle et al. 2016).
Functional role of CASQ in the regulation of RyR activation
The specific functional role of CASQ in the context of the quaternary protein complex in both skeletal and cardiac muscle has been investigated and debated for a long time. For example, CASQ was originally described to act as an endogenous activator of RyR in isolated heavy SR vesicles (Kawasaki and Kasai 1994). However, further studies based on lipid bilayer experiments suggested that, at low Ca2+ concentrations, CASQ1 may function as a Ca2+ sensor that prevents RyR1 to increase its activity in order to preserve the intracellular SR Ca2+ store (Beard et al. 2002, 2005; Wei et al. 2006, 2009a). Finally, in another study, CASQ1 was found not to exert any significant regulation of RyR1 channel opening (Qin et al. 2009). Distinct effects on channel regulation have been also reported when comparing different CASQ or RyR isoforms. Single channel lipid bilayer experiments showed that CASQ2 can increase the open probability of both RyR1 and RyR2 channels (Wei et al. 2009a). RyR2 channels show an increase in open probability in the presence of either CASQ1 or CASQ2 (Qin et al. 2009). Similar to what observed for CASQ1 in skeletal muscle, CASQ2 in cardiac muscle is believed to act as a SR Ca2+ sensor, inhibiting RyR2 opening at low Ca2+ concentrations (Györke et al. 2004a). More recently, Chen and collaborators reported that CASQ2 association with RyR2 determined a reduction in sensitivity to cytosolic Ca2+ activation, while RyR2 from CASQ2 knockout mice were significantly more sensitive to cytosolic Ca2+ activation and had significantly longer mean open times than RyR2 from control mice (Chen et al. 2013).
Finally, CASQ polymerization was shown to be a reversible process; however, since Ca2+-binding sites of different affinities are present in the CASQ1 polymers, under physiological conditions, CASQ1 can sequester and deliver Ca2+ ions without the need to completely depolymerize (Kumar et al. 2013). Nevertheless, in conditions of critical SR depletion, when the intraluminal Ca2+ concentration decreases in a significant manner, CASQ can actually depolymerize. Interestingly, in both cardiac and skeletal muscle, SR depletion is paralleled by a progressive closure of the ryanodine receptors (RyRs) and a consequent reduction in Ca2+ release, to prevent dangerous SR exhaustion (Canato et al. 2010; Sztretye et al. 2011; Zima et al. 2010); on this line, and it has been proposed that CASQ1 depolymerization may represent the intracellular switch to induce channel closing (Manno et al. 2017).
The combined role of CASQ, triadin and junctin in the regulation of RyR activation
As described at the beginning of this chapter, in striated muscles, CASQ is present at the junctional SR together with two other proteins, triadin and junctin. Hence, when considering the combined role of calsequestrin, triadin and junctin on RyR activation and in the regulation of the excitation–contraction coupling, the scenario becomes quite complex. Calsequestrin was found to increase [3H]ryanodine binding to solubilized heavy SR vesicles, but this effect was abolished by the presence of triadin (Ohkura et al. 1998). Similarly, addition of triadin and calsequestrin to purified RyRs resulted in channel inhibition, an effect that was not observed on channels depleted of triadin, suggesting that calsequestrin may reduce channel activity by binding the luminal domain of triadin (Beard et al. 2002; Györke et al. 2004b). Finally, more recently, Wei and collaborators showed that while both triadin and junctin appeared to increase the open probability of RyR1 channels, the presence of CASQ1 abolished the activation induced by junctin, but not that induced by triadin (Wei et al. 2009b). In conclusion, results from all these studies, although in some cases conflicting, converge to the idea that the regulation of RyR activity in physiological conditions may actually depend on a complex combination of biochemical, structural or functional components. A more detailed discussion on this point is reported in a comprehensive review by Gaburjakova and collaborators, who critically analyze the functional meaning of protein interactions examined under distinct experimental conditions (Gaburjakova et al. 2013).
Mouse models to study CASQ function in skeletal and cardiac muscle
CASQ1-related mouse models
To investigate the role of CASQ in striated muscles, different mouse models have been generated. CASQ1 knockout mice have a normal motor activity. Nevertheless, they show a profound remodeling of the excitation contraction coupling apparatus, which is characterized by narrower terminal cisternae and by the appearance of multilayered junctions, likely due to a compensatory effect in response to a reduction in Ca2+ storage capacity. Although muscle performance is not significantly impaired, measurement of Ca2+ release parameters revealed that the average amplitude of Ca2+ release following either low frequency electrical stimulation or caffeine treatment was reduced (Paolini et al. 2007; Tomasi et al. 2012). Nevertheless, CASQ1 knockout mice were unable to sustain prolonged muscle activity; actually, the time parameters of twitch were significantly longer compared to wild type mice, with a significant increased rate of SR Ca2+ depletion, likely due to a reduced inhibition of the RyR1 Ca2+ release activity (Canato et al. 2010). Together these observations suggest that ablation of CASQ1 correlated with a reduction in the SR Ca2+content associated with an impaired Ca2+ re-uptake by the SR, possibly due to a decreased efficiency of SERCA pumps and/or of the SOCE refilling process (Paolini et al. 2007, 2011; Protasi et al. 2011; Canato et al. 2010). In slow twitch skeletal muscle fibers, expression of CASQ2 in addition to CASQ1 may further support intraluminal Ca2+ buffering. However, depletion of both isoforms in double knockout mice resulted in a decrease in Ca2+ store amplitude and SR refilling, with kinetics similar to CASQ1 knockout mice, suggesting that the role of CASQ2 in slow twitch muscle fibers may not be determinant (Canato et al. 2010). On the contrary, in C2C12 cells CASQ2 seems to play a more relevant role than CASQ1 in determining the SR Ca2+ store, since depletion of CASQ1 following small interfering RNA transfection does not result in a significant depletion of the SR (Wang et al. 2006). It has to be mentioned, however, that muscle fibers devoid of CASQ1 or CASQ2 or both can still sustain Ca2+ release and muscle contraction, suggesting that other molecules, such as the Histidine Rich Calcium (HRC) Binding Protein may provide reversible Ca2+ binding and luminal Ca2+ buffering together with or in alternative to CASQ (Canato et al. 2010; Royer and Ríos 2009).
One of the most interesting phenotypic traits of CASQ1 knockout mice is their increased susceptibility to undergo hypermetabolic crises in response to halothane, heat-exposure and physical exertion, a syndrome remarkably similar to human malignant hyperthermia (MH) and environmental/exhertional heat-stroke (Paolini et al. 2011; Dainese et al. 2009; Protasi et al. 2009; Michelucci et al. 2015, 2017a, b; Guarnier et al. 2018). Loss of inhibition of RyR1 activity due to CASQ1 ablation (Beard et al. 2002) and SR Ca2+ depletion (Canato et al. 2010) may explain this MH-like phenotype in CASQ1 knockout mice, although the exact mechanisms still need to be elucidated.
However, despite evidence that CASQ1 knockout mice clearly show typical traits of human MH, no mutations in CASQ1 have been associated to human Malignant Hyperthermia, although reports of variants of unknown significance have detected in some patients (Kraeva et al. 2013; Lewis et al. 2015; Bjorksten et al. 2016).
CASQ2-related mouse models
CASQ2 knockout mice are viable and display normal SR Ca2+ release and contractile function under basal conditions. Nevertheless, these mice show an increased SR volume and absence of triadin and junctin. Exposure to catecholamines caused increased diastolic SR Ca2+ leak, resulting in premature spontaneous SR Ca2+ release, a condition that closely mimics human arrhythmias (Knollmann et al. 2006; Chopra et al. 2007). On the contrary, transgenic mice for CASQ2 showed cardiac hypertrophy and alteration of the beta-adrenergic receptor signaling, together with a reduction in Ca2+-induced-Ca2+ release and frequency of Ca2+ sparks (Jones et al. 1998; Cho et al. 1999; Wang et al. 2000; Schmidt et al. 2000). A more recent model of CASQ2 knockout mice showed that the same alterations in Ca2+ release can be detected both in the atrium and in ventricle, indicating a more extended role of CASQ2 in cardiac Ca2+ physiology (Gergs et al. 2017). Moreover, a role of CASQ2 in the cardiac conduction system was described. In particular, CASQ2 deletion was found to give rise to sinus and atrio-ventricular nodal dysfunctions, which, together with alterations occurring in ventricular cardiomyocytes, correlate with development of the catecholaminergic polymorphic ventricular tachycardia (CPVT) phenotype (Glukhov et al. 2015; Faggioni et al. 2013; Flores et al. 2018).
Novel roles of CASQ in muscle physiology and Ca 2+ homeostasis
Store-operated Ca2+ entry (SOCE) represents an important mechanism for the refilling of depleted intracellular-reticulum Ca2+ stores. In muscle fibers, this process is necessary to maintain the Ca2+ stores at steady state levels as well as for generation of long-lasting Ca2+ signals and contractile function during tetanic stimulation and fatigue (Launikonis and Ríos 2007; Stiber et al. 2008; Stiber and Rosenberg 2011; Pan et al. 2014; Michelucci et al. 2018). The SOCE pathway is operated by the coordinated activity of two main molecules: stromal interaction molecule 1 (STIM1) and calcium release-activated calcium channel protein 1 (Orai1) whose activation should be tightly regulated. The process that supports SOCE activation in non-excitable cells is slow (Lunz et al. 2019), as it requires a sequence of events starting from STIM1 sensing a drop of endoplasmic reticulum (ER) Ca2+ levels by intraluminal EF-end motifs that triggers structural rearrangement and oligomerization; the cytoplasmic C-terminus acquires an extended conformation that exposes a STIM–Orai1 activating region (SOAR) that, in turn, induces Orai1 cluster formation and channel activation. In skeletal muscle, a longer form of STIM1 (STIM1L) exists and forms permanent STIM1L–Orai1 clusters (Darbellay et al. 2011): this could explain the rapid SOCE activation that sustains repetitive bursts of tetanic stimulations by compensating for Ca2+ efflux occurring during normal muscle contraction (Launikonis et al. 2010). More recently, intense exercise was found to induce formation of intracellular junctions in skeletal muscle between stacks of SR cisternae and extensions of transverse-tubules; these were proposed to increase co-localization of proteins required for SOCE and thus to optimize Ca2+ entry and SR refilling during acute exercise (Boncompagni et al. 2017, 2018; Michelucci et al. 2019).
Given that CASQ has a key role in regulating the intraluminal Ca2+ concentration, it can be expected that it may also play a role in SOCE. Indeed, it was originally proposed that CASQ may provide a retrograde signal to regulate this process (Ma and Pan 2003; Shin et al. 2003). This early idea was later supported by experiments showing that knockdown of CASQ1 in flexor digitorum brevis (FDB) muscle fibers increased Ca2+ entry across the sarcolemma (Zhao et al. 2010). Additional evidences demonstrated that monomeric CASQ1 that forms following Ca2+ store depletion can actually interact with STIM1 and inhibit STIM1/Orai1 interaction (Wang et al. 2015; Zhang et al. 2016). In particular, amino acid residues 362–396 of CASQ1 were proposed to be responsible for CASQ1-STIM1 association, since expression of CASQ1 proteins deleted in this region did not result in SOCE inhibition (Zhang et al. 2016).
More recently, the ER stress response sensor IRE1α was identified as a novel interactor of CASQ in cardiac and skeletal muscle. Interaction with CASQ1 prevents IRE1α oligomerization and activation, suggesting that this may represent a cell approach to avoid IRE1α activation in response to the constant fluctuations in the Ca2+ concentration that occur in the SR during muscle contraction and relaxation cycles (Wang et al. 2019).
Calsequestrin and human diseases
CASQ2 mutations in cardiac diseases
The importance of calsequestrin in the regulation of Ca2+ handling in striated muscles is supported by identification of mutations in both CASQ1 and CASQ2 genes associated to human diseases (see Table 1). The first mutations in CASQ were identified in the CASQ2 gene in patients affected by CPVT, a form of cardiac arrhythmia which occurs in the absence of structural abnormalities (Lahat et al. 2001, 2003, 2004; Eldar et al. 2003; Viatchenko-Karpinski et al. 2004; Houle et al. 2004; Postma et al. 2002). At present, more than ten different mutations have been identified in both recessive and dominant forms of CPVT. The characterization of some of these mutations confirmed that CASQ2 regulates the SR Ca2+ store both by acting as an active Ca2+ buffer, which controls the amount of releasable Ca2+ upon each stimulation and by controlling SR re-charging and RyR2 channel sensitivity to luminal Ca2+ (Györke et al. 2004a, b). The first mutation in CASQ2, the D307H mutation, was identified in a patient affected by a recessive form of CPVT and was characterized in rat myocytes and, more recently, through generation of hIPSC-derived cardiomyocytes from affected patients (Maizels et al. 2017). Expression of the mutant protein resulted in a decrease in SR Ca2+ storage capacity and Ca2+ release, with signs of delayed afterdepolarization likely due to abnormal Ca2+ polymerization and buffering of the mutant protein at high Ca2+ concentrations (Viatchenko-Karpinski et al. 2004; Houle et al. 2004; Dirksen et al. 2007; Kalyanasundaram et al. 2009; Novak et al. 2012; Maizels et al. 2017). On the contrary, expression of the R33Q mutation in CASQ2 led to an increase in Ca2+ release and occurrence of spontaneous Ca2+ sparks, suggesting that the R33Q mutation lost its ability to inhibit RyR2 at low Ca2+ concentrations (Terentyev et al. 2006). Accordingly, an enhanced sensitivity of RYR2 to cytosolic [Ca2+] and longer RyR2 open times were observed in CASQ2 R33Q knock-in mice (Chen et al. 2013). An even more significant reduction in the ability of CASQ2 to inhibit RyR2 activity was observed for the L167H mutation (Qin et al. 2008). Cristal structures obtained for the D307H and the R33Q mutant proteins revealed that they had a reduced Ca2+ binding capacity due to altered polymerization. In particular, R33Q and D307H mutants did not form properly oriented dimers, while L167H mutant resulted in formation of high molecular weight aggregates (Kim et al. 2007). Molecular conformation studies confirmed that the R33Q mutation resides in a region of the protein responsible for the bidirectional transition from monomer to dimer (Bal et al. 2010). Finally, the K206N mutations creates an additional N-glycosylation site and it is associated with a reduction in oligomer formation and Ca2+ binding capacity. In cardiomyocytes, these alterations caused a reduction of the SR Ca2+ load and an increase in spontaneous RyR2 opening (Kirchhefer et al. 2010).
CASQ1 mutations in skeletal muscle myopathies
A mutation in CASQ1, D244G, was first described in patients affected by a mild myopathy characterized by the presence of inclusions containing an excess of SR proteins, including CASQ1 (Tomelleri et al. 2006; Rossi et al. 2014b; D’Adamo et al. 2016). The mutation affects a conserved high-affinity Ca2+ binding sites of CASQ1 and alters the kinetics of CASQ1 polymerization and Ca2+ release in muscle fibers and primary myotubes (Rossi et al. 2014b). In particular, D244G CASQ1 forms large aggregates, with atypical dimer interactions (Lewis et al. 2015) (Fig. 1). Additional CASQ1 mutations were later identified in patients affected by Tubular Aggregate Myopathy (TAM). TAMs were originally associated with mutations in STIM1, mostly in the Ca2+ sensing EF hand in the N-terminal luminal region (Bohm et al. 2013), or in Orai1 (Nesin et al. 2014; Lacruz and Feske 2015; Bohm et al. 2017; Bohm and Laporte 2018a). All these mutations resulted in SOCE hyperactivation; on the contrary, a mutation, in the cytoplasmic C-terminal inhibitory domain of STIM1, identified in a family with TAM, induced a decrease in Ca2+ influx (Okuma et al. 2016), suggesting a general dysregulation of SOCE and Ca2+ homeostasis in the pathogenesis of TAM (Lee and Noguchi 2016). Concerning CASQ1, four mutations, D44N, N56Y, G103D, and I385T have been identified in patient with TAM. Similar to the D244G mutation, alteration of CASQ1 polymerization was a typical trait of these mutations, resulting in a reduced ability to store Ca2+ (Barone et al. 2017; Bohm et al. 2018b). The altered Ca2+-dependent aggregation of some of these mutated CASQ1 proteins was also associated with altered SOCE. In particular, two of these mutants (D44N and I385T), were shown to have lost the ability to inhibit SOCE, while the G103D mutant was still able to inhibit Ca2+ influx (Barone et al. 2017), further supporting that a general dysregulation of SOCE and Ca2+ homeostasis, either due to mutations in STIM1 and Orai1, or to mutations in CASQ1 can be associated to development of TAM (see Table 1).

Expression and localization of wild type and D244G CASQ1 in COS7 cells and in adult muscle fibers. Wild type CASQ1 distributed in regular linear structures resembling CASQ1 polymers in COS7 cells (a) and localized at triads in adult skeletal muscle fibers (c). The D244G CASQ1 mutation identified in patients affected by vacuolar myopathy affects CASQ1 polymerization. Expression of the D244G CASQ1 mutant in COS7 cells resulted in protein assembly in puncta (b), while in skeletal muscle fibers it formed large intracellular aggregates (d) (Tomelleri et al. 2006; Rossi et al. 2014b). Bar = 3.5 µm
Additional proposed CASQ-related diseases
In addition to typical muscle diseases, the genomic region where the CASQ1 gene maps has been associated to type 2 diabetes susceptibility. CASQ1 is localized to human chromosome 1 (Fujii et al. 1990). In particular, two SNPs in intron two of the CASQ1 gene were found to be strongly associated in the Caucasian population in North America with type 2 diabetes; other SNPs were also found to be associated with the same disease in the Amish population. However, the molecular mechanisms explaining the correlation between CASQ1 and diabetes have not been defined yet (Das et al. 2004; Fu et al. 2004). In addition, in the Danish population this correlation apparently does not exist, suggesting that association of CASQ1 with type 2 diabetes may be a coincidence or may be restricted to specific populations (Sparsø et al. 2007).
Finally, CASQ1 was found to be associated with Graves’ ophthalmopathy, a disease associated with the thyroid autoimmune disorder Graves’ disease. Significant levels of antibodies against CASQ1 or CASQ2 are present in the serum of patients affected by this disease; although the exact correlation with the pathological phenotype of these patients has not been defined, it has been suggested that these antibodies may contribute to eye muscle damage and/or cardiac complications associated with the disease (De Haan et al. 2010).
Concluding remarks
Since its initial identification as an intraluminal Ca2+ buffering protein, the role of CASQ in striated muscle has been continuously updated and novel functional aspects are being depicted. In this respect, the involvement of CASQ in the regulation of SOCE and ER stress response as well as in the cardiac muscle conduction system open new perspectives in the pathophysiology of striated muscle cells. Next generation sequencing approaches are constantly in progress to identify novel mutations in CASQ1 and CASQ2 genes associated with skeletal or cardiac myopathies. No target-specific therapeutic approaches have been identified so far. On this line, recent studies on the development and characterization of induced pluripotent stem cell lines generated from patients carrying CASQ2 mutations show that cardiomyocytes derived from these cells appear to recapitulate the main features of disease specific phenotypes thus representing a valuable model for personalized drug development and mutation screening.
References
Bal NC, Sharon A, Gupta SC, Jena N, Shaikh S, Gyorke S, Periasamy M (2010) The catecholaminergic polymorphic ventricular tachycardia mutation R33Q disrupts the N-terminal structural motif that regulates reversible calsequestrin polymerization. J Biol Chem 28(22):17188–17196. https://doi.org/10.1074/jbc.M109.096354
Bal NC, Jena N, Chakravarty H, Kumar A, Chi M, Balaraju T, Rawale SV, Rawale JS, Sharon A, Periasamy M (2015) The C-terminal calcium-sensitive disordered motifs regulate isoform-specific polymerization characteristics of calsequestrin. Biopolymers 103(1):15–22. https://doi.org/10.1002/bip.22534
Barone V, Del Re V, Gamberucci A, Polverino V, Galli L, Rossi D, Costanzi E, Toniolo L, Berti G, Malandrini A, Ricci G, Siciliano G, Vattemi G, Tomelleri G, Pierantozzi E, Spinozzi S, Volpi N, Fulceri R, Battistutta R, Reggiani C, Sorrentino V (2017) Identification and characterization of three novel mutations in the CASQ1 gene in four patients with tubular aggregate myopathy. Hum Mutat 38(12):1761–1773. https://doi.org/10.1002/humu.23338
Beard NA, Sakowska MM, Dulhunty AF, Laver DR (2002) Calsequestrin is an inhibitor of skeletal muscle ryanodine receptor calcium release channels. Biophys J 82(1 Pt 1):310–320
Beard NA, Casarotto MG, Wei L, Varsányi M, Laver DR, Dulhunty AF (2005) Regulation of ryanodine receptors by calsequestrin: effect of high luminal Ca2+ and phosphorylation. Biophys J 88(5):3444–3454
Beard NA, Wei L, Cheung SN, Kimura T, Varsányi M, Dulhunty AF (2008) Phosphorylation of skeletal muscle calsequestrin enhances its Ca2+ binding capacity and promotes its association with junctin. Cell Calcium 44(4):363–373
Beard NA, Dulhunty AF (2015) C-terminal residues of skeletal muscle calsequestrin are essential for calcium binding and for skeletal ryanodine receptor inhibition. Skelet Muscle 22:5:6. https://doi.org/10.1186/s13395-015-0029-7
Beltrán M, Barrientos G, Hidalgo C (2006) Fast kinetics of calcium dissociation from calsequestrin. Biol Res 39(3):493–503
Bohm J, Chevessier F, Maues De Paula A, Koch C, Attarian S, Feger C, Hantaï D, Laforet P, Ghorab K, Vallat JM, Fardeau M, Figarella-Branger D, Pouget J, Romero NB, Koch M, Ebel C, Levy N, Krahn M, Eymard B, Bartoli M, Laporte J (2013) Constitutive activation of the calcium sensor STIM1 causes tubular-aggregate myopathy. Am J Hum Genet 7;92(2):271–278
Bohm J, Bulla M, Urquhart JE, Malfatti E, Williams SG, O’Sullivan J, Szlauer A, Koch C, Baranello G, Mora M, Ripolone M, Violano R, Moggio M, Kingston H, Dawson T, DeGoede CG, Nixon J, Boland A, Deleuze JF, Romero N, Newman WG, Demaurex N, Laporte J (2017) ORAI1 mutations with distinct channel gating defects in tubular aggregate myopathy. Hum Mut 38(4):426–438
Bohm J, Laporte J (2018a) Gain-of-function mutations in STIM1 and ORAI1 causing tubular aggregate myopathy and Stormorken syndrome. Cell Calcium 76:1–9
Bohm J, Lornage X, Chevessier F, Birck C, Zanotti S, Cudia P, Bulla M, Granger F, Bui M, Sartori M, Schneider-Gold C, Malfatti E, Romero NB, Mora M, Laporte J (2018b) CASQ1 mutations impair calsequestrin polymerization and cause tubular aggregate myopathy. Acta Neuropathol 135:149–151
Boncompagni S, Thomas M, Lopez JR, Allen PD, Yuan Q, Kranias EG, Franzini-Armstrong C, Perez CF (2012) Triadin/Junctin double null mouse reveals a differential role for Triadin and Junctin in anchoring CASQ to the jSR and regulating Ca(2+) homeostasis. PLoS ONE 7(7):e39962. https://doi.org/10.1371/journal.pone.0039962
Boncompagni S, Michelucci A, Pietrangelo L, Dirksen RT, Protasi F (2017) Exercise-dependent formation of new junctions that promote STIM1-Orai1 assembly in skeletal muscle. Sci Rep 7(1):14286
Boncompagni S, Michelucci A, Pietrangelo L, Dirksen RT, Protasi F (2018) Addendum: exercise- dependent formation of new junctions that promote STIM1-Orai1 assembly in skeletal muscle. Sci Rep 8(1):17463
Bjorksten AR, Gillies RL, Hockey BM, Du Sart D (2016) Sequencing of genes involved in the movement of calcium across human skeletal muscle sarcoplasmic reticulum: continuing the search for genes associated with malignant hyperthermia. Anaesth Intensive Care 44(6):762–768
Cacheux M, Fauconnier J, Thireau J, Osseni A, Brocard J, Roux-Buisson N, Brocard J, Fauré J, Lacampagne A, Marty I (2020) Interplay between triadin and calsequestrin in the pathogenesis of CPVT in the mouse. Mol Ther 28(1):171–179. https://doi.org/10.1016/j.ymthe.2019.09.012
Canato M, Scorzeto M, Giacomello M, Protasi F, Reggiani C, Stienen GJ (2010) Massive alterations of sarcoplasmic reticulum free calcium in skeletal muscle fibers lacking calsequestrin revealed by a genetically encoded probe. Proc Natl Acad Sci USA 107(51):22326–22331. https://doi.org/10.1073/pnas.1009168108
Chen H, Valle G, Furlan S, Nani A, Gyorke S, Fill M, Volpe P (2013) Mechanism of calsequestrin regulation of single cardiac ryanodine receptor in normal and pathological conditions. J Gen Physiol 142(2):127–136. https://doi.org/10.1085/jgp.201311022
Cho MC, Rapacciuolo A, Koch WJ, Kobayashi Y, Jones LR, Rockman HA (1999) Defective beta-adrenergic receptor signaling precedes the development of dilated cardiomyopathy in transgenic mice with calsequestrin overexpression. J Biol Chem 6(32):22251–22256
Cho JH1, Ko KM, Singaruvelu G, Lee W, Kang GB, Rho SH, Park BJ, Yu JR, Kagawa H, Eom SH, Kim DH, Ahnn J (2007) Functional importance of polymerization and localization of calsequestrin in C. elegans. J Cell Sci 120(1):1551–1558
Chopra N, Kannankeril PJ, Yang T, Hlaing T, Holinstat I, Ettensohn K, Pfeifer K, Akin B, Jones LR, Franzini-Armstrong C, Knollmann BC (2007) Modest reductions of cardiac calsequestrin increase sarcoplasmic reticulum Ca2+ leak independent of luminal Ca2+ and trigger ventricular arrhythmias in mice. Circ Res 14(6):617–626
D’Adamo MC, Sforna L, Visentin S, Grottesi A, Servettini L, Guglielmi L, Macchioni L, Saredi S, Curcio M, De Nuccio C, Hasan S, Corazzi L, Franciolini F, Mora M, Catacuzzeno L, Pessia M (2016) A calsequestrin-1 mutation associated with a skeletal muscle disease alters sarcoplasmic Ca2+ release. PLoS ONE 11(5):e0155516. https://doi.org/10.1371/journal.pone.0155516
Dainese M, Quarta M, Lyfenko AD, Paolini C, Canato M, Reggiani C, Dirksen RT, Protasi F (2009) Anesthetic- and heat-induced sudden death in calsequestrin-1-knockout mice. FASEB J 23(6):1710–1720. https://doi.org/10.1096/fj.08-121335
Damiani E, Volpe P, Margreth A (1990) Coexpression of two isoforms of calsequestrin in rabbit slow-twitch muscle. J Muscle Res Cell Motil 11(6):522–530
Darbellay B, Arnaudeau S, Bader CR, Konig S, Bernheim L (2011) STIM1L is a new actin-binding splice variant involved in fast repetitive Ca2+ release. J Cell Biol 25(2):335–346
Das SK, Chu W, Zhang Z, Hasstedt SJ, Elbein SC (2004) Calsquestrin 1 (CASQ1) gene polymorphisms under chromosome 1q21 linkage peak are associated with type 2 diabetes in Northern European Caucasians. Diabetes 53(12):3300–3006
de Haan S, Lahooti H, Morris O, Wall JR (2010) Epitopes, immunoglobulin classes and immunoglobulin G subclasses of calsequestrin antibodies in patients with thyroid eye disease. Autoimmunity 43(8):698–703. https://doi.org/10.3109/08916931003774954
de la Fuente S, Van Langen IM, Postma AV, Bikker H, Meijer A (2008) A case of catecholaminergic polymorphic ventricular tachycardia caused by two calsequestrin 2 mutations. Pacing Clin Electrophysiol 31(7):916–919. https://doi.org/10.1111/j.1540-8159.2008.01111.x
di Barletta MR, Viatchenko-Karpinski S, Nori A, Memmi M, Terentyev D, Turcato F, Valle G, Rizzi N, Napolitano C, Gyorke S, Volpe P, Priori SG (2006) Clinical phenotype and functional characterization of CASQ2 mutations associated with catecholaminergic polymorphic ventricular tachycardia. Circulation 5(10):1012–1019
Dirksen WP, Lacombe VA, Chi M, Kalyanasundaram A, Viatchenko-Karpinski S, Terentyev D, Zhou Z, Vedamoorthyrao S, Li N, Chiamvimonvat N, Carnes CA, Franzini-Armstrong C, Györke S, Periasamy M (2007) A mutation in calsequestrin, CASQ2D307H, impairs sarcoplasmic reticulum Ca2+ handling and causes complex ventricular arrhythmias in mice. Cardiovasc Res 75(1):69–78
Dulhunty A, Wei L, Beard N (2009) Junctin—the quiet achiever. J Physiol 587:3135–3137
Eldar M, Pras E, Lahat H (2003) A missense mutation in the CASQ2 gene is associated with autosomal-recessive catecholamine-induced polymorphic ventricular tachycardia. Trends Cardiovasc Med 13(4):148–151
Faggioni M, Hwang HS, van der Werf C, Nederend I, Kannankeril PJ, Wilde AAM, Knollmann BC (2013) Accelerated sinus rhythm prevents catecholaminergic polymorphic ventricular tachycardia in mice and in patients. Circ Res 112:689–697
Flores DJ, Duong T, Brandenberger LO, Mitra A, Shiral A, Johnson JC, Springer D, Noguchi A, Y Z, Ebert SN, Ludwig A, Knollmann BC, Levin MD, Pfeifer K (2018) Conditional ablation and conditional rescue models for Casq2 elucidate the role of development and of cell-type specific expression of Casq2 in the CPVT2 phenotype. Hum Mol Gen 27:1533–1544
Franzini-Armstrong C, Kennery LJ, Varriano-Martson E (1987) The structure of calsequestrin in triads of vertebrate skeletal muscle: a deep-etch study. J Cell Biol 105(1):49–56
Fu M, Damcott CM, Sabra M, Pollin TI, Ott SH, Wang J, Garant MJ, O’Connell JR, Mitchell BD, Shuldiner AR (2004) Polymorphism in the calsequestrin 1 (CASQ1) gene on chromosome 1q21 is associated with type 2 diabetes in the old order Amish. Diabetes 53(12):3292–3299
Furlan S, Mosole S, Murgia M, Nagaraj N, Argenton F, Volpe P, Nori A (2016) Calsequestrins in skeletal and cardiac muscle from adult Danio rerio. J Muscle Res Cell Motil 37(1–2):27–39. https://doi.org/10.1007/s10974-015-9432-2.
Fujii J, Willard HF, MacLennan DH (1990) Characterization and localization to human chromosome 1 of human fast-twitch skeletal muscle calsequestrin gene. Somat Cell Mol Genet 16(2):185–189
Fujisawa T, Aizawa Y, Katsumata Y, Udo A, Ito S, Hatakeyama K, Hirose M, Miyama H, Nakajima K, Nishiyama T, Kimura T, Nitta M, Misumi K, Takatsuki S, Kosaki K, Fukuda K (2019) A homozygous CASQ2 mutation in a Japanese patient with catecholaminergic polymorphic ventricular tachycardia. Case Rep Genet 8:9056596. https://doi.org/10.1155/2019/9056596
Gaburjakova M, Bal NC, Gaburjakova J, Periasamy M (2013) Functional interaction between calsequestrin and ryanodine receptor in the heart. Cell Mol Life Sci 70(16):2935–2945. https://doi.org/10.1007/s00018-012-1199-7.
Gatti G, Podini P, Meldolesi J (1997) Overexpression of calsequestrin in L6 myoblasts: formation of endoplasmic reticulum subdomains and their evolution into discrete vacuoles where aggregates of the protein are specifically accumulated. Mol Biol Cell 8(9):1789–1803
Gatti G, Trifari S, Mesaeli N, Parker JM, Michalak M, Meldolesi J (2001) Head-to-tail oligomerization of calsequestrin: a novel mechanism for heterogeneous distribution of endoplasmic reticulum luminal proteins. J Cell Biol 6(3):525–534
Gergs U, Fahrion CM, Bock P, Fisher M, Wache H, Hauptmann S, Schmits W, Neumann J (2017) Evidence for a functional role of calsequestrin 2 in mouse atrium. Acta Physiol 219:671–684
Györke S, Györke I, Terentyev D, Viatchenko-Karpinski S, Williams SC (2004a) Modulation of sarcoplasmic reticulum calcium release by calsequestrin in cardiac myocytes. Biol Res 37(4):603–607
Györke I, Hester N, Jones LR, Györke S (2004b) The role of calsequestrin, triadin, and junctin in conferring cardiac ryanodine receptor responsiveness to luminal calcium. Biophys J 86(4):2121–2128
Glover L, Quinn S, Ryan M, Pette D, Ohlendieck K (2002) Supramolecular calsequestrin complex. Eur J Biochem 269(18):4607–4616
Glukhov AV, Kalyanasundaram A, Lou Q, Hage LT, Hansen BJ, Belevych AE, Mohler PJ, Knollmann BC, Periasamy M, Gyorke S, Fedorov VV (2015) Calsequestrin 2 deletion causes sinoatrial node dysfunction and atrial arrhythmias associated with altered sarcoplasmic reticulum calcium cycling and degenerative fibrosis within the mouse atrial pacemaker complex. Eur Heart J 36:686–697. https://doi.org/10.1093/eurheartj/eht452
Gray B, Bagnall RD, Lam L, Ingles J, Turner C, Haan E, Davis A, Yang PC, Clancy CE, Sy RW, Semsarian C (2016) A novel heterozygous mutation in cardiac calsequestrin causes autosomal dominant catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 13(8):1652–1660. https://doi.org/10.1016/j.hrthm.2016.05.004
Guarnier FA, Michelucci A, Serano M, Pietrangelo L, Pecorai C, Boncompagni S, Protasi F (2018) Aerobic training prevents heatstrokes in calsequestrin-1 knockout mice by reducing oxidative stress. Oxid Med Cell Longev. https://doi.org/10.1155/2018/4652480
Guo W, Campbell KP (1995) Association of triadin with the ryanodine receptor and calsequestrin in the lumen of the sarcoplasmic reticulum. J Biol Chem 21(16):9027–9030
Handhle A, Ormonde CE, Thomas NL, Bralesford C, Williams AJ, Lai FA, Zissimopoulos S (2016) Calsequestrin interacts directly with the cardiac ryanodine receptor luminal domain. J Cell Sci 1(21):3983–3988
Hayakawa K, Swenson L, Baksh S, Wei Y, Michalak M, Derewenda ZS (1994) Crystallization of canine cardiac calsequestrin. J Mol Biol 7(1):357–360
Henson JH, Begg DA, Beaulieu SM, Fishkind DJ, Bonder EM, Terasaki M, Lebeche D, Kaminer B (1989) A calsequestrin-like protein in the endoplasmic reticulum of the sea urchin: localization and dynamics in the egg and first cell cycle embryo. J Cell Biol 109(1):149–161
Herzog A, Szegedi C, Jona I, Herberg FW, Varsanyi M (2000) Surface plasmon resonance studies prove the interaction of skeletal muscle sarcoplasmic reticular Ca2+ release channel/ryanodine receptor with calsequestrin. FEBS Lett 472:73–77
Houle TD, Ram ML, Cala SE (2004) Calsequestrin mutant D307H exhibits depressed binding to its protein targets and a depressed response to calcium. Cardiovasc Res 64(2):227–233
Houle TD, Ram ML, McMurray WJ, Cala SE (2006) Different endoplasmic reticulum trafficking and processing pathways for calsequestrin (CSQ) and epitope-tagged CSQ. Exp Cell Res 312(20):4150–4161
Jones LR, Zhang L, Sanborn K, Jorgensen AO, Kelley J (1995) Purification, primary structure, and immunological characterization of the 26-kDa calsequestrin binding protein (junctin) from cardiac junctional sarcoplasmic reticulum. J Biol Chem 22(51):30787–30796
Jones LR, Suzuki YJ, Wang W, Kobayashi YM, Ramesh V, Franzini-Armstrong C, Cleemann L, Morad M (1998) Regulation of Ca2+ signaling in transgenic mouse cardiac myocytes overexpressing calsequestrin. J Clin Invest 1(7):1385–1385
Josephs K, Patel K, Janson CM, Montagna C, McDonald TV (2017) Compound heterozygous CASQ2 mutations and long-term course of catecholaminergic polymorphic ventricular tachycardia. Mol Genet Genom Med 5(6):788–794. https://doi.org/10.1002/mgg3.323
Kraeva N, Zvaritch E, Frodis W, Sizova O, Kraev A, MacLennan DH, Riazi S. (2013) CASQ1 gene is an unlikely candidate for malignant hyperthermia susceptibility in the North American population. Anesthesiology 118:344–349
Kalyanasundaram A, Bal NC, Franzini-Armstrong C, Knollmann BC, Periasamy M (2009) The calsequestrin mutation CASQ2D307H does not affect protein stability and targeting to the junctional sarcoplasmic reticulum but compromises its dynamic regulation of calcium buffering. J Biol Chem 29(5):3076–3083. https://doi.org/10.1074/jbc.M109.053892
Kawamura M, Ohno S, Naiki N, Nagaoka I, Dochi K, Wang Q, Hasegawa K, Kimura H, Miyamoto A, Mizusawa Y, Itoh H, Makiyama T, Sumitomo N, Ushinohama H, Oyama K, Murakoshi N, Aonuma K, Horigome H, Honda T, Yoshinaga M, Ito M, Horie M (2013) Genetic background of catecholaminergic polymorphic ventricular tachycardia in Japan. Circ J 77(7):1705–1713
Kawasaki T, Kasai M (1994) Regulation of calcium channel in sarcoplasmic reticulum by calsequestrin. Biochem Biophys Res Commun 30(3):1120–1127
Kim E, Youn B, Kemper L, Campbell C, Milting H, Varsanyi M, Kang C (2007) Characterization of human cardiac calsequestrin and its deleterious mutants. J Mol Biol 2(4):1047–1057
Kirchhefer U, Wehrmeister D, Postma AV, Pohlentz G, Mormann M, Kucerova D, Müller FU, Schmitz W, Schulze-Bahr E, Wilde AA, Neumann J (2010) The human CASQ2 mutation K206N is associated with hyperglycosylation and altered cellular calcium handling. J Mol Cell Cardiol 49(1):95–105. https://doi.org/10.1016/j.yjmcc.2010.03.006
Knollmann BC, Chopra N, Hlaing T, Akin B, Yang T, Ettensohn K, Knollmann BE, Horton KD, Weissman NJ, Holinstat I, Zhang W, Roden DM, Jones LR, Franzini-Armstrong C, Pfeifer K (2006) Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2 + release, and catecholaminergic polymorphic ventricular tachycardia. J Clin Invest 116(9):2510–2520
Krause KH, Chou M, Thomas MA, Sjolund RD, Campbell KP (1989) Plant cells contain calsequestrin. J Biol Chem 15(8):4269–4272
Kobayashi YM, Alseikhan BA, Jones LR (2000) Localization and characterization of the calsequestrin-binding domain of triadin 1. Evidence for a charged beta-strand in mediating the protein-protein interaction. J Biol Chem 275(23):17639–17646
Kumar A, Chakravarty H, Bal NC, Balaraju T, Jena N, Misra G, Bal C, Pieroni E, Periasamy M, Sharon A (2013) Identification of calcium binding sites on calsequestrin 1 and their implications for polymerization. Mol Biosyst 9(7):1949–1957. https://doi.org/10.1039/c3mb25588c
Lacruz RS, Feske S (2015) Diseases caused by mutations in ORAI1 and STIM1. Ann N Y Acad Sci 1356:45–79
Lahat H, Pras E, Olender T, Avidan N, Ben-Asher E, Man O, Levy-Nissenbaum E, Khoury A, Lorber A, Goldman B, Lancet D, Eldar M (2001) A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am J Hum Genet 69(6):1378–1384
Lahat H, Pras E, Eldar M (2003) RYR2 and CASQ2 mutations in patients suffering from catecholaminergic polymorphic ventricular tachycardia. Circulation 28(3):e29
Lahat H, Pras E, Eldar M (2004) A missense mutation in CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Ann Med 36(Suppl 1):87–91
Launikonis BS, Ríos E (2007) Store-operated Ca2+ entry during intracellular Ca2+ release in mammalian skeletal muscle. J Physiol 583(1):81–97
Launikonis BS, Murphy RM, Edwards JN (2010) Toward the roles of store-operated Ca2+ entry in skeletal muscle. Pflug Arch 460(5):813–823
Lee JM, Noguchi S (2016) Calcium dyshomeostasis in tubular aggregate myopathy. Int J Mol Sci 22(11):e1952
Lewis KM, Ronish LA, Ríos E, Kang C (2015) Characterization of two human skeletal calsequestrin mutants implicated in malignant hyperthermia and vacuolar aggregate myopathy. J Biol Chem 290(48):28665–28674. https://doi.org/10.1074/jbc.M115.686261
Lunz V, Romanin C, Frischauf I (2019) STIM1 activation of Orai1. Cell Calcium 77:29–38
Ma J, Pan Z (2003) Retrograde activation of store-operated calcium channel. Cell Calcium 33(5–6):375–384
MacLennan DH, Wong PTS (1971) Isolation of a calcium-sequestering protein from sarcoplasmic reticulum. Proc Natl Acad Sci 68(6):1231–1235
Maguire PB, Briggs FN, Lennon NJ, Ohlendieck K (1997) Oligomerization is an intrinsic property of calsequestrin in normal and transformed skeletal muscle. Biochem Biophys Res Commun 26(3):721–727
Maguire PB, Lennon NJ, Ohlendieck K (1998) Oligomerisation of calsequestrin from rabbit skeletal muscle. Biochem Soc Trans 26(3):S292
Maizels L, Huber I, Arbel G, Tijsen AT, Gepstein A, Khoury A, Gepstein L (2017) Patient-specific drug screening using a human induced pluripotent stem cell model of catecholaminergic polymorphic ventricular tachycardia type 2. Circ Arrhythm Electrophysiol 10:e004725
Manno C, Figueroa LC, Gillespie D, Fitts R, Kang C, Franzini-Armstrong C, Rios E (2017) Calsequestrin depolymerizes when calcium is depleted in the sarcoplasmic reticulum of working muscle. Proc Natl Acad Sci USA 114(4):E638–E647
McFarland TP, Sleiman NH, Yaeger DB, Cala SE (2011) The cytosolic protein kinase CK2 phosphorylates cardiac calsequestrin in intact cells. Mol Cell Biochem 353(1–2):81–91. https://doi.org/10.1007/s11010-011-0777-6
Michelucci A, Paolini C, Canato M, Wei-Lapierre L, Pietrangelo L, De Marco A, Reggiani C, Dirksen RT, Protasi F (2015) Anti-oxidants protects calsequestrin-1 knockout mice from halothane- and heat- induced sudden death. Anesthesiology 123(3):603–617. https://doi.org/10.1097/ALN.0000000000000748
Michelucci A, Paolini C, Boncompagni S, Canato M, Reggiani C, Protasi F (2017a) Strenuous exercise triggers a life-threatening response in mice susceptible to Malignant Hyperthermia. FASEB J 31(8):3649–3662. https://doi.org/10.1096/fj.201601292R
Michelucci A, De Marco A, Guarnier F, Protasi F, Boncompagni S (2017b) Antioxidant treatment reduces formation of structural cores and improves muscle function in RYR1Y522S/WT mice. Oxid Med Cell Longev 2017:6792694. https://doi.org/10.1155/2017/6792694
Michelucci A, García-Castañeda M, Boncompagni S, Dirksen RT (2018) Role of STIM1/ORAI1-mediated store-operated Ca2+ entry in skeletal muscle physiology and disease. Cell Calcium 76:101–115
Michelucci A, Boncompagni S, Pietrangelo L, García-Castañeda M, Takano T, Malik S, Dirksen RT, Protasi F (2019) Transverse tubule remodeling enhances Orai1-dependent Ca2+ entry in skeletal muscle. Elife 28(8):e47576. https://doi.org/10.7554/eLife.47576
Mitchell RD, Simmerman HK, Jones LR (1988) Ca2+ binding effects on protein conformation and protein interactions of canine cardiac calsequestrin. J Biol Chem 25(3):1376–1381
Nesin V, Wiley G, Kousi M, Ong EC, Lehmann T, Nicholl DJ, Suri M, Shahrizaila N, Katsanis N, Gaffney PM, Wierenga KJ, Tsiokas L (2014) Activating mutations in STIM1 and ORAI1 cause overlapping syndromes of tubular myopathy and congenital miosis. Proc Natl Acad Sci USA 111(11):4197–4202
Nori A, Gola E, Tosato S, Cantini M, Volpe P (1999) Targeting of calsequestrin to sarcoplasmic reticulum after deletions of its acidic carboxy terminus. Am J Physiol 277(5):C974–C981. https://doi.org/10.1152/ajpcell.1999.277.5.C974
Nori A, Furlan S, Patiri F, Cantini M, Volpe P (2000) Site-directed mutagenesis and deletion of three phosphorylation sites of calsequestrin of skeletal muscle sarcoplasmic reticulum. Effects on intracellular targeting. Exp Cell Res 10(1):40–49
Nori A, Valle G, Massimino ML, Volpe P (2001) Targeting of calsequestrin to the sarcoplasmic reticulum of skeletal muscle upon deletion of its glycosylation site. Exp Cell Res 15(1):104 –104
Nori A, Bortoloso E, Frasson F, Valle G, Volpe P (2004) Vesicle budding from endoplasmic reticulum is involved in calsequestrin routing to sarcoplasmic reticulum of skeletal muscles. Biochem J 15(Pt 2):505–512
Nori A, Valle G, Bortoloso E, Turcato F, Volpe P (2006) Calsequestrin targeting to sarcoplasmic reticulum of skeletal muscle fibers. Am J Physiol Cell Physiol 291(2):C245–C253
Novak A, Barad L, Zeevi-Levin N, Shick R, Shtrichman R, Lorber A, Itskovitz-Eldor J, Binah O (2012) Cardiomyocytes generated from CPVT D307H patients are arrhythmogenic in response to β-adrenergic stimulation. J Cell Mol Med 16(3):468–482. https://doi.org/10.1111/j.1582-4934.2011.01476.x
Oddoux S, Brocard J, Schweitzer A, Szentesi P, Giannesini B, Brocard J, Fauré J, Pernet-Gallay K, Bendahan D, Lunardi J, Csernoch L, Marty I (2009) Triadin deletion induces impaired skeletal muscle function. J Biol Chem 11(50):34918–34929. https://doi.org/10.1074/jbc.M109.022442
Ohkura M, Furukawa K, Fujimori H, Kuruma A, Kawano S, Hiraoka M, Kuniyasu A, Nakayama H, Ohizumi Y (1998) Dual regulation of the skeletal muscle ryanodine receptor by triadin and calsequestrin. Biochemistry 15(37):12987–12993
Okuma H, Saito F, Mitsui J, Hara Y, Hatanaka Y, Ikeda M, Shimizu T, Matsumura K, Shimizu J, Tsuji S, Sonoo M (2016) Tubular aggregate myopathy caused by a novel mutation in the cytoplasmic domain of STIM1. Neurol Genet 2(1):e50
Pan Z, Brotto M, Ma J (2014) Store-operated Ca2+ entry in muscle physiology and diseases. BMB Rep 47(2):69–79
Paolini C, Quarta M, Nori A, Boncompagni S, Canato M, Volpe P, Allen PD, Reggiani C, Protasi F (2007) Reorganized stores and impaired calcium handling in skeletal muscle of mice lacking calsequestrin-1. J Physiol 583(Pt 2):767–784
Paolini C, Quarta M, D’Onofrio L, Reggiani C, Protasi F (2011) Differential effect of calsequestrin ablation on structure and function of fast and slow skeletal muscle fibers. J Biomed Biotechnol. https://doi.org/10.1155/2011/634075
Pape PC, Fénelon K, Lamboley CR, Stachura D (2007) Role of calsequestrin evaluated from changes in free and total calcium concentrations in the sarcoplasmic reticulum of frog cut skeletal muscle fibres. J Physiol 15:319–367
Park H, Wu S, Dunker AK, Kang C (2003) Polymerization of calsequestrin. Implications for Ca2+ regulation. J Biol Chem 2(18):16176–16182
Park H, Park IY, Kim E, Youn B, Fields K, Dunker AK, Kang C (2004) Comparing skeletal and cardiac calsequestrin structures and their calcium binding: a proposed mechanism for coupled calcium binding and protein polymerization. J Biol Chem 23(17):18026–18033
Perni S, Close M, Franzini-Armstrong C (2013) Novel details of calsequestrin gel conformation in situ. J Biol Chem 25(43):31358–31362. https://doi.org/10.1074/jbc.M113.507749
Postma AV, Denjoy I, Hoorntje TM, Lupoglazoff JM, Da Costa A, Sebillon P, Mannens MM, Wilde AA, Guicheney P (2002) Absence of calsequestrin 2 causes severe forms of catecholaminergic polymorphic ventricular tachycardia. Circ Res 91(8):e21–26
Protasi F, Paolini C, Dainese M (2009) Calsequestrin-1: a new candidate gene for malignant hyperthermia (MH) and environmental heat stroke (EHS). J Physiol 587:3095–3100. https://doi.org/10.1113/jphysiol.2009.171967
Protasi F, Paolini C, Canato M, Reggiani C, Quarta M (2011) Lessons from calsequestrin-1 ablation in vivo: much more than a Ca(2+) buffer after all. J Muscle Res Cell Motil 32(4–5):257–720. https://doi.org/10.1007/s10974-011-9277-2
Qin J, Valle G, Nani A, Nori A, Rizzi N, Priori SG, Volpe P, Fill M (2008) Luminal Ca2+ regulation of single cardiac ryanodine receptors: insights provided by calsequestrin and its mutants. J Gen Physiol 131(4):325-325 34. https://doi.org/10.1085/jgp.200709907
Qin J, Valle G, Nani A, Chen H, Ramos-Franco J, Nori A, Volpe P, Fill M (2009) Ryanodine receptor luminal Ca2+ regulation: swapping calsequestrin and channel isoforms. Biophys J 7(7):1961–1970. https://doi.org/10.1016/j.bpj.2009.07.030
Ram ML, Kiarash A, Marsh JD, Cala SE (2004) Phosphorylation and dephosphorylation of calsequestrin on CK2-sensitive sites in heart. Mol Cell Biochem 266(1–2):209–217
Rani S, Park CS, Sreenivasaiah PK, Kim DH (2016) Characterization of Ca(2+)-dependent protein-protein interactions within the Ca(2+) release units of cardiac sarcoplasmic reticulum. Mol Cells 39(2):149–515. https://doi.org/10.14348/molcells.2016.2284
Rossi D, Bencini C, Maritati M, Benini F, Lorenzini S, Pierantozzi E, Scarcella AM, Paolini C, Protasi F, Sorrentino V (2014a) Distinct regions of triadin are required for targeting and retention at the junctional domain of the sarcoplasmic reticulum. Biochem J 458(2):407–17. https://doi.org/10.1042/BJ20130719
Rossi D, Vezzani B, Galli L, Paolini C, Toniolo L, Pierantozzi E, Spinozzi S, Barone V, Pegoraro E, Bello L, Cenacchi G, Vattemi G, Tomelleri G, Ricci G, Siciliano G, Protasi F, Reggiani C, Sorrentino V (2014b) A mutation in the gene causes a vacuolar myopathy with accumulation of sarcoplasmic reticulum protein aggregates. Hum Mutat 35(10):1163–1170
Royer L, Ríos E (2009) Deconstructing calsequestrin. Complex buffering in the calcium store of skeletal muscle. J Physiol 587(Pt 13):3101–3111. https://doi.org/10.1113/jphysiol.2009.171934
Sacchetto R, Volpe P, Damiani E, Margreth A (1993) Postnatal development of rabbit fast-twitch skeletal muscle: accumulation, isoform transition and fibre distribution of calsequestrin. J Muscle Res Cell Motil 14(6):646–653
Sanchez EJ, Munske GR, Criswell A, Milting H, Dunker AK, Kang C (2011) Phosphorylation of human calsequestrin: implications for calcium regulation. Mol Cell Biochem 353(1–2):195–204. https://doi.org/10.1007/s11010-011-0787-4
Sanchez EJ, Lewis KM, Danna BR, Kang C (2012a) High-capacity Ca2+ binding of human skeletal calsequestrin. J Biol Chem 30(14):11592–11601. https://doi.org/10.1074/jbc.M111.335075
Sanchez EJ, Lewis KM, Munske GR, Nissen MS, Kang C (2012b) Glycosylation of skeletal calsequestrin: implications for its function. J Biol Chem 27(5):3042–3050. https://doi.org/10.1074/jbc.M111.326363
Sato Y, Ferguson DG, Sako H, Dorn GW II, Kadambi VJ, Yatani A, Hoit BD, Walsh RA, Kranias EG (1998) Cardiac-specific overexpression of mouse cardiac calsequestrin is associated with depressed cardiovascular function and hypertrophy in transgenic mice. J Biol Chem. 273(43):28470–28477
Schmidt AG, Kadambi VJ, Ball N, Sato Y, Walsh RA, Kranias EG, Hoit BD (2000) Cardiac-specific overexpression of calsequestrin results in left ventricular hypertrophy, depressed force-frequency relation and pulsus alternans in vivo. J Mol Cell Cardiol 32(9):1735–1744
Shin DW, Ma J, Kim DH (2000) The asp-rich region at the carboxyl-terminus of calsequestrin binds to Ca(2+) and interacts with triadin. FEBS Lett 8(2):178–182
Shin DW, Pan Z, Kim EK, Lee JM, Bhat MB, Parness J, Kim DH, Ma J (2003) A retrograde signal from calsequestrin for the regulation of store-operated Ca2+ entry in skeletal muscle. J Biol Chem 278(5):3286–3292
Sparsø T, Hussain MS, Borch-Johnsen K, Jørgensen T, Madsbad S, Hansen T, Pedersen O, Andersen G (2007) Studies of association of the CASQ1 rs2275703 polymorphism in relation to type 2 diabetes and related quantitative metabolic traits among 7088 Danish whites. Mol Genet Metab 92(3):278-278 82
Stiber JA, Hawkins A, Zhang Z-S, Wang S, Burch J, Graham V, Ward CC, Seth M, Finch E, Malouf N, Williams RS, Eu JP, Rosenberg P (2008) STIM1 signaling controls store operated calcium entry required for development and contractile function in skeletal muscle. Nat Cell Biol 10(6):688–697
Stiber JA, Rosenberg PB (2011) The role of store-operated calcium influx in skeletal muscle signaling. Cell Calcium 49(5):341–349
Sztretye M, Yi J, Figueroa L, Zhou J, Royer L, Allen P, Brum G, Ríos E (2011) Measurement of RyR permeability reveals a role of calsequestrin in termination of SR Ca2+ release in skeletal muscle. J Gen Physiol 138(2):231–247. https://doi.org/10.1085/jgp.201010592
Terentyev D, Nori A, Santoro M, Viatchenko-Karpinski S, Kubalova Z, Gyorke I, Terentyeva R, Vedamoorthyrao S, Blom NA, Valle G, Napolitano C, Williams SC, Volpe P, Priori SG, Gyorke S (2006) Abnormal interactions of calsequestrin with the ryanodine receptor calcium release channel complex linked to exercise-induced sudden cardiac death. Circ Res 98(9):1151–1158
Thomas K, Navarro J, Benson RJ, Campbell KP, Rotundo RL, Fine RE (1989) Newly synthesized calsequestrin, destined for the sarcoplasmic reticulum, is contained in early/intermediate Golgi-derived clathrin-coated vesicles. J Biol Chem 25(6):3140–3145
Tijskens P, Jones LR, Franzini-Armstrong C (2003) Junctin and calsequestrin overexpression in cardiac muscle: the role of junctin and the synthetic and delivery pathways for the two proteins. J Mol Cell Cardiol 35(8):961–974
Tomasi M, Canato M, Paolini C, Dainese M, Reggiani C, Volpe P, Protasi F, Nori A (2012) Calsequestrin (CASQ1) rescues function and structure of calcium release units in skeletal muscles of CASQ1-null mice. Am J Physiol Cell Physiol 1(3):C575–C586. https://doi.org/10.1152/ajpcell.00119.2011
Tomelleri G, Palmucci L, Tonin P, Mongini T, Marini M, L’erario R, Rizzuto N, Vattemi G (2006) SERCA1 and calsequestrin storage myopathy: a new surplus protein myopathy. Brain 129(8):2085–2092
Viatchenko-Karpinski S, Terentyev D, Györke I, Terentyeva R, Volpe P, Priori SG, Napolitano C, Nori A, Williams SC, Györke S (2004) Abnormal calcium signaling and sudden cardiac death associated with mutation of calsequestrin. Circ Res 5(4):471–477
Volpe P, Simon BJ (1991) The bulk of Ca2+ released to the myoplasm is free in the sarcoplasmic reticulum and does not unbind from calsequestrin. FEBS Lett 28(2):274–278
Wang S, Trumble WR, Liao H, Wesson CR, Dunker AK, Kang CH (1998) Crystal structure of calsequestrin from rabbit skeletal muscle sarcoplasmic reticulum. Nat Struct Biol 5(6):476–476
Wang W, Cleemann L, Jones LR, Morad M (2000) Modulation of focal and global Ca2+ release in calsequestrin-overexpressing mouse cardiomyocytes. J Physiol 15(524 Pt 2):399–414
Wang Y, Xu L, Duan H, Pasek DA, Eu JP, Meissner G (2006) Knocking down type 2 but not type 1 calsequestrin reduces calcium sequestration and release in C2C12 skeletal muscle myotubes. J Biol Chem 2(22):15572–15581
Wang L, Zhang L, Li S, Zheng Y, Yan X, Chen M, Wang H, Putney JW, Luo D (2015) Retrograde regulation of STIM1-Orai1 interaction and store-operated Ca2+ entry by calsequestrin. Sci Rep 18(5):11349. https://doi.org/10.1038/srep11349
Wang Q, Groenendyk J, Paskevicius T, Qin W, Kor KC, Liu Y, Hiess F, Knollmann BC, Wayne Chen SR, Tang J, Chen X, Agellon LB, Michalak M (2019) Two pools of IRE1a in cardiac and skeletal muscle cells. FASEB J 33:8892–8904
Wei L, Varsányi M, Dulhunty AF, Beard NA (2006) The conformation of calsequestrin determines its ability to regulate skeletal ryanodine receptors. Biophys J 15(4):1288–1301
Wei L, Hanna AD, Beard NA, Dulhunty AF (2009a) Unique isoform-specific properties of calsequestrin in the heart and skeletal muscle. Cell Calcium 45(5):474–484. https://doi.org/10.1016/j.ceca.2009.03.006
Wei L, Gallant EM, Dulhunty AF, Beard NA (2009b) Junctin and triadin each activate skeletal ryanodine receptors but junctin alone mediates functional interactions with calsequestrin. Int J Biochem Cell Biol 41(11):2214–2224. https://doi.org/10.1016/j.biocel.2009.04.017
Zima AV, Bovo E, Bers DM, Blatter LA (2010) Ca²+ spark-dependent and -independent sarcoplasmic reticulum Ca²+ leak in normal and failing rabbit ventricular myocytes. J Physiol 588:4743–4757. https://doi.org/10.1113/jphysiol.2010.197913
Zhang L, Kelley J, Schmeisser G, Kobayashi YM, Jones LR (1997) Complex formation between junctin, triadin, calsequestrin, and the ryanodine receptor. Proteins of the cardiac junctional sarcoplasmic reticulum membrane. J Biol Chem 12(37):23389–23397
Zhang L, Wang L, Li S, Xue J, Luo D (2016) Calsequestrin-1 regulates store-operated Ca2+ entry by inhibiting STIM1 aggregation. Cell Physiol Biochem 8(6):2183–2193. https://doi.org/10.1159/000445574
Zhao X, Min CK, Ko JK, Parness J, Kim DH, Weisleder N, Ma J (2010) Increased store-operated Ca2+ entry in skeletal muscle with reduced calsequestrin-1 expression. Biophys J 99(5):1556–1564. https://doi.org/10.1016/j.bpj.2010.06.050
Funding
Funding was provided by Telethon Grant No. GGP19231A and PRIN 2015 Grant No. 2015ZZR4W3 to VS.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare not to have conflicts of interest or competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Rossi, D., Gamberucci, A., Pierantozzi, E. et al. Calsequestrin, a key protein in striated muscle health and disease. J Muscle Res Cell Motil 42, 267–279 (2021). https://doi.org/10.1007/s10974-020-09583-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10974-020-09583-6