
Abstract
Evidence obtained in the last two decades indicates that calsequestrin (CSQ2), as the major Ca2+-binding protein in the sarcoplasmic reticulum of cardiac myocytes, communicates changes in the luminal Ca2+ concentration to the cardiac ryanodine receptor (RYR2) channel. This review summarizes the major aspects in the interaction between CSQ2 and the RYR2 channel. The single channel properties of RYR2 channels, discussed here in the context of structural changes in CSQ2 after Ca2+ binding, are particularly important. We focus on five important questions concerning: (1) the method for reliable detection of CSQ2 on the reconstituted RYR2 channel complex; (2) the power of the procedure to strip CSQ2 from the RYR2 channel complex; (3) structural changes in CSQ2 upon binding of Ca2+ which cause CSQ2 dissociation; (4) the potential role of CSQ2-independent regulation of the RYR2 activity by luminal Ca2+; and (5) the vizualization of CSQ2 dissociation from the RYR2 channel complex on the single channel level. We discuss the potential sources of the conflicting experimental results which may aid detailed understanding of the CSQ2 regulatory role. Although we mainly focus on the cardiac isoform of the proteins, some aspects of more extensive work carried out on the skeletal isoform are also discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
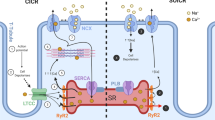
The cardiac ryanodine receptor (RYR2) channel plays a key role in excitation–contraction (EC) coupling in the heart, providing the pathway for release of Ca2+ from the sarcoplasmic reticulum (SR) required for activation of cardiac contraction. It is becoming increasingly evident that the Ca2+ release by RYR2 channels is governed not only by cytosolic Ca2+ but also by Ca2+ in the lumen of the SR (luminal Ca2+). The first evidence of a regulatory role of luminal Ca2+ was reported by Fabiato and Fabiato [1, 2]. They showed that the magnitude of Ca2+ release was scaled by the Ca2+ loading of the SR. These findings are easily explained by an increase in the Ca2+ gradient across the SR membrane; and thus, increased amplitude in the Ca2+ current. However, studies on muscle cells and isolated vesicles have revealed that the rate of Ca2+ release was increased by luminal Ca2+ too steeply (non-linearly) to result merely from the enhanced Ca2+ current (for review, see [3, 4]). Therefore, the additional mechanism that obviously comes into play has been debated; and the regulation of the RYR2 channel by luminal Ca2+ appears the most feasible explanation. Indeed, experiments on the RYR2 channel reconstituted in the planar lipid membrane (BLM) revealed that luminal Ca2+ significantly enhanced the channel activity [5–11]. Considering the fact that Ca2+ could permeate the channel, the observed effect of luminal Ca2+ was interpreted by two principal mechanisms. The “true luminal” model attributed luminal regulation to the Ca2+ binding sites located on the luminal face of the RYR2 channel, or on associated proteins; whereas, the “feed-through” model proposed that luminal Ca2+ passes the channel pore in the lumen-to-cytosol direction and binds to Ca2+ sites located on the cytoplasmic channel face. Although the pioneering single channel studies selectively supported only either one of these suggested mechanisms; recently Laver [12–14] has developed a hybrid model where interplay between the “true luminal” and “feed-through” mechanisms has been proposed (Fig. 1). The molecular nature of a luminal Ca2+ sensor of the RYR2 channel has been extensively studied in the last two decades, and currently, it is accepted that calsequestrin (CSQ), as one of the major Ca2+-binding proteins in the SR, could play this role. CSQ was first identified in the early 1970s in skeletal muscle (CSQ1) [15, 16] and then later in cardiac muscle (CSQ2) [17]. The CSQ2 is the solely expressed isoform in the cardiac muscle [18] and has been detected as filamentous matrices anchored to the junctional face membrane in the vicinity of the RYR2 channel [19–21]. Thus, it is feasible that the CSQ2 proximity to the RYR2 channel ensures that high Ca2+ concentrations are stored quite close to the site of Ca2+ release [22]. CSQ2 interacts with the RYR2 channel either directly [23, 24] or via scaffolding proteins such as triadin and junctin [20]. Both of these proteins span the SR membrane and bind by their luminal domains to both CSQ2 [25, 26] and the RYR2 channel [20]. In addition, triadin and junctin also interact with each other, forming a quaternary complex between themselves, the RYR2 channel and CSQ2 [20] (Fig. 1). As a low-affinity and high-capacity Ca2+ binding protein, CSQ2 was originally thought to play a role as a Ca2+ storage site in the SR, however, extensive experimental evidence has shown that the role of CSQ2 extends well beyond that of a simple Ca2+ buffer. It is well recognized that Ca2+ in the lumen of the SR extensively regulates the RYR2 activity, and as CSQ2 is Ca2+ dependent and localized in the vicinity of this channel, it has been suggested to be its putative luminal Ca2+ sensor [9, 10, 20, 27]. This new regulatory role of CSQ2 was originally proposed by Ikemoto et al. [28], who suggested that the binding of Ca2+ to CSQ produces a conformational change in this protein, which subsequently causes a conformational change in the RYR channel leading to variations in SR Ca2+ release. This hypothesis has recently been refined, since it was found that Ca2+ predominantly controls binding between CSQ2 and triadin/junctin by weakening the electrostatic interactions [9, 20]; and thus, CSQ2-triadin/junctin interaction is thought to provide a molecular basis for regulation of the RYR2 channel by luminal Ca2+ [27, 29]. The question of whether triadin and junctin equally transmit the signal from CSQ2 to the RYR2 channel is still unresolved. Some studies have concluded that the CSQ2-triadin binding is essential for regulation of the RYR2 channel by luminal Ca2+ [9, 30]; however, direct evidence from the BLM experiments which can reliably separate triadin and junctin roles in the CSQ2-RYR2 functional interaction is still lacking. It is noteworthy that this type of study was performed on the skeletal isoform of the RYR channel (RYR1), and therein it was clearly shown that junctin alone, but not triadin, mediated CSQ1 inhibition of the reconstituted RYR1 channel [31]. As further support for the role of CSQ2 as a Ca2+ sensor, it has been shown that mutations in CSQ2 are equally deleterious as mutations in the RYR2 channel in causing catecholaminergic polymorphic ventricular tachycardia (CPVT) by changing the RYR2 sensitivity to luminal Ca2+ [32–35]. CVPT is a familial disorder characterized by adrenergically mediated arrhythmias which often lead to sudden cardiac failure and death (reviewed by [36]).

Proposed mechanisms of the RYR2 regulation by luminal Ca2+. The first (a), “true-luminal” model proposes the existence of luminally located Ca2+-binding activation sites on the RYR2 channel, or on associated proteins such as calsequestrin (CSQ2) that could play the role of a luminal Ca2+ sensor. CSQ2 is anchored in the vicinity of the RYR2 channel by two membrane proteins: junctin (J) and triadin (T). The second (b), “feed-through” model goes without Ca2+ binding sites on the luminal domain and explains the activation effect of luminal Ca2+ by binding of Ca2+ ions which permeate from the SR lumen to Ca2+ sites located on the cytosolic face of the RYR2 channel. The third (c), “luminal triggered Ca2+ feed-through” model combines both the previously suggested mechanisms into one. This model suggests that both luminal and cytosolic Ca2+ sites mediate the channel activation, and that these sites are functionally coupled by the flow of Ca2+ from the SR lumen. T—triadin, J—junctin, CSQ2 cardiac calsequestrin, red dot—Ca2+ ion, SR lumen —lumen of sarcoplasmic reticulum, RYR2—cardiac ryanodine receptor
Here we provide a brief overview of what we know, and do not know, about the functional role of CSQ2 in the sensitization of the RYR2 channel to luminal Ca2+. The single channel properties of RYR2 channels are of particular importance, and are discussed in the context of structural changes in CSQ2 following Ca2+ binding. Our aim is to highlight current controversies in this field and endeavour to identify further ones. Discussion of potential sources of conflicting results will be beneficial in detailed understanding of the role CSQ2 plays in the responsiveness of the RYR2 channel to luminal Ca2+. Herein, we focus on the cardiac isoform of the proteins; however, due to more specific work done on the skeletal isoform, we also discuss some aspects of this isoform.
In our review, we focus on five inter-related questions concerning the CSQ2-RYR2 interaction which can potentially form the basis for future studies:
-
1.
How can the presence of CSQ2 on the RYR2 channel be reliably assessed in BLM experiments?
-
2.
How effectively can we strip CSQ2 from the RYR2 channel complex?
-
3.
What structural changes in CSQ2 cause the dissociation following Ca2+ binding?
-
4.
What is the role of CSQ2-independent regulation of the RYR2 channel by luminal Ca2+?
-
5.
How can the CSQ2 dissociation process be visualized on the single channel level?
Unresolved questions
How can the presence of CSQ2 on the RYR2 channel be reliably assessed in BLM experiments?
A biochemical approach has documented that CSQ2 can be completely released from the RYR2 channel complex by a high concentration of luminal Ca2+ ([Ca2+]L) [20]. Although a similar situation has been found for the CSQ1-RYR1 complex [37, 38], here increased ionic strength was defined as an additional effective CSQ stripping factor [37]. Therefore, CSQ2 dissociation from the RYR2 channel complex can easily occur when either [Ca2+]L is widely manipulated during BLM experiments or the ionic strength of solutions used to isolate SR crude microsomes is substantially increased. This can bias interpretation of experimental results, and several groups have documented that regulation of the RYR2 channel by luminal Ca2+ is considerably influenced by CSQ2 [9, 10, 24, 27, 39]. Here, it is important to note that the SR crude microsomes isolated by the standard procedure with moderate ionic strength of isolation buffers contain a detectable amount of CSQ2, and this has been confirmed in several studies by Western blotting [8, 24, 40]. According to a recent study, this portion of CSQ2 represents only approximately 5 % of the total amount originally present in the ventricular tissue [40]. This indicates that CSQ2 found in the SR microsomes was most likely tightly bound to RYR2 channels. Importantly, this implies that the CSQ2-RYR2 functional interaction could be preserved in the SR crude microsomes, at least under some conditions. From this, one can assume that RYR2 channels could be incorporated into the BLM with associated CSQ2, and it is not inevitably to add this protein either as recombinant or purified in each BLM experiment to restore the natural CSQ2 status of reconstituted RYR2 channels. However, at this point we must again emphasize that CSQ2 can be lost during the isolation procedure or the BLM experiment whenever the appropriate, and so far not completely identified, conditions for CSQ2 stripping are met. Supporting this concept, Qin et al. [9] stated that in their hands a discrete population of control RYR2 channels did not initially have CSQ2 attached. This conclusion, however, was based on comparison between the activity of control RYR2 channels which were never exposed to the CSQ2 stripping procedure and those with added CSQ2 upon the stripping. This issue was also discussed in the work of Beard et al. [37] for the RYR1 channel. These authors argued that since a considerable number of RYR1 channels failed to respond functionally to the CSQ1 stripping procedure, they all lacked associated CSQ1. However, in both cases, the absence of CSQ was determined only intuitively without direct evidence. This clearly indicates that a reliable molecular tool for detection of CSQ2 on the particular RYR2 channel reconstituted in the BLM should be established. One promising, but still not ideal approach, which has been partly tested on the RYR1 channel is the effect of CSQ antibody on channel activity [37]. The antibody added to the luminal face of the RYR1 channel substantially reduced the channel activity, most likely by binding to attached CSQ1. This inhibition did not occur when channels were stripped of CSQ1 and this was observed in all cases when CSQ1 was reassociated upon the previous stripping. In our opinion, it is extremely worthwhile to also test this method on the RYR2 channel complex, because it is likely that binding of the antibody to CSQ2 complexed with the RYR2 channel will lead to similar changes in the channel activity. This method using a potential molecular probe to detect CSQ2 in the particular RYR2 channel complex should be further explored in sufficient detail, and the experimental conditions necessary to detect the effect on the channel activity must be strictly specified. Undoubtedly, this is critical for making an objective conclusion about the presence of CSQ2 in the RYR2 channel complex which will consequently eliminate conflicting interpretations of the experimental results on the regulation of the RYR2 channel by luminal Ca2+.
How effectively can we strip CSQ2 from the RYR2 channel complex?
For both skeletal and cardiac muscles, two common conditions under which CSQ completely dissociates from the RYR channel complex have been identified. The first is high [Ca2+]L and the second is increased ionic strength of solutions [20, 37, 38]. When the RYR2 channel complexes were only solubilized by detergent, a considerable fraction of CSQ2 remained associated with triadin and junctin at [Ca2+]L < 1 mM. Moreover, 20 mM [Ca2+]L was shown to sufficiently disrupt all CSQ2 protein-binding interactions [20]. This biochemical study provided direct evidence of the stripping power of high [Ca2+]L. However, interpretation of the outcomes requires ultimate caution when applying this approach to the BLM experiments. Specifically, the biochemical study did not monitor the time course of the stripping procedure, and the ionic composition of buffers used for biochemistry is not similar to that used for BLM experiments. All of these factors can easily modify the stripping power of high [Ca2+]L to an unknown extent. These conditions which could influence the CSQ-RYR interaction are certainly better explored in the CSQ1-RYR1 interaction, where the precise correlation between biochemical and BLM experiments has been determined [37, 38]. Specifically, [Ca2+]L ≥ 4 mM effectively dissociated CSQ1 from the RYR1 channel complex, and this therefore prevented its reassociation; whereas CSQ1 always remained attached in the range of 1–3 mM [Ca2+]L. The BLM experiments further, although indirectly, confirmed these results. The RYR1 channels were irreversibly activated after at least 3 min exposure to 5 mM [Ca2+]L and at least 9 min after exposure to 4 mM [Ca2+]L. In contrast, exposing the channels to 2 mM and 3 mM [Ca2+]L did not cause irreversible change in channel activity within the time frame of these experiments. Similar results were obtained when increased ionic strength of solution was used as the dominant stripping factor [37]. Here, the reconstituted RYR1 channels were visibly activated in 2–5 min when [Cs+]L was increased from 250 to 500 mM. For the RYR2 channel complex, efficacy of CSQ2 stripping by high [Ca2+]L or high ionic strength employed in several recent studies was tested only at the single channel level, and no biochemical evidence supporting the functional data was provided. It appears from these measurements that 5 mM [Ca2+]L [27, 41] or 500 mM [Cs+]L [24] were strong enough to release CSQ2 from the RYR2 channel complex incorporated in the BLM; however, the exposure to 10 mM instead of 5 mM [Ca2+]L within 10 min seemed a more reliable stripping method [9, 10]. The CSQ2 dissociation was attributed to irreversible changes in channel activity and this intuitive idea was further supported by adding CSQ2 to the RYR2 channel complex when [Ca2+]L was decreased to a level favouring CSQ2 reassociation. This manoeuvre completely reversed the effect of high [Ca2+]L and channel activity returned to its initial level. Combining results obtained for the RYR1 and RYR2 channels, it can be concluded that 5–10 mM [Ca2+]L or 500 mM [Cs+]L is sufficient to dissociate all CSQ from the channel complex. However, our recent results obtained from BLM experiments do not fully support this conclusion [11]. In order to correlate the functional effects of luminal Ca2+ (0.005, 1, 8, 15, 26, 53 mM) with the presence of CSQ2, we tested whether 8 mM [Ca2+]L removed CSQ2 from the RYR2 channel complex. On the basis of observations mentioned above, we suggested that 8 mM [Ca2+]L has only limited power to strip CSQ2 from all studied RYR2 channel complexes under our experimental conditions. It could be argued that some RYR2 channel complexes did not respond to the used stripping procedure because CSQ2 had been dissociated from the channel complexes during SR microsomes preparation. However, we can conclude that it is unlikely, because all studied channels were activated by cytosolic and luminal Ca2+ in a similar manner and no heterogeneity in channel properties was observed which otherwise could have pointed to a different CSQ status of studied RYR2 channels [9, 39]. In order to identify the source of this CSQ2 stripping power controversy, we compared the composition and the ionic strength of the solutions used. Specifically in our study, the ionic strength of the luminal solution was 216 mM and in other studies this varied from 29 to 380 mM. Therefore, it is unlikely that the ionic strength enhanced the power of luminal Ca2+ to dissociate CSQ2. Importantly, we found that in contrast to previous studies, we did not mix luminal Ca2+ with Cs+ to increase the signal/noise ratio. Whether Cs+ ions can boost the CSQ2 stripping process remains to be elucidated, but in this regard it may be beneficial to test if it is possible to strip residual CSQ2 with other metals such as Cu2+ or Mn2+ which have greater affinity than Ca2+, as discussed below.
Here, it is important to note that the third CSQ stripping condition has also been recognized. In this case, although CSQ1 binding to the RYR1 channel complex is eliminated by low [Ca2+]L of approximately 100 nM–100 μM because of CSQ1 depolymerization, a residual amount of CSQ1 in the monomeric state remains attached [24, 42]. This specific stripping method can reasonably be applied only to the RYR1 channel complex because CSQ1 is mostly polymerized [9, 42, 43], while CSQ2 is mostly monomeric at 1 mM [Ca2+]L and physiological ionic strength [24]. Furthermore, the RYR1 regulation by CSQ1 depends on its association with CSQ1 polymers and not with any residual monomers [24, 42], while the RYR2 channel appears to be predominantly regulated by monomeric CSQ2 [9, 24].
What structural changes in CSQ2 cause the dissociation following Ca2+ binding?
Structural changes in CSQ upon Ca2+ binding have been studied by several groups [16, 17, 44]. The discovery of CPVT mutations in CSQ2 has triggered many recent studies focused on CSQ2 structure but there are still many gaps in knowledge which need to be addressed [36, 45–47]. Since CSQ has a dual role as a Ca2+-buffer and modulator of RYR function, its structure and metal binding properties appear very closely intertwined. It has been suggested that CSQ2 undergoes polymerization just like the CSQ1 isoform although it requires higher [Ca2+] in vitro [43, 48–50]. Circular Dichroism (CD) studies show that at a very low [Ca2+] of <0.1 mM, CSQ2 has low CD ellipticity and it increases with increasing [Ca2+] [51]. This indicates that CSQ2 exists in a less structured, most likely a molten globular state up to ~0.1 mM [Ca2+] and at around ~0.3 mM [Ca2+] CSQ2 undergoes a key structural transition forming compact monomers ready to undergo polymerization with further increase in [Ca2+] beyond 1 mM. It has been proposed that with increasing [Ca2+], CSQ2 monomers first form a front-to-front dimer and then two such dimers undergo back-to-back stacking leading to a polymeric structure [43, 48, 49, 52, 53]. The physiological role of these Ca2+-mediated structural changes is further supported by the fact that CPVT mutations affect CSQ2-Ca2+ interaction and its polymerization/aggregation properties [48, 49, 54–56]. The unique structural properties of CSQ2 which could have implications for RYR2 function are discussed below.
Sites for interaction with Ca2+ and RYR2 channel on CSQ2
The CSQ class of proteins is distinct in its Ca2+ interaction as they lack any rigid Ca2+-binding motif such as EF-hand [52] or double-clamp [57]. Recent structural studies have shown that Ca2+ binds to the acidic residues on the surface without any uniform Ca2+-binding geometry, and this varies depending on local structure [43, 50]. Such Ca2+-binding mode with low affinity makes dissociation suitable to facilitate Ca2+-release during contraction. However, the exact role of the unique highly acidic C-terminus composed of aspartate and glutamate residues is not currently known, as the X-ray crystal structures published to date do not include this region [50, 52]. The highly acidic C-terminus might play a key role in either Ca2+-binding/release or interaction with the RYR2 channel complex. Another important structural feature which has not been clarified is monomeric compaction. Although structural data from various groups has indicated that CSQ2 monomers undergo a key structural change with increasing [Ca2+] before their 3-dimensional conformation becomes favourable for polymerization [17, 48, 54, 56], detailed structural analysis is still lacking. This conformational transition is a Ca2+-mediated hydrophilic collapse occurring at ~300 μM Ca2+, which is in the physiological range of Ca2+ oscillation. This transition cannot be visualized by crystallography, but it could most likely be resolved by nuclear magnetic resonance (NMR) although NMR application to CSQ2 is very challenging due to its large size, aggregation/polymerization and dynamicity in the presence of metal ions. Furthermore, the site(s) on CSQ2 for its interaction with the RYR2 channel complex has not been clearly defined.
Physiological importance of CSQ2 polymerization and depolymerization
Few recent structural studies have indicated that cation mediated CSQ2 polymerization is dynamic and that CSQ2 can undergo reversible polymerization and depolymerization [43, 49, 58]. Furthermore, the critical role of CSQ2 in the dynamic buffering capacity of the cardiac SR is also supported by studies using the CSQ2 knockout mouse model [55, 59, 60]. In a recent study, Lee et al. [61] showed at the cellular level that CSQ2 undergoes dynamic polymerization-depolymerization and that this is influenced by junctin. In contrast, based on in vitro studies it has recently been suggested that CSQ2 polymers may not exist in normal hearts [40] and also only play a limited role in RYR2 function regulation. However, earlier EM studies suggest the presence of CSQ2 in dense thread-like bodies in the T-tubules [62–64] which can be formed only by CSQ2 polymers or aggregates. One of the major complications in understanding CSQ2 polymerization and depolymerization is that this occurs in vitro at high [Ca2+] (above 1.0 mM) [43, 49, 54]. However, this could indicate that dynamic polymerization of CSQ2 is important during higher physiological demand. As there is currently no clear consensus concerning [Ca2+] range (both free as well as bound) inside the cardiac SR, a physiological role for the CSQ2 polymer, or at least a tetramer or octamer or aggregate, cannot be ruled out. In addition, it is currently uncertain whether it is the monomer or polymer structural form of CSQ2 which interacts with the RYR2 channel complex. The Ca2+-dependent structural changes of CSQ2 are illustrated in the model in Fig. 2. If the function of CSQ2 is to keep Ca2+ in close proximity to the RYR2 channel complex for release, then the CSQ2 polymeric form with a high amount of bound Ca2+ should interact with the RYR2 channel complex. Alternatively, the benefit of CSQ2 monomer binding to the RYR2 channel complex may help to keep the RYR2 gate closed during the refractory period after Ca2+ release [65].

Proposed model of CSQ2 polymerization. The centre of each domain of the CSQ2 molecule is hydrophobic and must undergo hydrophobic collapse to form the domains. The core region of CSQ2 at the interface between the three domains is acidic and can only be neutralized by Ca2+ before the three domains can come together to form a compact monomer. The C-terminal acid rich region of CSQ2 is ~28 amino acids long and can most likely bind more than 15 Ca2+ ions (CSQ2 has the capacity to bind 40–50 Ca2+ per monomer). High affinity sites on the CSQ2 surface bind Ca2+ first, and then low affinity sites will bind Ca2+. The “Physiological range of Ca2+ oscillation” indicates the range in which CSQ2-RYR2 interaction may occur, and dotted lines indicate extreme Ca2+ concentrations that might be important only during higher physiological demand
Role of other metal ions in the CSQ2-RYR2 interaction
Structural changes in CSQ2 upon interaction with many cations other than Ca2+ have been reported by several groups [17, 48, 54, 56, 66]. Many cations such as Lanthanum (La2+), Cadmium (Cd2+), Manganese (Mn2+) and Zinc (Zn2+) interfere with Ca2+ binding to CSQ1, and this indicates that they may have higher affinity for cation-binding sites on CSQ1 than Ca2+ [16, 67]. Detailed study on the effect of various metal ions on CSQ2 structure is lacking. High concentrations ≥300 mM of monovalent metal ions such as K+ and Na+ can alter CSQ2 conformation at the monomer level but not lead to polymerization/aggregation [54, 56]. The Cs+ has been shown to affect CSQ1 structure, but its effect on CSQ2 has not yet been studied. Although divalent cations, including Mg2+, Ca2+, Sr2+, and Ba2+, can cause monomeric compaction of CSQ2, only Ca2+ can cause aggregation/polymerization of CSQ2 [56]. However, many polyvalent cations such as Mn2+, Ni2+, Cu2+, and Zn2+ can change both CSQ2 conformation at the monomer level and also cause aggregation/polymerization [56]. Although Cu2+ and Mn2+ are specifically unique causing CSQ2 aggregation/polymerization at very low concentrations [56], competition between Cu2+/Mn2+ and Ca2+ ions in causing structural changes in CSQ2 has not been studied. Here, it must be pointed out that metal ions which can cause CSQ2 aggregation/polymerization at lower concentrations than Ca2+ may be more powerful in stripping residual CSQ2 bound to the RYR2 channel complex in BLM experiments. Several studies have found that CPVT mutations alter Ca2+ selectivity and polymerization of CSQ2 [48, 54, 56]. The D307H and P308L mutations showed a biphasic aggregation pattern (wildtype CSQ2 is monophasic) with increasing [Ca2+] and in addition showed the sensitivity to increasing [Mg2+] [56]. The L167H and R33Q mutations largely impaired Ca2+ sensitivity, while they can aggregate by many transition metals including Cu2+ and Mn2+. The role of Ca2+ selectivity of CSQ2 under normal physiological conditions and in the above CPVT situation is currently not understood. Furthermore, the role of cations other than Ca2+ in the CSQ2-RYR2 interaction has not been investigated. This lack has most likely resulted from the fact that it has been generally assumed that the SR does not contain other cations, even though some studies have suggested that the SR may contain Mg2+, Zn2+, and Fe3+ [68–70].
CSQ2 may influence CSQ2-independent luminal Ca2+-mediated RYR2 function
Although many recent studies have shown that the modulation of RYR2 activity by luminal Ca2+ could be mediated by CSQ2 [9, 10, 27], it has been proposed that luminal Ca2+ can also regulate RYR2 function in a CSQ2-independent manner. However, it has to be pointed out that by virtue of its low affinity Ca2+-binding and rapid dissociation, CSQ2 can modulate luminal Ca2+. Therefore, the RYR2 function can be influenced; (1) directly by structural changes in CSQ2 and (2) indirectly by Ca2+ binding to CSQ2, thus altering free [Ca2+] inside the SR and modulating RYR2 function. Importantly, there is also a body of evidence, mostly derived from knockout and mutation studies, which implicates triadin and junctin in direct regulation of the RYR2 channel; thus, it seems that the roles of triadin and junctin are more complex, and not limited only to anchoring CSQ2 and transmitting signals from it [21, 31, 71–75]. Although only three studies were found which examined the RYR2-triadin/junctin functional interaction using the BLM technique, when summarized these showed that triadin and junctin added together or alone to the luminal face of the purified RYR2 channel activated it in a [Ca2+]L independent manner [27, 39]. Furthermore, the RYR2 channel derived from junctin null mice exhibited the altered sensitivity to luminal Ca2+, being more inhibited at [Ca2+]L < 1 mM and more activated at [Ca2+]L > 1 mM compared to the wild type RYR2 channel. This was explained by the direct activator-inhibitor action of junctin on the RYR2 channel with crossover at [Ca2+]L, similar to that in resting cardiomyocytes [75]. This finding thus indicates the new role for junctin as an additional luminal Ca2+ sensor. It is clear that future detailed studies are needed to systematically describe the direct effect of both triadin and junctin on RYR2 function, in order to understand the physiological importance of these effects and their contribution to overall CSQ2-independent luminal Ca2+-mediated regulation of the RYR2 function. This is dealt with in more detail below.
What is the role of CSQ2-independent regulation of the RYR2 channel by luminal Ca2+?
Although it is strongly supported that CSQ2 communicates the changes in [Ca2+]L to the RYR2 channel, the direct activation of RYR2 channels by luminal Ca2+ in a CSQ2-independent manner is still debated. In particular, single channel studies have demonstrated that even purified RYR2 channels lacking associated triadin, junctin and CSQ2 responded to increased [Ca2+]L from 10 μM to 2 mM by significant activation [3]. This clearly indicates that the luminal Ca2+ sensor is also intrinsic to the RYR2 channel and it must be located directly on the luminal loops. Recently, Dulhunty et al. [39] have published new data consistent with this idea. These authors showed that CSQ2-stripped RYR2 channels were substantially activated by luminal Ca2+ manipulated in the similar range of 0.3–1.5 mM. In contrast, Gyorke et al. [27] demonstrated that the purified RYR2 channels were not sensitive to luminal Ca2+ ranging from 20 μM to 5 mM, when cardiac triadin, junctin and CSQ2 were not added to the luminal solution to recover the luminal Ca2+-sensing complex. A possible explanation for this apparent controversy could be the presence of Mg2+ in the cytosolic solution, which we have recently shown diminished effects of luminal Ca2+ even at 53 mM [Ca2+]L when CSQ2 was most likely absent from the native RYR2 channel [11]. An interesting study presented by Qin et al. [9] has clearly identified potential sources of these experimental controversies. They revealed that activation of CSQ2-stripped RYR2 channels was dependent on the concentration of cytosolic Ca2+ ([Ca2+]C) used as a sole activator or, as we thought, on the extent of channel activation. More specifically, CSQ2-free RYR2 channels were responsive to luminal Ca2+ ranging from 10 μM to 10 mM only when the [Ca2+]C was increased from 1 μM to 100 μM. This led to dramatic RYR2 activation from almost the minimum to the maximum achievable level under the given experimental conditions. From this, it can be concluded that the action of luminal Ca2+ on the CSQ2-free RYR2 channel is apparent only under certain experimental conditions that enhance channel activity. However, when we compared all published studies, we found that the situation is more complex. While the effect of luminal Ca2+ was observable only for CSQ2-free RYR2 channels maximally activated solely by cytosolic Ca2+, in other cases when [Ca2+]C ranged from 100 pM to 6 μM and additional activators such as ATP or sulmazole were present the stimulatory effect of luminal Ca2+ was also observed for negligibly activated channels. Taken together, although all the studies outlined above collectively provide support for the existence of CSQ2-independent regulation of the RYR2 channel;, the physiological relevance of this process remains to be elucidated. Qin et al. [9, 10] have speculated that this mechanism is not of major physiological consequence because it does not discriminate between luminal Ca2+ and Mg2+. This is in sharp contrast to the CSQ2-dependent mechanism. In this respect, it is noteworthy that the absence of CSQ2 in humans and CSQ2-null transgenic animals has been associated with susceptibility to CPVT arrhythmias [76–78]. At the single channel level, this could be explained by the recent observation of Dulhunty et al. [39], who found that the CSQ2-stripped RYR2 channels were activated more strongly by luminal Ca2+ than when CSQ2 was attached to the channels. This indicates that RYR2 channels without CSQ2 appear to be more sensitive to the activation effect exerted by luminal Ca2+, and this was suggested to be the primary culprit for delayed-after-depolarizations leading, as large enough, to extra action potentials and severe cardiac arrhythmias [79, 80].
How can the CSQ2 dissociation process be visualized on the single channel level?
On the single channel level, several studies of CSQ2-dependent regulation of the RYR2 channel by luminal Ca2+ have been done, yielding variable and in some aspects, controversial results [9, 10, 27, 39]. As a first, Gyorke et al. [27] showed that native RYR2 channels were irreversibly activated when luminal Ca2+ was increased from 20 μM to 5 mM. The channel activity declined to its initial level solely after the addition of recombinant CSQ2 to the luminal bath. Thus, this observed potentiation of the native RYR2 channel was likely related to the dissociation of CSQ2 from the channel complex. In contrast, Qin et al. [10] showed in a similar study that the dissociation of CSQ2 resulted in the RYR2 inhibition and reassociation of CSQ2 in channel activation, and they attributed this discrepancy to the different activation status of the CSQ2-free channel. Indeed, in the first study, the CSQ2-stripped RYR2 channels exhibited moderate activity (P o = 0.36) induced by 6 μM [Ca2+]C, 3 mM ATP and 0.9 mM Mg2+, while in the later one, CSQ2-free RYR2 channels were activated only negligibly (P o = 0.001) solely by 0.75 μM [Ca2+]C. Importantly, we found an additional aspect that should be considered in this regard and it is [Ca2+]L. Gyorke et al. [27] added CSQ2 to the luminal bath when [Ca2+]L = 20 μM, while Qin et al. [10] performed their experiments at 1 mM [Ca2+]L. Considering that Ca2+ regulates CSQ2 protein folding in a monomeric state [43], it is likely in the aforementioned studies that CSQ2 acquired different conformations and could have regulated the RYR2 channel differently. Our proposal is further supported by the attractive idea of Beard et al. [38] for the CSQ1-RYR1 interaction. Particularly, these authors suggested that the changes in RYR1 activity induced by the increase in [Ca2+]L from 1 to 2 mM, which apparently did not lead to CSQ1 dissociation from the channel complex, could be partly attributed to changes in the CSQ1 conformation induced by luminal Ca2+. This hypothesis has potential physiological significance, and should be further tested focusing on the detailed description of the CSQ2-RYR2 interaction from both functional and structural aspects. Noteworthy, we found that some preliminary results have already been shown in the work of Qin et al. [9]. They clearly observed the activation of the RYR2 channel when luminal Ca2+ was manipulated from 10 μM up to 1 mM, which most likely did not disrupt the CSQ2-RYR2 interaction; and furthermore, this behavior had not previously been observed for CSQ2-stripped RYR2 channels, thus pointing to the CSQ2-dependent mechanism.
As mentioned above, CSQ2-dependent regulation of the RYR2 channel by luminal Ca2+ has resulted in certain experimental discrepancies. It is surprising that a similar situation is noted for the RYR1 channel, where also both activation and inhibitory effects of the CSQ1 dissociation have been reported [37, 38, 81].
These differences in CSQ1-RYR1 functional interaction have been explained by the existence of two mechanisms. Firstly, RYR1 inhibition is induced via interactions of CSQ1 with skeletal triadin and junctin in a phosphorylation-independent manner; and secondly, RYR1 activation is induced by binding of phosphorylated CSQ1 directly to the channel [23, 81]. It is currently unknown if both mechanisms of CSQ1 regulation operate in vivo; however, in the context of the RYR2 channel, the phosphorylation status of CSQ2 has not previously been considered as a potential source of controversial results.
Summary
During the past two decades, several new observations have indicated that CSQ2 is important in communicating the SR Ca2+ load to the RYR2 channel and, as a result of this regulation, it is important in determining Ca2+ release from the SR, and cardiac contraction. This conclusion has been drawn from a variety of experiments examining Ca2+ release in cardiomyocytes, the Ca2+ release from SR microsomes and the activity of the RYR2 channel reconstituted in the BLM. The most reliable evidence has come from the BLM experiments, although we are still unable to directly detect the presence of CSQ2 on the RYR2 channel complex reconstituted in the BLM. These studies have demonstrated that the significant changes in RYR2 gating caused by transient increase in [Ca2+]L were reversible only following addition of CSQ2 to the luminal channel face. Combined with biochemical observations highlighting the Ca2+ dependence of the CSQ2-RYR2 interaction, these results indicate that the changes in RYR2 activity caused by luminal Ca2+ may be presumably attributed to the CSQ2 dissociation from the channel complex; and thus, it is highly likely that CSQ2 plays an active role in communicating changes in [Ca2+]L to the RYR2 channel. In this regard it is important to emphasize that the precise molecular mechanism involved in this process is still not fully understood. New observations often lead to additional questions, mainly due to apparent contradictions; and therefore, the determination of the functional interaction between the RYR2 channel and CSQ2 together with its modification in normal and in disease states, is likely to be a prominent focus of research over the next few years.
As a first, it is important to consistently describe the different biophysical aspects of CSQ2-RYR2 channel interaction from both functional and structural point of view at [Ca2+]L less than 1 mM, and further carefully consider the interpretation of these obtained results in a physiological context, since the BLM situation is not exactly equivalent to the intact cell. Currently, it is becoming evident that CSQ2 dissociation-induced changes in RYR2 activity may not be of physiological importance; because it is highly unlikely that the dissociation of CSQ2 from the junctional face membrane is caused by normal physiological changes in [Ca2+]L (<1 mM). However, during Ca2+ overload (metabolic inhibition, ischemia/reperfusion, digitalis poisoning, and the early stage of heart failure), when the [Ca2+]L is increased above the normal level, the contribution of CSQ2 dissociation to the observed enhanced Ca2+ release could be significant, and this remains to be investigated. On the other hand, when the SR Ca2+ load is normal, the effect of CSQ2 on RYR2 function may depend only on the Ca2+ binding status of still associated CSQ2, and, thus, the CSQ2 conformation, and this provocative scenario suggested by Beard et al. [38] for CSQ1-RYR1 interaction, has to be seriously considered. Finally, the direct effect of luminal Ca2+ on the activity of the RYR2 channel that is not mediated by CSQ2 should be explored in greater detail, because of its possible physiological importance. In this regard, we should focus on the effect of physiological [Ca2+]L (<1 mM) on the activity of the native RYR2 channel complex, including triadin, junctin and/or CSQ2.
References
Fabiato A, Fabiato F (1975) Contractions induced by a calcium-triggered release of calcium from the sarcoplasmic reticulum of single skinned cardiac cells. J Physiol 249(3):469–495
Fabiato A, Fabiato F (1978) Effects of pH on the myofilaments and the sarcoplasmic reticulum of skinned cells from cardiac and skeletal muscles. J Physiol 276:233–255
Sitsapesan R, Williams AJ (1997) Regulation of current flow through ryanodine receptors by luminal Ca2+. J Membr Biol 159(3):179–185
Gyorke S, Gyorke I, Lukyanenko V, Terentyev D, Viatchenko-Karpinski S, Wiesner TF (2002) Regulation of sarcoplasmic reticulum calcium release by luminal calcium in cardiac muscle. Front Biosci 7:d1454–d1463
Sitsapesan R, Williams AJ (1994) Regulation of the gating of the sheep cardiac sarcoplasmic reticulum Ca(2+)-release channel by luminal Ca2+. J Membr Biol 137(3):215–226
Gyorke I, Gyorke S (1998) Regulation of the cardiac ryanodine receptor channel by luminal Ca2+ involves luminal Ca2+ sensing sites. Biophys J 75(6):2801–2810
Xu L, Meissner G (1998) Regulation of cardiac muscle Ca2+ release channel by sarcoplasmic reticulum lumenal Ca2+. Biophys J 75(5):2302–2312
Gaburjakova J, Gaburjakova M (2006) Comparison of the effects exerted by luminal Ca2+ on the sensitivity of the cardiac ryanodine receptor to caffeine and cytosolic Ca2+. J Membr Biol 212(1):17–28
Qin J, Valle G, Nani A, Nori A, Rizzi N, Priori SG, Volpe P, Fill M (2008) Luminal Ca2+ regulation of single cardiac ryanodine receptors: insights provided by calsequestrin and its mutants. J Gen Physiol 131(4):325–334
Qin J, Valle G, Nani A, Chen H, Ramos-Franco J, Nori A, Volpe P, Fill M (2009) Ryanodine receptor luminal Ca2+ regulation: swapping calsequestrin and channel isoforms. Biophys J 97(7):1961–1970
Tencerova B, Zahradnikova A, Gaburjakova J, Gaburjakova M (2012) Luminal Ca2+ controls activation of the cardiac ryanodine receptor by ATP. J Gen Physiol 140(2):93–108
Laver DR (2007) Ca2+ stores regulate ryanodine receptor Ca2+ release channels via luminal and cytosolic Ca2+ sites. Biophys J 92(10):3541–3555
Laver DR (2007) Ca2+ stores regulate ryanodine receptor Ca2+ release channels via luminal and cytosolic Ca2+ sites. Clin Exp Pharmacol Physiol 34(9):889–896
Laver DR (2009) Luminal Ca(2+) activation of cardiac ryanodine receptors by luminal and cytoplasmic domains. Eur Biophys J 39(1):19–26
Ikemoto N, Bhatnagar GM, Nagy B, Gergely J (1972) Interaction of divalent cations with the 55,000-dalton protein component of the sarcoplasmic reticulum. Studies of fluorescence and circular dichroism. J Biol Chem 247(23):7835–7837
MacLennan DH, Wong PT (1971) Isolation of a calcium-sequestering protein from sarcoplasmic reticulum. Proc Natl Acad Sci U S A 68(6):1231–1235
Campbell KP, MacLennan DH, Jorgensen AO, Mintzer MC (1983) Purification and characterization of calsequestrin from canine cardiac sarcoplasmic reticulum and identification of the 53,000 dalton glycoprotein. J Biol Chem 258(2):1197–1204
Scott BT, Simmerman HK, Collins JH, Nadal-Ginard B, Jones LR (1988) Complete amino acid sequence of canine cardiac calsequestrin deduced by cDNA cloning. J Biol Chem 263(18):8958–8964
Guo W, Jorgensen AO, Campbell KP (1996) Triadin, a linker for calsequestrin and the ryanodine receptor. Soc Gen Physiol Ser 51:19–28
Zhang L, Kelley J, Schmeisser G, Kobayashi YM, Jones LR (1997) Complex formation between junctin, triadin, calsequestrin, and the ryanodine receptor. Proteins of the cardiac junctional sarcoplasmic reticulum membrane. J Biol Chem 272(37):23389–23397
Chopra N, Yang T, Asghari P, Moore ED, Huke S, Akin B, Cattolica RA, Perez CF, Hlaing T, Knollmann-Ritschel BE, Jones LR, Pessah IN, Allen PD, Franzini-Armstrong C, Knollmann BC (2009) Ablation of triadin causes loss of cardiac Ca2+ release units, impaired excitation-contraction coupling, and cardiac arrhythmias. Proc Natl Acad Sci U S A 106(18):7636–7641
Beard NA, Laver DR, Dulhunty AF (2004) Calsequestrin and the calcium release channel of skeletal and cardiac muscle. Prog Biophys Mol Biol 85(1):33–69
Herzog A, Szegedi C, Jona I, Herberg FW, Varsanyi M (2000) Surface plasmon resonance studies prove the interaction of skeletal muscle sarcoplasmic reticular Ca(2+) release channel/ryanodine receptor with calsequestrin. FEBS Lett 472(1):73–77
Wei L, Hanna AD, Beard NA, Dulhunty AF (2009) Unique isoform-specific properties of calsequestrin in the heart and skeletal muscle. Cell Calcium 45(5):474–484
Shin DW, Ma J, Kim DH (2000) The asp-rich region at the carboxyl-terminus of calsequestrin binds to Ca(2+) and interacts with triadin. FEBS Lett 486(2):178–182
Kobayashi YM, Alseikhan BA, Jones LR (2000) Localization and characterization of the calsequestrin-binding domain of triadin 1. Evidence for a charged beta-strand in mediating the protein–protein interaction. J Biol Chem 275(23):17639–17646
Gyorke I, Hester N, Jones LR, Gyorke S (2004) The role of calsequestrin, triadin, and junctin in conferring cardiac ryanodine receptor responsiveness to luminal calcium. Biophys J 86(4):2121–2128
Ikemoto N, Ronjat M, Meszaros LG, Koshita M (1989) Postulated role of calsequestrin in the regulation of calcium release from sarcoplasmic reticulum. Biochemistry 28(16):6764–6771
Terentyev D, Cala SE, Houle TD, Viatchenko-Karpinski S, Gyorke I, Terentyeva R, Williams SC, Gyorke S (2005) Triadin overexpression stimulates excitation-contraction coupling and increases predisposition to cellular arrhythmia in cardiac myocytes. Circ Res 96(6):651–658
Terentyev D, Viatchenko-Karpinski S, Vedamoorthyrao S, Oduru S, Gyorke I, Williams SC, Gyorke S (2007) Protein protein interactions between triadin and calsequestrin are involved in modulation of sarcoplasmic reticulum calcium release in cardiac myocytes. J Physiol 583(Pt 1):71–80
Wei L, Gallant EM, Dulhunty AF, Beard NA (2009) Junctin and triadin each activate skeletal ryanodine receptors but junctin alone mediates functional interactions with calsequestrin. Int J Biochem Cell Biol 41(11):2214–2224
Terentyev D, Nori A, Santoro M, Viatchenko-Karpinski S, Kubalova Z, Gyorke I, Terentyeva R, Vedamoorthyrao S, Blom NA, Valle G, Napolitano C, Williams SC, Volpe P, Priori SG, Gyorke S (2006) Abnormal interactions of calsequestrin with the ryanodine receptor calcium release channel complex linked to exercise-induced sudden cardiac death. Circ Res 98(9):1151–1158
di Barletta MR, Viatchenko-Karpinski S, Nori A, Memmi M, Terentyev D, Turcato F, Valle G, Rizzi N, Napolitano C, Gyorke S, Volpe P, Priori SG (2006) Clinical phenotype and functional characterization of CASQ2 mutations associated with catecholaminergic polymorphic ventricular tachycardia. Circulation 114(10):1012–1019
Jiang D, Chen W, Wang R, Zhang L, Chen SR (2007) Loss of luminal Ca2+ activation in the cardiac ryanodine receptor is associated with ventricular fibrillation and sudden death. Proc Natl Acad Sci U S A 104(46):18309–18314
Jiang D, Chen W, Xiao J, Wang R, Kong H, Jones PP, Zhang L, Fruen B, Chen SR (2008) Reduced threshold for luminal Ca2+ activation of RyR1 underlies a causal mechanism of porcine malignant hyperthermia. J Biol Chem 283(30):20813–20820
Priori SG, Chen SR (2011) Inherited dysfunction of sarcoplasmic reticulum Ca2+ handling and arrhythmogenesis. Circ Res 108(7):871–883
Beard NA, Sakowska MM, Dulhunty AF, Laver DR (2002) Calsequestrin is an inhibitor of skeletal muscle ryanodine receptor calcium release channels. Biophys J 82(1 Pt 1):310–320
Beard NA, Casarotto MG, Wei L, Varsanyi M, Laver DR, Dulhunty AF (2005) Regulation of ryanodine receptors by calsequestrin: effect of high luminal Ca2+ and phosphorylation. Biophys J 88(5):3444–3454
Dulhunty AF, Wium E, Li L, Hanna AD, Mirza S, Talukder S, Ghazali NA, Beard NA (2012) Proteins within the intracellular calcium store determine cardiac RyR channel activity and cardiac output. Clin Exp Pharmacol Physiol 39(5):477–484
Murphy RM, Mollica JP, Beard NA, Knollmann BC, Lamb GD (2011) Quantification of calsequestrin 2 (CSQ2) in sheep cardiac muscle and Ca2+ -binding protein changes in CSQ2 knockout mice. Am J Physiol Heart Circ Physiol 300(2):H595–H604
Viatchenko-Karpinski S, Terentyev D, Gyorke I, Terentyeva R, Volpe P, Priori SG, Napolitano C, Nori A, Williams SC, Gyorke S (2004) Abnormal calcium signaling and sudden cardiac death associated with mutation of calsequestrin. Circ Res 94(4):471–477
Wei L, Varsanyi M, Dulhunty AF, Beard NA (2006) The conformation of calsequestrin determines its ability to regulate skeletal ryanodine receptors. Biophys J 91(4):1288–1301
Park H, Park IY, Kim E, Youn B, Fields K, Dunker AK, Kang C (2004) Comparing skeletal and cardiac calsequestrin structures and their calcium binding: a proposed mechanism for coupled calcium binding and protein polymerization. J Biol Chem 279(17):18026–18033
Mitchell RD, Simmerman HK, Jones LR (1988) Ca2+ binding effects on protein conformation and protein interactions of canine cardiac calsequestrin. J Biol Chem 263(3):1376–1381
Chopra N, Knollmann BC (2009) Cardiac calsequestrin: the new kid on the block in arrhythmias. J Cardiovasc Electrophysiol 20(10):1179–1185
MacLennan DH (1813) Zvaritch E (2011) Mechanistic models for muscle diseases and disorders originating in the sarcoplasmic reticulum. Biochim Biophys Acta 5:948–964
Faggioni M, Knollmann BC (2012) Calsequestrin 2 and arrhythmias. Am J Physiol Heart Circ Physiol 302(6):H1250–H1260
Kim E, Youn B, Kemper L, Campbell C, Milting H, Varsanyi M, Kang C (2007) Characterization of human cardiac calsequestrin and its deleterious mutants. J Mol Biol 373(4):1047–1057
Bal NC, Sharon A, Gupta SC, Jena N, Shaikh S, Gyorke S, Periasamy M (2010) The catecholaminergic polymorphic ventricular tachycardia mutation R33Q disrupts the N-terminal structural motif that regulates reversible calsequestrin polymerization. J Biol Chem 285(22):17188–17196
Sanchez EJ, Lewis KM, Danna BR, Kang C (2012) High-capacity Ca2+-binding of human skeletal calsequestrin. J Biol Chem 287:11592–11601
Slupsky JR, Ohnishi M, Carpenter MR, Reithmeier RA (1987) Characterization of cardiac calsequestrin. Biochemistry 26(20):6539–6544
Wang S, Trumble WR, Liao H, Wesson CR, Dunker AK, Kang CH (1998) Crystal structure of calsequestrin from rabbit skeletal muscle sarcoplasmic reticulum. Nat Struct Biol 5(6):476–483
Park H, Wu S, Dunker AK, Kang C (2003) Polymerization of calsequestrin. Implications for Ca2+ regulation. J Biol Chem 278(18):16176–16182
Valle G, Galla D, Nori A, Priori SG, Gyorke S, de Filippis V, Volpe P (2008) Catecholaminergic polymorphic ventricular tachycardia-related mutations R33Q and L167H alter calcium sensitivity of human cardiac calsequestrin. Biochem J 413(2):291–303
Kalyanasundaram A, Bal NC, Franzini-Armstrong C, Knollmann BC, Periasamy M (2010) The calsequestrin mutation CASQ2D307H does not affect protein stability and targeting to the junctional sarcoplasmic reticulum but compromises its dynamic regulation of calcium buffering. J Biol Chem 285(5):3076–3083
Bal NC, Jena N, Sopariwala D, Balaraju T, Shaikh S, Bal C, Sharon A, Gyorke S, Periasamy M (2011) Probing cationic selectivity of cardiac calsequestrin and its CPVT mutants. Biochem J 435(2):391–399
Mishra A, Suman SK, Srivastava SS, Sankaranarayanan R, Sharma Y (2012) Decoding the molecular design principles underlying Ca(2+) binding to betagamma-crystallin motifs. J Mol Biol 415(1):75–91
Launikonis BS, Zhou J, Royer L, Shannon TR, Brum G, Rios E (2006) Depletion “skraps” and dynamic buffering inside the cellular calcium store. Proc Natl Acad Sci U S A 103(8):2982–2987
Chopra N, Kannankeril PJ, Yang T, Hlaing T, Holinstat I, Ettensohn K, Pfeifer K, Akin B, Jones LR, Franzini-Armstrong C, Knollmann BC (2007) Modest reductions of cardiac calsequestrin increase sarcoplasmic reticulum Ca2+ leak independent of luminal Ca2+ and trigger ventricular arrhythmias in mice. Circ Res 101(6):617–626
Stevens SC, Terentyev D, Kalyanasundaram A, Periasamy M, Gyorke S (2009) Intra-sarcoplasmic reticulum Ca2+ oscillations are driven by dynamic regulation of ryanodine receptor function by luminal Ca2+ in cardiomyocytes. J Physiol 587(Pt 20):4863–4872
Lee KW, Maeng JS, Choi JY, Lee YR, Hwang CY, Park SS, Park HK, Chung BH, Lee SG, Kim YS, Jeon H, Eom SH, Kang C, Kim do H, Kwon KS (2012) Role of Junctin protein interactions in cellular dynamics of calsequestrin polymer upon calcium perturbation. J Biol Chem 287(3):1679–1687
Franzini-Armstrong C, Protasi F, Tijskens P (2005) The assembly of calcium release units in cardiac muscle. Ann N Y Acad Sci 1047:76–85
Jones LR, Suzuki YJ, Wang W, Kobayashi YM, Ramesh V, Franzini-Armstrong C, Cleemann L, Morad M (1998) Regulation of Ca2+ signaling in transgenic mouse cardiac myocytes overexpressing calsequestrin. J Clin Invest 101(7):1385–1393
Zhang L, Franzini-Armstrong C, Ramesh V, Jones LR (2001) Structural alterations in cardiac calcium release units resulting from overexpression of junctin. J Mol Cell Cardiol 33(2):233–247
Terentyev D, Viatchenko-Karpinski S, Valdivia HH, Escobar AL, Gyorke S (2002) Luminal Ca2+ controls termination and refractory behavior of Ca2+ -induced Ca2+ release in cardiac myocytes. Circ Res 91(5):414–420
Baksh S, Spamer C, Oikawa K, McCubbin WD, Heilmann C, Kay CM, Michalak M (1995) Zn2+ binding to cardiac calsequestrin. Biochem Biophys Res Commun 209(1):310–315
Ikemoto N, Nagy B, Bhatnagar GM, Gergely J (1974) Studies on a metal-binding protein of the sarcoplasmic reticulum. J Biol Chem 249(8):2357–2365
Xie H, Chen KY, Zhu PH (2004) Effect of Zn2+ ions on ryanodine binding to sarcoplasmic reticulum of striated muscles in the presence of pyrithione. Acta Pharmacol Sin 25(12):1647–1651
Palmer BM, Vogt S, Chen Z, Lachapelle RR, Lewinter MM (2006) Intracellular distributions of essential elements in cardiomyocytes. J Struct Biol 155(1):12–21
Laver DR, Honen BN (2008) Luminal Mg2+, a key factor controlling RYR2-mediated Ca2+ release: cytoplasmic and luminal regulation modeled in a tetrameric channel. J Gen Physiol 132(4):429–446
Kirchhefer U, Neumann J, Bers DM, Buchwalow IB, Fabritz L, Hanske G, Justus I, Riemann B, Schmitz W, Jones LR (2003) Impaired relaxation in transgenic mice overexpressing junctin. Cardiovasc Res 59(2):369–379
Tijskens P, Jones LR, Franzini-Armstrong C (2003) Junctin and calsequestrin overexpression in cardiac muscle: the role of junctin and the synthetic and delivery pathways for the two proteins. J Mol Cell Cardiol 35(8):961–974
Kirchhefer U, Hanske G, Jones LR, Justus I, Kaestner L, Lipp P, Schmitz W, Neumann J (2006) Overexpression of junctin causes adaptive changes in cardiac myocyte Ca(2+) signaling. Cell Calcium 39(2):131–142
Yuan Q, Fan GC, Dong M, Altschafl B, Diwan A, Ren X, Hahn HH, Zhao W, Waggoner JR, Jones LR, Jones WK, Bers DM, Dorn GW 2nd, Wang HS, Valdivia HH, Chu G, Kranias EG (2007) Sarcoplasmic reticulum calcium overloading in junctin deficiency enhances cardiac contractility but increases ventricular automaticity. Circulation 115(3):300–309
Altschafl BA, Arvanitis DA, Fuentes O, Yuan Q, Kranias EG, Valdivia HH (2011) Dual role of junctin in the regulation of ryanodine receptors and calcium release in cardiac ventricular myocytes. J Physiol 589(Pt 24):6063–6080
Postma AV, Denjoy I, Hoorntje TM, Lupoglazoff JM, Da Costa A, Sebillon P, Mannens MM, Wilde AA, Guicheney P (2002) Absence of calsequestrin 2 causes severe forms of catecholaminergic polymorphic ventricular tachycardia. Circ Res 91(8):e21–e26
Knollmann BC, Chopra N, Hlaing T, Akin B, Yang T, Ettensohn K, Knollmann BE, Horton KD, Weissman NJ, Holinstat I, Zhang W, Roden DM, Jones LR, Franzini-Armstrong C, Pfeifer K (2006) Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J Clin Invest 116(9):2510–2520
Song L, Alcalai R, Arad M, Wolf CM, Toka O, Conner DA, Berul CI, Eldar M, Seidman CE, Seidman JG (2007) Calsequestrin 2 (CASQ2) mutations increase expression of calreticulin and ryanodine receptors, causing catecholaminergic polymorphic ventricular tachycardia. J Clin Invest 117(7):1814–1823
Jiang D, Wang R, Xiao B, Kong H, Hunt DJ, Choi P, Zhang L, Chen SR (2005) Enhanced store overload-induced Ca2+ release and channel sensitivity to luminal Ca2+ activation are common defects of RyR2 mutations linked to ventricular tachycardia and sudden death. Circ Res 97(11):1173–1181
Jones PP, Jiang D, Bolstad J, Hunt DJ, Zhang L, Demaurex N, Chen SR (2008) Endoplasmic reticulum Ca2+ measurements reveal that the cardiac ryanodine receptor mutations linked to cardiac arrhythmia and sudden death alter the threshold for store-overload-induced Ca2+ release. Biochem J 412(1):171–178
Szegedi C, Sarkozi S, Herzog A, Jona I, Varsanyi M (1999) Calsequestrin: more than ‘only’ a luminal Ca2+ buffer inside the sarcoplasmic reticulum. Biochem J 337(Pt 1):19–22
Acknowledgments
This work was supported by the Slovak Grant Agency (VEGA) (Grants 2/0102/12 to J.G. and 2/0033/11 to M.G.), and National Institutes of Health (Grant R01 HL64014 to M. P). N.C.B. was supported by postdoctoral fellowships (10POST3360007) from the American Heart Association.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
M. Gaburjakova and N. C. Bal contributed equally to this work.
Rights and permissions
About this article
Cite this article
Gaburjakova, M., Bal, N.C., Gaburjakova, J. et al. Functional interaction between calsequestrin and ryanodine receptor in the heart. Cell. Mol. Life Sci. 70, 2935–2945 (2013). https://doi.org/10.1007/s00018-012-1199-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-012-1199-7