
Abstract
Cardiac hypertrophy (CH) is an adaptive process that exists in two distinct forms and allows the heart to adequately respond to an organism’s needs. The first form of CH is physiological, adaptive and reversible. The second is pathological, irreversible and associated with fibrosis and cardiomyocyte death. CH involves multiple molecular mechanisms that are still not completely defined but it is now accepted that physiological CH is associated more with the PI3-K/Akt pathway while the main signaling cascade activated in pathological CH involves the Calcineurin-NFAT pathway. It was recently demonstrated that the TRPM4 channel may act as a negative regulator of pathological CH by regulating calcium entry and thus the Cn-NFAT pathway. In this study, we examined if the TRPM4 channel is involved in the physiological CH process. We evaluated the effects of 4 weeks endurance training on the hearts of Trpm4 +/+ and Trpm4 −/− mice. We identified an elevated functional expression of the TRPM4 channel in cardiomyocytes after endurance training suggesting a potential role for the channel in physiological CH. We then observed that Trpm4 +/+ mice displayed left ventricular hypertrophy after endurance training associated with enhanced cardiac function. By contrast, Trpm4 −/− mice did not develop these adaptions. While Trpm4 −/− mice did not develop gross cardiac hypertrophy, the cardiomyocyte surface area was larger and associated with an increase of Tunel positive cells. Endurance training in Trpm4 +/+ mice did not increase DNA fragmentation in the heart. Endurance training in Trpm4 +/+ mice was associated with activation of the classical physiological CH Akt pathway while Trpm4 −/− favored the Calcineurin pathway. Calcium studies demonstrated that TRPM4 channel negatively regulates calcium entry providing support for activation of the Cn-NFAT pathway in Trpm4 −/− mice. In conclusion, we provide evidence for the functional expression of TRPM4 channel in response to endurance training. This expression may help to maintain the balance between physiological and pathological hypertrophy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cardiac hypertrophy (CH) reflects an adaptive enlargement of the heart in order to maintain or increase its output. CH can be classified by the functional outcomes as either physiological (normal) or pathological (detrimental) (Bernardo et al. 2010; Shimizu and Minamino 2016). Physiological CH develops in response to endurance training or pregnancy. The morphological cardiac growth and associated enhanced cardiac contractility are reversible (Eghbali et al. 2005; Umar et al. 2012; Mann and Rosenzweig 2012). In exercise-induced physiological CH, the canonical intracellular signaling pathway phosphatidylinositol-3 kinase–protein kinase B (PI3K-Akt) pathway is involved (Kemi et al. 2008; Kim et al. 2008; Ikeda et al. 2009). Upstream stimulation of molecules such as the β-adrenergic receptors, insulin receptor or insulin-like growth factor one activates Akt through PI3Kα. Akt, the key molecule for physiological CH, also named protein kinase B, is a serine/threonine kinase, which phosphorylates downstream targets such as glucose transporter (GLUT1) and mTOR (mammalian target of rapamycin). While three isoforms of Akt (Akt1, Akt2 and Akt3) exist, only Akt2 and Akt1 are expressed in heart. It is now well established that Akt1 mediates cardiac growth and that Akt2 is important for cell metabolism (DeBosch et al. 2006). Finally, the major effect of the activation of this signaling pathway in cardiomyocytes is an increase of cell size, inhibition of apoptosis, and regulation of metabolism (Tao et al. 2015).
By contrast, pathological CH develops in response to disease-associated stimuli such as hypertension, myocardial infarction or genetic mutations. Activation of the neuro-endocrine system provokes alterations in signal-transduction pathways associated with increased cell size, increased apoptosis, and fibrosis (Bernardo et al. 2010). The literature is profuse on the involvement of the pathway involving the calcium (Ca2+) dependent phosphatase, calcineurin (Cn) and nuclear factor of activated T-cells (NFAT). This signaling pathway is sufficient to induce pathological CH (Molkentin et al. 1998; Wilkins et al. 2004; Wilkins and Molkentin 2004). Cn dephosphorylates NFAT, allowing its nuclear translocation, and thus inducing a hypertrophic gene expression adaption (Wilkins and Molkentin 2004). Ca2+ entry in the cardiomyocytes is critically involved in pathological CH (Bers 2008; Cho et al. 2016). It has been hypothesized that members of the transient receptor potential (TRP) channels family could induce signal transduction via Ca2+ elevation and Ca2+ store refill, prolonging the pathological hypertrophic pathway (Nakayama et al. 2006; Bush et al. 2006; Onohara et al. 2006; Kuwahara et al. 2006; Makarewich et al. 2014). By contrast, physiological CH, induced by exercise or pregnancy, is not associated with an increase of the Cn-NFAT pathway (Wilkins et al. 2004).
The TRPM4 channel (Ca2+-activated TRP subfamily melastatin, member 4) is a voltage-sensitive channel that selectively conducts Na+ and K+ but not Ca2+ ions thereby negatively regulating Ca2+ entry from store-operated Ca2+ channels (Launay et al. 2002, 2004; Nilius et al. 2003; Vennekens et al. 2007; Park et al. 2008; Barbet et al. 2008). TRPM4 channel is widely expressed and functional in numerous tissues and has been implicated in Ca2+-dependent functions (Barbet et al. 2008; Guinamard et al. 2010; Earley 2013; Li et al. 2014). This channel is expressed in the left ventricle in various forms of pathological CH (Colquhoun et al. 1981; Guinamard et al. 2002, 2006). In particular, TRPM4 channel expression correlated positively with the heart weight to body weight ratio in spontaneously hypertensive rats while no expression was detected in control rats (Guinamard et al. 2006). Conversely, other groups report no modification of its expression in an experimental murine CH model (Jacobs et al. 2015). A putative protective role of TRPM4 in the development of pathological CH has also been suggested by preventing the activation of the Cn-NFAT pathway (Kecskés et al. 2015).
The role of TRPM4 in physiological CH is currently unknown. In the present study, we investigated the potential role of the TRPM4 channel in the normal adaptive CH in response to endurance training using wild-type mice (Trpm4 +/+) and Trpm4 knockout mice (Trpm4 −/− mice). We found an increased TRPM4 channel expression and function, associated with a functional regulation of Ca2+ entry through store-operated channels (SOC), and activation of the PI3K-Akt pathway in Trpm4 +/+ mice in response to endurance training. By contrast, the combination of endurance training and the absence of the TRPM4 channel was associated with increased apoptosis and the activation of the pathological associated CH Cn-NFAT signaling pathway. Our results suggest that expression of TRPM4 channel, due to hypertrophic remodeling, may modulate a balance between physiological and pathological CH signaling pathways.
Results
Functional expression of TRPM4 channel is promoted by endurance training
We investigated whether TRPM4 channel expression could be detected in response to 4 week endurance training (Tr) when compared to sedentary mice (Sed). At the transcript level, assessed by quantitative RT-PCR, TRPM4 channel was detected in the left ventricle (LV) of Sed-Trpm4 +/+ mice and significantly increased after endurance training (Tr-Trpm4 +/+mice) (Fig. 1a). In order to determine if this expression correlated with a functional TRPM4-like current in single LV myocytes, we performed electrophysiological analyses. We used caffeine (5 µmol/L) to induce an intracellular Ca2+ rise and to thus unmask the Ca2+-activated native counterpart of the TRPM4-like current (Köster et al. 1999). The current–voltage relationship of the caffeine-induced current in isolated myocytes from Sed-Trpm4 +/+ mice represents a small inward current probably due to a Ca2+ entry through the L-type calcium channel (Fig. 1b). In LV cardiomyocytes from Tr-Trpm4 +/+, the current–voltage relationship of the caffeine-induced current displayed an outward current slightly rectifying in positives voltages. The outward current was sensitive to the TRPM4 channel inhibitor 9-phenanthrol (9-P; 10 µmol/L). Indeed, the 9-P sensitive-current recorded in Tr-Trpm4 +/+ cardiomyocytes also displayed an outward rectification in positive voltages and a reversal potential close to −10 mV (Fig. 1c). The 9-P application induced a 75% reduction of the current at a membrane potential of +40 mV (Fig. 1d, black histogram). By contrast, cardiomyocytes from Sed-Trpm4 +/+ mice displayed a negligible sensitivity to 9-P (Fig. 1c, d). The caffeine-induced current shares the electrophysiological properties described for the TRPM4 current (Launay et al. 2002, 2004; Nilius et al. 2003, 2005; Grand et al. 2008). The rectification observed at positive voltages in the whole cell patch clamp condition is explained by a voltage dependence of the TRPM4 channel associated with an increased open probability. Both currents are 9-P sensitive. The observed reversal potential follows the pore selectivity of TRPM4 for cesium as well as lithium ions (Köster et al. 1999; Nilius et al. 2003). Our electrophysiological studies support an increase of TRPM4 channel function in LV cardiomyocytes after endurance training, consistent with the quantitative RT-PCR analysis.

Endurance training leads to TRPM4 channel functional expression in mice. a Relative Trpm4 expression in the left ventricle from Sed- and Tr-Trpm4 +/+ related to Gapdh housekeeping gene. Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001. Student’s t-test. b Current–voltage (I/V) relationships obtained in ventricular myocytes in whole cell configuration of the patch-clamp technique from Sed- (black square) and Tr- (open square) Trpm4 +/+ mice. Red open circle represent the subtraction Tr-Sed. Upper panel represents the voltage clamp protocol. (Sed- Trpm4 +/+ n = 28 cells, Tr-Trpm4 +/+ n = 21 cells). c I/V relationship of the 9-P sensitive current on the caffeine-induced current recorded on cardiomyocytes from Sed-Trpm4 −/− and Tr-Trpm4 +/+ mice (n = 7 and 18 cells, respectively). d Average outward current density recorded at +40 mV in Sed- (black bar graph) and Tr- (open bar graph) Trpm4 +/+ ventricular myocytes before and after caffeine (5 mM) application and with 9-Phenanthrol treatments (10 µmol/L) (n = 18 for each conditions). Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001. Mann–Witney test was used. (Color figure online)
Endurance training induced cellular hypertrophy but did not modify gross heart size in Tr-Trpm4 −/− mice
To clarify the potential role of TRPM4 channel expression with training, we used Trpm4 −/− mice as a negative control for the TRPM4 channel expression after training. Trpm4 +/+ and Trpm4 −/− mice were separated into two groups, a sedentary group (Sed-Trpm4 +/+ and Sed-Trpm4 −/− mice), which underwent only treadmill familiarization, and the trained group, which followed endurance training for 4 weeks (Tr-Trpm4 +/+ and Tr-Trpm4 −/− mice). Endurance training has been described to increase cardiac function and is associated with physiologic cardiac growth via cardiomyocyte hypertrophy due to the activation of several signaling pathways (Tao et al. 2015), including the IGF-1-PI3K-Akt pathway. Training-induced cardiac remodeling was assessed in Trpm4 +/+ and Trpm4 −/− groups before and after training by echocardiography. We found an increase of the left ventricle mass to body weight ratio (LV Mass/BW) in Tr-Trpm4 +/+ hearts when compared to pre-training. This was associated with an increased fractional shortening and ejection fraction, demonstrating enhanced cardiac function (Table 1). This result was confirmed by an increase of whole heart to body weight ratio (Fig. 2a, lower histogram) with no modification of body weight (Fig. 2a, upper histogram). Surprisingly, we observed no changes in LV mass or functional parameters in response to training in Tr-Trpm4 −/− mice (Table 2). These results were validated by an unchanged heart weight to body weight ratio in Tr-Trpm4 −/− mice (Fig. 2c). Of note, and as previously described, Trpm4 −/− mice display left ventricle hypertrophy in basal conditions compared to Trpm4 +/+ due to hyperplasia (Demion et al. 2014). As CH is commonly associated with an increase of cardiomyocytes size (Bernardo et al. 2010), we measured cardiomyocyte surface area after endurance training. Cardiomyocytes membranes in cryosections were labeled with wheat germ agglutinin (WGA) to visualize and measure cell surface area (CSA). CSA was increased in cardiomyocytes from Tr-Trpm4 +/+ mice when compared to Sed-Trpm4 +/+ (Fig. 2b). Similarly, the CSA of cardiomyocytes in Tr-Trpm4 −/− mice was elevated compared to Sed-Trpm4 −/− (Fig. 2d). Measurement of cell capacitance by patch-clamp in freshly isolated cardiomyocytes confirmed the increased size of cardiomyocytes after training in both Trpm4 +/+ and Trpm4 −/− mice (Fig. 2b, d, lower histogram).

Endurance training induced cardiac hypertrophy in Trpm4 +/+ mice but not in Trpm4 −/− mice, while both had cardiomyocytes growth. a Upper histogram represents the body weight comparison between Sed and Tr-Trpm4 +/+ mice. Lower histogram represents the heart weight on body weight ratio (HW/BW) comparison between Sed and Tr-Trpm4 +/+ mice. b Cell surface labelling: in red, WGA labelling on cryosections from Sed and Tr-Trpm4 +/+ mice and staining with 4,6-diamidino-2-phenylindole (DAPI) in blue. Upper histogram represents the calculated cell surface area (CSA) from longitudinal plans (Sed-Trpm4 +/+ n = 44 cells from six mice, Tr-Trpm4 +/+ n = 52 cells from seven mice). Lower histogram represents cell capacitance measurement on freshly isolated cardiomyocytes. c Upper histogram represents the body weight comparison between Sed and Tr-Trpm4 −/− mice (Sed- Trpm4 −/− n = 10 mice, Tr-Trpm4 −/− n = 11 mice). Lower histogram represents the heart weight on body weight ratio (HW/BW) comparison between Sed and Tr-Trpm4 +/+ mice. d In red, WGA labelling on cryosections from Sed and Tr-Trpm4 −/− mice and DAPI labeling in blue. Upper histogram represents the calculated CSA from longitudinal plans (Sed- Trpm4 −/− n = 45 cells from six mice, Tr-Trpm4 −/− n = 42 cells from four mice). Lower histogram represents cell capacitance measurement on freshly isolated cardiomyocytes. Data are presented as mean ± SEM. **p < 0.01, ***p < 0.001. Student’s t-test
Our data suggest that the gross cardiac remodeling that occurs with endurance training in Trpm4 +/+ mice was absent in Trpm4 −/− mice. However, cardiomyocyte hypertrophy was comparable in trained Trpm4 −/− and Trpm4 +/+ mice. We therefore hypothesized that Trpm4 −/− have an increase in cell death in response to training that could mask the heart size increase that should normally be correlated with cardiomyocyte hypertrophy.
DNA fragmentation is increased in Trpm4 −/− mice in response to endurance training
As DNA fragmentation represents a hallmark of apoptosis, we performed a TUNEL assay on heart cryosections to determine the number of TUNEL+ cells (Fig. 3a, b). Endurance training did not modify the number of TUNEL+ cells in Trpm4 +/+ mice (left panels) whereas the number of TUNEL+ cells was increased after 4 weeks of endurance training in Trpm4 −/− mice (right panels). Death of cardiomyocytes is a characteristic of pathological CH, which normally does not occur in physiological CH. Due to this, we postulated that Trpm4 −/− mice may adapt to endurance training by using a pathological signaling pathway, instead of a physiological one.

Endurance training induced increased apoptosis in Trpm4 −/− mice. a Representative staining of apoptotic cells in cardiac sections performed with DAPI (blue) for nuclei, with WGA for the plasma membrane (red) and terminal deoxynucleotidyl transferase dUTP-mediated nick-end labeling (TUNEL) assay (green). The images of the top are the negative and positive control, respectively. b The bar graphs represent the number of TUNEL+ nuclei per section divided by the total area (mm2). Data are presented as mean ± SEM. *p < 0.05. Student’s t-test. (Color figure online)
Calcineurin but not IGF-1-PI3K-Akt is enhanced in hearts from Tr-Trpm4 −/−
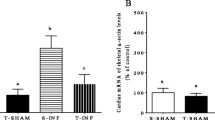
We tested the two canonical hypertrophic pathways (IGF-1-PI3K-Akt and Calcineurin) by Western blot analysis in order to characterize the type of cellular hypertrophy induced by endurance training in both Trpm4 +/+ and Trpm4 −/− mice. The P-Akt/Akt ratio, an indicator of IGF-1-PI3K-Akt pathway activation and physiological CH, was increased in Tr-Trpm4 +/+ LV when compared to Sed-Trpm4 +/+ (Fig. 4a, left panel and histogram). A recent study demonstrated that TRPM4 channel expression was associated with the chaperone protein 14-3-3 as a physical partner at the c-terminus domain (Cho et al. 2014). This chaperone protein is also activated by the IGF-1-PI3K-Akt signaling pathway (Qi et al. 2010). We found an increase of 14-3-3 protein expression in the LV from Tr-Trpm4 +/+ when compared to Sed-Trpm4 +/+ (Fig. 4a, middle panel and histogram), confirming activation of the IGF-1-PI3K-Akt pathway. Cn protein expression, a marker of pathological CH, was unchanged in the LV from Tr-Trpm4 +/+ when compared to Sed-Trpm4 +/+ (Fig. 4a, right panel and histogram). Finally, qPCR analysis of Serca2a, a marker of beneficial remodeling, was found to be increased in the LV from Tr-Trpm4 +/+ mice when compared to Sed-Trpm4 +/+ mice (Fig. 4b). Altogether, these data indicate that the cellular hypertrophy observed in response to endurance training in Tr-Trpm4 +/+ mice was associated with activation of the physiological IGF-1-PI3K-Akt signaling pathway.

Characterization of the pro-hypertrophic pathway activated by endurance training in Trpm4 +/+ and Trpm4 −/− mice. a Relative P-Akt/Akt ratio, Pan-14.3.3 and Pan CnA proteins expression assessed by Western-blot analysis normalized to loading control GAPDH. b Relative expression of Serca2a mRNA assessed by quantitative PCR. c Relative P-Akt/Akt ratio, Pan-14.3.3 and Pan Cn proteins expression assessed by Western-blot analysis normalized to loading control GAPDH. d Relative expression of Serca2a mRNA assessed by quantitative PCR. Data are presented as mean ± SEM. *p < 0.05, ***p < 0.001. Student’s t-test
By contrast, the P-Akt/Akt ratio was unchanged in the LV from Trpm4 −/− mice after endurance training (Fig. 4c, left panel). The 14-3-3 protein expression was also unchanged after training (Fig. 4c, middle panel). Surprisingly, the Cn protein level was increased in Tr-Trpm4 −/− LV lysate when compared to Sed-Trpm4 −/− (Fig. 4c, right panel). The activated Cn-NFAT pathway is associated with pathological CH, heart failure and decreased Serca2a transcript (Molkentin et al. 1998). Validating this in our Trpm4 −/− mice, we observed a reduction of Serca2a mRNA expression of the LV from Tr-Trpm4 −/− mice in comparison with Sed-Trpm4 −/− (Fig. 4d). Thus, endurance training in Trpm4 −/− mice is associated with an increase in Cn expression, a hallmark of pathological CH.
TRPM4 channel regulates SOCE with endurance training
TRPM4 is a negative regulator of store-operated-Ca2+-entry (SOCE) in the immune system (Launay et al. 2004; Vennekens et al. 2007; Barbet et al. 2008; Serafini et al. 2012), as well as in cardiomyocytes (Kecskés et al. 2015), allowing a negative control of the activation of the Cn-NFAT signalling pathway in pathological hypertrophy. We investigated whether this regulation is also present in isolated LV cardiomyocytes from Sed- and Tr-Trpm4 +/+ mice. After depletion of sarcoplasmic reticulum Ca2+ content by caffeine (5 mmol/L) in a Ca2+-free Tyrode solution, SOCE was measured following the addition of 1 mmol/L of free Ca2+. SOCE was similar between isolated LV cardiomyocytes from Sed-Trpm4 +/+ and Tr-Trpm4 +/+ mice (Fig. 5a). To detect whether TRPM4 channel is involved in the regulation of calcium entry as was demonstrated in the immune system, we pre-incubate the cardiomyocytes with 10 µM 9-P during SOCE measurement (Vennekens et al. 2007; Barbet et al. 2008; Serafini et al. 2012). Pre-incubation with 9-P (Fig. 5b) increased SOCE in LV myocytes from Tr-Trpm4 +/+ mice only and not from Sed-Trpm4 +/+ mice. Calculation of the area under the curve (AUC) demonstrated the significance of this observation (Fig. 5c). These results suggest that the TRPM4 channel is open during SOCE. Increased SOCE in the presence of 9-P suggests that the TRPM4 channel, when opened, negatively regulates Ca2+ entry. Altogether, this data suggests a potential involvement of the TRPM4 channel in CH adaptions via the regulation of Ca2+ entry.

Store-operated calcium entry measurement in freshly isolated cardiomyocytes from Trpm4 +/+ and Trpm4 −/− mice a Mean fluorescence evolution during the application of 1 mM free Ca2+ solution in Sed (in black) and Tr-Trpm4 +/+ (in grey) freshly isolated myocytes. b Same experience as in a. In presence of 9-Phenanthrol (9P) in Sed- and Tr-Trpm4 −/− freshly isolated myocytes. c The calculated areas under the curve after calcium application from single cell recording are presented in histograms. d Mean fluorescence evolution during the application of 1 mM free Ca2+ solution in Sed (in black) and Tr-Trpm4 −/− (in grey) freshly isolated myocytes. e Same experience as in d. in presence of 9P in Sed and Tr-Trpm4 −/− freshly isolated myocytes. f The calculated areas under the curve after calcium application from single cell recording are presented in histograms. Data are presented as mean ± SEM. ***p < 0.001. Two-way ANOVA analysis
SOCE was also measured in isolated cardiomyocytes from Sed-Trpm4 −/− and Tr-Trpm4 −/− mice. No changes in SOCE amplitude were identified between basal conditions and 9-P treatment in isolated myocytes from either Sed-Trpm4 −/− or Tr-Trpm4 −/− mice (Fig. 5d, e). The absence of a difference in AUC calculations (Fig. 5f) reinforces the observation that 9-P unmasked the TRPM4 functional expression in Tr-Trpm4 +/+ mice and strengthened the SOCE negative regulation by TRPM4 channel in cardiomyocytes.
Discussion
In this study, as expected, Trpm4 +/+ mice displayed increased LV heart muscle mass associated with cardiomyocyte hypertrophy in response to endurance training. We confirmed activation of the pro-survival PI3K-Akt signal transduction pathway in the hearts of Tr-Trpm4 +/+ mice by an increased P-Akt/Akt ratio, the branch point of the physiological CH signaling cascade (Kemi et al. 2008; Bernardo et al. 2010). We revealed a concomitant increase of the functional expression of the TRPM4 channel after 4 weeks of endurance training in Trpm4 +/+ mice that was not evident in sedentary mice. When incubated in the presence of 9-P, a TRPM4 channel inhibitor, cardiomyocytes from Tr-Trpm4 +/+ mice exhibited an increased Ca2+-entry through store-operated Ca2+ channels, consistent with the negative regulation of SOCE by the TRPM4 channel. By contrast, in the absence of TRPM4 in Trpm4 −/− mice, no change on SOCE was detected with or without 9-P incubation, validating the direct involvement of TRPM4 channel activity in cardiomyocytes from Tr-Trpm4 +/+ mice.
Surprisingly, in the TRPM4 channel deficient mice, despite their basal hypertrophy due to an increased number of cardiomyocytes (Demion et al. 2014), we did not observe an increase of heart size or functional modifications in response to training while cardiomyocytes hypertrophied. We hypothesized that this phenomenon could be related to an increased number of apoptotic cells. Apoptosis, or programmed cell death, when increased, is an indicator of heart failure (Cho et al. 2016). In Tr-Trpm4 −/− mice, the increase of TUNEL+ nuclei, a marker of apoptotic cells, correlated with the activation of the pathological CH Cn-NFAT pathway without any modifications of PI3K-Akt pathway. Altogether, these data suggest that the IGF-1-PI3K-Akt pathway activation by endurance training positively correlates with TRPM4 channel functional expression. This functional expression could, in turn, contribute to the negative control of Ca2+-entry and thus prevent the pathological, pro-hypertrophic Ca2+-dependent Cn-NFAT signaling pathway (Fig. 6). Consistent with our data, it was recently demonstrated that TRPM4 channel regulates the pathological hypertrophy induced by Angiotensin II infusion (Kecskés et al. 2015). Specifically, transgenic mice with a cardiac specific Trpm4 gene deletion displayed a worsened CH associated with an augmented activation of the Cn-NFAT signaling pathway (Kecskés et al. 2015).

Proposed model of TRPM4 channel rolein CH signalling. The IGF-1-PI3K-Akt pathway activation by endurance training induces the activation of the chaperone protein 14-3-3. This chaperone protein may induce increased expression of TRPM4 channel at the plasma membrane. Once expressed at the membrane, TRPM4 channel can negatively regulates SOCE and thus the Cn-NFAT pathway. This may be beneficial for Ph-CH as well as Pa-CH remodelling due to a favouring of the IGF-1-PI3K-Akt pathway. When TRPM4 channel expression is blunted, there is no inhibitory regulation of Cn-NFAT pathway. Due to this, the IGF-1-PI3K-Akt pathway is counterbalanced by the Cn-NFAT pathway, leading to no adaptions to training. Abbreviations: SR sarcoplasmic reticulum; IGF insulin growth factor; IGFR IGF receptor; Et-1 endothelin-1; PI3K phosphoinositide 3 kinase; SOCs store operated channels; SACs stretch activated channels; ROCs receptor operated channels; CaM calmodulin; Cn calcineurin; NFAT nuclear factor of activated T cell; PMI post-myocardial infarction
TRPM4 channel belongs to the TRPM (transient receptor potential melastatin) channel family. This heterogeneous family includes ionic channels involved in melanoma aggressiveness (TRPM1) (Dalal et al. 2013) to channels involved in menthol and cold sensation (TRPM8) (Palkar et al. 2015). TRPM6 and TRPM7 are very particular members of the family due to a kinase domain capable of phosphorylating itself and other substrates (Runnels et al. 2001; Runnels 2011). While most of TRP channels are calcium permeable, TRPM4 and TRPM5 channels are the exception by their ability to be selective to sodium and potassium but not calcium (Ullrich et al. 2005). Although TRPM4 and TRPM5 channels have similar actions, they do not share the same pattern of expression and cannot compensate each other’s expression (Barbet et al. 2008). While it was demonstrated that TRPM7 channel was important in the cardiogenesis and automaticity (Sah et al. 2013a, b), TRPM6 was involved in atrial fibrillation (Zhang et al. 2015). TRPM4 channel is the most studied channel in the heart function, particularly in conduction disorders (Liu et al. 2010, 2013; Simard et al. 2012, 2013; Abriel et al. 2012; Mathar et al. 2014; Jacobs et al. 2015; Hof et al. 2016).
TRPM4 channel is known to negatively regulate SOCE in several cell types including non-excitable and excitable cells, but also in heterologous expression systems (Launay et al. 2004; Vennekens et al. 2007; Barbet et al. 2008; Serafini et al. 2012; Kecskés et al. 2015). For example, single ventricular cardiomyocytes from cardiac-specific TRPM4 KO mice displayed a significant increase of SOCE when compared to cardiomyocytes from WT mice after angiotensin II treatment (Kecskés et al. 2015). In line with this, in the present study, using 9-P, we unmasked a Ca2+ entry regulation by TRPM4 activity in cardiomyocytes from Tr-Trpm4 +/+ mice. This regulation is absent in cardiomyocytes from Tr-Trpm4 −/−. These results, in combination with previous studies, demonstrate that the TRPM4 channel negatively regulates Ca2+ entry in hypertrophied cardiomyocytes. By regulating Ca2+-entry into the cardiomyocytes, TRPM4 channel might thus be able to regulate the Ca2+-dependent Cn-NFAT activation pathway.
In hearts from patients with heart failure (HF) and presenting CH, Cn activity is increased (Haq et al. 2001; Diedrichs et al. 2004). Regular physical activity can improve the quality of life in patients with chronic HF and reverse pathological cardiac remodeling. In mice presenting HF with CH and activated Cn-NFAT signaling pathway, exercise training can prevent the activation of the pathological CH Cn-NFAT pathway (Oliveira et al. 2009). Consistently, we demonstrate here that, concomitant to TRPM4 channel functional expression, the physiological cardiac hypertrophy signaling pathway IGF-1-PI3K-Akt is favored by endurance training, whereas Cn expression remains unchanged. The opposite results were identified when TRPM4 is absent, an elevated Cn expression without enhanced Akt phosphorylation. A balance between these CH signaling pathways similar to that in the current study has been reported previously. In cultured cardiomyocytes from rat, endothelin-1 induced hypertrophy was inhibited by the peroxisome proliferator activated receptor alpha (PPAR-α) activation via Akt phosphorylation and NFAT nuclear translocation prevention (Li et al. 2007). These evidences suggest that a counter balance may exist in the regulation of these signaling pathways.
We also correlated an increased expression of the 14-3-3 protein with Akt activation after training. The 14-3-3 protein is a chaperone protein, activated by the Akt pathway, and involved in the response to IGF-1. 14-3-3 recognizes and conducts Serine/threonine phosphorylated proteins to their target destination. A recent study demonstrated that a physical interaction exists between the 14-3-3 protein and the TRPM4 channel protein in the brain (Cho et al. 2014). Our data suggest that during endurance training, the activation of Akt by phosphorylation is associated with an increase of the 14-3-3 protein expression (Qi et al. 2010). This increased 14-3-3 protein may in turn favor TRPM4 channel expression at the plasma membrane and thus its function (Cho et al. 2014). Finally, this improved TRPM4 channel functional expression may negatively regulate Ca2+-entry through the plasma membrane and thus the activation of the Cn-NFAT pathway. However, in the absence of TRPM4 channel expression during endurance training, cellular hypertrophy pairs with elevated Cn expression, suggesting that TRPM4 expression may be linked to the physiological CH signaling pathway.
Additional studies have correlated the Akt pathway to TRP channel function. Indeed, it was demonstrated that in the absence of TRPM4 channel in macrophages, impaired calcium mobilization and down-regulated Akt signaling pathway were correlated (Serafini et al. 2012). It was also shown that TRPC channels are involved in the IGF1R-PI3K-Akt pathway via calcium signal in epithelial cells (Dai et al. 2014). Moreover, PI3K pathway activation was shown to favor TRPC6 expression by increasing its exocytosis (Xie et al. 2012).
In summary, these data suggest that TRPM4 expression may be protective by correlating with the pro-survival effects of the IGF-1-PI3K-Akt signal transduction pathway. This may in turn involve a regulatory effect of enhanced 14.3.3 protein expression in association with the TRPM4 channel. In addition, these finding further support a negative regulation of TRPM4 in the Cn pathway. In conclusion, these findings strengthen the importance of the TRPM4 channel in the cardiac physiopathology.
Materials and methods
All procedures conformed to the European parliament directive 2010/63/EU and were approved by the local institutional committee (CEEA-LR-12110).
Animals
Knock-out mice (Trpm4 −/−) and wild-type littermate controls (Trpm4 +/+) obtained as previously described (Barbet et al. 2008; Demion et al. 2014) were randomly separated into four groups: Sedentary-Trpm4 +/+ (Sed-Trpm4 +/+), Trained-Trpm4 +/+ (Tr-Trpm4 +/+), Sedentary-Trpm4 −/− (Sed-Trpm4 −/−) and Trained- Trpm4 −/− (Tr-Trpm4 −/−). Mice were sacrificed at 14 weeks of age by cervical dislocation.
Exercise training protocol
Trpm4 +/+ and Trpm4 −/− mice were assigned randomly to the sedentary or trained group. Sedentary mice at 12 weeks old were subjected to 2 weeks of familiarization on the rodent treadmill (15 min at 12 m/min, 5% incline, 5 days/week) and with the experimenter. Trained mice, at 8 weeks old, were subjected to the same 2 weeks of treadmill familiarization, followed an endurance training of 4 weeks (70% of their maximal aerobic speed, from 30 min/day to 75 min/day with a 15 min increase /week, 5 days/week, 5% incline). Before the beginning of the 4 weeks endurance training, all mice underwent an exhaustive exercise to determine maximal aerobic speed of each individual mouse. Briefly, exhaustive exercise consisted of: 10 min at 10 m/min, followed by increased treadmill speed by 2 m/min every 2 min until exhaustion and fatigue of the mouse. Exhaustion was defined as the mouse’s inability to maintain running and 5 × 5 sec on the electrical grid stimulus. The speed at exhaustion was recorded as their maximal aerobic speed (Supplementary Fig. 1).
Echocardiography
Trpm4 +/+ and Trpm4 −/− mice were anesthetized with isoflurane and echocardiography was performed using Vevo 2100 ultrasound system (VisualSonics, Toronto, Canada) equipped with a real-time micro-visualization scan head probe (MS-550D) operating at a frame rate ranging from 740 frames per sec and a nosepiece-transducer used with a central frequency of 22–50 MHz, as previously described (Demion et al. 2014). All measurements and calculations were performed with the Vevo 2100 Software and were averaged from a minimum of four cycles and followed the standards of the American society of echocardiology and the Vevo 2100 protocol-based measurements and calculations guidelines. Echocardiography was performed in each trained animal at the end of the treadmill familiarization and following 4 weeks of endurance training. Before the beginning of the 4 weeks endurance training, to determine maximal aerobic speed of mice, all mice underwent an exhaustive exercise. Briefly, exhaustive exercise consists in: after 10 min at 10 m/min, treadmill speed was increased by 2 m/min every 2 min until exhaustion and fatigue of the mouse. Exhaustion was defined as the mouse inability to maintain running and 5 × 5 sec on the electrical grid stimulus. The speed of exhaustion was recorded as their maximal aerobic speed (Supplementary Fig. 1).
Cardiomyocytes isolation
The mouse was euthanized by cervical dislocation, after which the heart was rapidly excised and retrogradely perfused (Langendorff) at 37 °C for 6–8 min with a modified Tyrode solution (in mmol/L: NaCl 113; KCL 4.7; KH2PO4 0.6; Na2HPO4 0.6; MgSO4 1.2; NaHCO3 12; KHCO3 10; HEPES 10; Taurine 30; pH 7.4) containing 0.1 g/mL liberase dispase high research grade (Roche, France). Cells were kept in 1.8 mmol/L Ca2+ (20–22 °C) before starting recordings experiments.
Patch clamp recordings
Whole-cell patch-clamp recordings were performed (Axon Instruments, Foster City, CA) at room temperature (22–24 °C) to record cell membrane capacitance in LV myocytes enzymatically isolated from Trpm4 +/+ and Trpm4 −/− mice. Cell capacitance was measured following brief voltage steps (±10 mV) from the holding potential to provide an estimation of cell size.
TRPM4-like current recording were performed using the whole cell configuration of the patch-clamp technique using an Axopatch 200B (Axon instruments). Extracellular solution contained (mmol/L): 140 LiCl, 1.8 CaCl2, 1 MgCl2, 10 HEPES, 10 glucose, 20 tetraethylammonium chloride (TEA) buffered at pH 7.4 with LiOH. Pipette solution contained (mmol/L): 140 CsCl, 20 TEA-Cl, 1 MgCl2, 10 HEPES and 10 EGTA, pH 7.2 with CsOH. The Ca2+ concentration in the pipette was adjusted to 60 nmol/L by adding appropriate amounts of CaCl2 calculated by the CaBuf program. Patch electrodes had a DC resistance between 2 and 4 MΩ when filled with the different recording solutions. Currents were sampled at 2.5 kHz and filtered at 10 kHz. The protocol consisted of test pulses which were preceded by five depolarizing prepulses from −60 to +10 mV in order to load the SR with Ca2+ to a similar extent, followed by 5 s pulse from −40 to 40 mV with 20 mV ΔVm. Data were analyzed using the pClamp 10.0 software (molecular devices corporation).
Calcium measurements
Changes in intracellular Ca2+ concentration [(Ca2+)i] were determined in ventricular cardiomyocytes using the ratiometric fluorescent Ca2+ indicator Fura-2 probe. Cells were loaded in MaTteck by incubation with 2.5 µM Fura-2AM (Tef-labs) plus 0.02% pluronic acid F-127 (Molecular Probes Inc) in Ringer EGTA solution (in mM: 145 NaCl, 5.4 KCl, 1 MgCl2, 10 glucose, 10 N-(hydroxyethyl)piperazine-N′-2-ethanesulfonic acid, 5 mM EGTA; pH was adjusted to 7.5 with NaOH) during 30 min at 37 °C in a humidified air atmosphere. The loaded cells were then washed and placed with Ringer EGTA solution without Ca2+ and containing Nifedipine 10 µM and KB-R7943 5 µM and mounted on the microscope stage at room temperature for 15 min to allow Fura-2AM de-esterification. Measurements of the Fura-2 probe fluorescence ratio were made from images captured digitally, every second, by a cooled CCD camera (Photometrics) with a X20 objective lens mounted on an inverted microscope (Axiovert, Zeiss), and acquired after excitations at 340- and 380 nm using a lambda-DG4 excitation system (Sutter Instrument Company, USA). Ca2+ signals were then analyzed by using the Metafluor software (Universal Imaging Corporation, USA). The results are given as the variation from basal values of the ratio of 340/380 nm recordings with fluorescence values corrected for background and dark currents.
Western blot analysis
Frozen left ventricle (LV) tissue (≈50 mg) was homogenized using a manual polytron instrument. LV tissue was lysed under rotating agitation during 45 min in an extraction buffer containing protease and phosphatase inhibitors (Roche). After 5 min of centrifugation at 10,000 g at 4 °C, proteins were collected from the supernatant. Protein expression was assessed by immunoblot analysis with the primary antibodies anti-PhosphoSer473-Akt (P-Akt; #4060), anti-Akt (#9171), Pan-CalcineurinA (#2614) and Pan 14-3-3 (#8312) (Cell Signaling, Ozyme, Montigny-le-Bretonneux, France). Each protein was quantified using glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (PA1-913, Fisher Scientific, Illkirch, France) as a loading control with ImageJ software. All immunoblots were visualized using the Odyssey® infrared imaging system (LI-COR Biosciences, Lincoln, Nebraska, USA) coupled to infrared-labeled anti-goat, anti-mouse or anti-rabbit IgG secondary antibodies (1/30,000 dilution) (LI-COR Biosciences, Courtaboeuf, France).
TUNEL assay method
Apoptotic nuclei were detected in heart sections by in situ terminal deoxynucleotidyl transferase (TdT)-mediated digoxigenin-conjugated deoxyuridine triphosphate (dUTP) nick end-labeling (TUNEL), with the In Situ Cell Death Detection Kit (Roche Diagnostics, Mannheim, Germany) in accordance with manufacturer recommendations. The total number of TUNEL-positive cardiomyocytes, indicated by a green nucleus, per section was quantified. dUTP labelling was not observed when TdT was omitted from the reaction (negative control). The number of TUNEL+ cells was determined as the number of labeled positive cardiomyocytes per section divided by the total area (mm²).
Wheat germ agglutinin labelling
Adult heart 10 µm cryosections were stained with wheat germ agglutinin (WGA 1:250; Vector Labs) counterstained with 4′,6-diamidino-2-phenylindole (DAPI, 1:1000 from Sigma–Aldrich), and examined using AxioVision Rel.4.8 software.
PCR
Genomic PCR was performed on tail DNA with primers (Eurofin genomics, Ebersberg, Deutschland) specific for the wild-type and null alleles as described before (Barbet et al. 2008). Total RNA was isolated from a minimum of five samples per group using a Nucleospin total RNA isolation kit (Macherey–Nagel, Hoerdt, France) according to the manufacturer’s instructions. Total RNA, oligo-dT and random hexamer primers were used to generate cDNA using a Verso enzyme kit (ThermoFischer, Illkirch, France). RT-PCR for the evaluation of the expression of Trpm4, Serca2a and Gapdhwas performed using gene- specific primers and performed in duplicate. Reactions were achieved using SYBR green Mix (Roche Applied System, Meylan, France) and commercially prepared primers (Sigma-Aldrich, Saint-Quentin Fallavier, France) (see primer sequences in Table 1).
Primer | Forward | Reverse |
|---|---|---|
mTrpm4 | CCAGGGCTCATTCCCCGCAAGGT | ATTCACCTGCCTCTTCCTCCCG |
mSerca2a | AGTTCATCCGCTACCTCATCTCA | CACCAGATTGACCCAGAGTAACTG |
Statistics
All statistical analyses and artworks were performed using Prism (GraphPad) software. Data are expressed as mean ± SEM. Differences between groups were compared by two-sample t test and two-way repeated measurement ANOVA. When assumptions were not valid, non-parametric tests were performed (Mann–Whitney test). For comparison of echocardiographic parameters recorded in the same mice before and after training, called pre- versus post-training respectively, a paired t-test was used. P-values of 0.05 or less indicated significant difference between groups. *P < 0.05; **P < 0.01; ***P < 0.001; ns no significant.
References
Abriel H, Syam N, Sottas V et al (2012) TRPM4 channels in the cardiovascular system: physiology, pathophysiology, and pharmacology. Biochem Pharmacol 84:873–881. doi:10.1016/j.bcp.2012.06.021
Barbet G, Demion M, Moura IC et al (2008) The calcium-activated nonselective cation channel TRPM4 is essential for the migration but not the maturation of dendritic cells. Nat Immunol 9:1148–1156. doi:10.1038/ni.1648
Bernardo BC, Weeks KL, Pretorius L, McMullen JR (2010) Molecular distinction between physiological and pathological cardiac hypertrophy: experimental findings and therapeutic strategies. Pharmacol Ther 128:191–227. doi:10.1016/j.pharmthera.2010.04.005
Bers DM (2008) Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol 70:23–49. doi:10.1146/annurev.physiol.70.113006.100455
Bush EW, Hood DB, Papst PJ et al (2006) Canonical transient receptor potential channels promote cardiomyocyte hypertrophy through activation of calcineurin signaling. J Biol Chem 281:33487–33496. doi:10.1074/jbc.M605536200
Cho C-H, Kim E, Lee Y-S et al (2014) Depletion of 14-3-3γ reduces the surface expression of transient receptor potential melastatin 4b (TRPM4b) channels and attenuates TRPM4b-mediated glutamate-induced neuronal cell death. Mol Brain 7:52. doi:10.1186/s13041-014-0052-3
Cho GW, Altamirano F, Hill JA (2016) Chronic heart failure: Ca(2+), catabolism, and catastrophic cell death. Biochim Biophys Acta 1862:763–777. doi:10.1016/j.bbadis.2016.01.011
Colquhoun D, Neher E, Reuter H, Stevens CF (1981) Inward current channels activated by intracellular Ca in cultured cardiac cells. Nature 294:752–754
Dai W, Bai Y, Hebda L et al (2014) Calcium deficiency-induced and TRP channel-regulated IGF1R-PI3K-Akt signaling regulates abnormal epithelial cell proliferation. Cell Death Differ 21:568–581. doi:10.1038/cdd.2013.177
Dalal MD, Morgans CW, Duvoisin RM et al (2013) Diagnosis of occult melanoma using transient receptor potential melastatin 1 (TRPM1) autoantibody testing: a novel approach. Ophthalmology 120:2560–2564. doi:10.1016/j.ophtha.2013.07.037
DeBosch B, Treskov I, Lupu TS et al (2006) Akt1 is required for physiological cardiac growth. Circulation 113:2097–2104. doi:10.1161/CIRCULATIONAHA.105.595231
Demion M, Thireau J, Gueffier M et al (2014) Trpm4 gene invalidation leads to cardiac hypertrophy and electrophysiological alterations. PLoS ONE 9:e115256. doi:10.1371/journal.pone.0115256
Diedrichs H, Chi M, Boelck B et al (2004) Increased regulatory activity of the calcineurin/NFAT pathway in human heart failure. Eur J Heart Fail 6:3–9. doi:10.1016/j.ejheart.2003.07.007
Earley S (2013) TRPM4 channels in smooth muscle function. Pflugers Arch 465:1223–1231. doi:10.1007/s00424-013-1250-z
Eghbali M, Deva R, Alioua A et al (2005) Molecular and functional signature of heart hypertrophy during pregnancy. Circ Res 96:1208–1216. doi:10.1161/01.RES.0000170652.71414.16
Grand T, Demion M, Norez C et al (2008) 9-phenanthrol inhibits human TRPM4 but not TRPM5 cationic channels. Br J Pharmacol 153:1697–1705. doi:10.1038/bjp.2008.38
Guinamard R, Rahmati M, Lenfant J, Bois P (2002) Characterization of a Ca2+-activated nonselective cation channel during dedifferentiation of cultured rat ventricular cardiomyocytes. J Membr Biol 188:127–135. doi:10.1007/s00232-001-0180-4
Guinamard R, Demion M, Magaud C et al (2006) Functional expression of the TRPM4 cationic current in ventricular cardiomyocytes from spontaneously hypertensive rats. Hypertension 48:587–594. doi:10.1161/01.HYP.0000237864.65019.a5
Guinamard R, Demion M, Launay P (2010) Physiological roles of the TRPM4 channel extracted from background currents. Physiology 25:155–164. doi:10.1152/physiol.00004.2010
Haq S, Choukroun G, Lim H et al (2001) Differential activation of signal transduction pathways in human hearts with hypertrophy versus advanced heart failure. Circulation 103:670–677
Hof T, Sallé L, Coulbault L et al (2016) TRPM4 non-selective cation channels influence action potentials in rabbit Purkinje fibres. J Physiol 594:295–306. doi:10.1113/JP271347
Ikeda H, Shiojima I, Ozasa Y et al (2009) Interaction of myocardial insulin receptor and IGF receptor signaling in exercise-induced cardiac hypertrophy. J Mol Cell Cardiol 47:664–675. doi:10.1016/j.yjmcc.2009.08.028
Jacobs G, Oosterlinck W, Dresselaers T et al (2015) Enhanced β-adrenergic cardiac reserve in Trpm4−/− mice with ischaemic heart failure. Cardiovasc Res 105:330–339. doi:10.1093/cvr/cvv009
Kecskés M, Jacobs G, Kerselaers S et al (2015) The Ca(2+)-activated cation channel TRPM4 is a negative regulator of angiotensin II-induced cardiac hypertrophy. Basic Res Cardiol 110:43. doi:10.1007/s00395-015-0501-x
Kemi OJ, Ceci M, Wisloff U et al (2008) Activation or inactivation of cardiac Akt/mTOR signaling diverges physiological from pathological hypertrophy. J Cell Physiol 214:316–321. doi:10.1002/jcp.21197
Kim J, Wende AR, Sena S et al (2008) Insulin-like growth factor I receptor signaling is required for exercise-induced cardiac hypertrophy. Mol Endocrinol 22:2531–2543. doi:10.1210/me.2008-0265
Köster OF, Szigeti GP, Beuckelmann DJ (1999) Characterization of a [Ca2+]i-dependent current in human atrial and ventricular cardiomyocytes in the absence of Na+ and K+. Cardiovasc Res 41:175–187
Kuwahara K, Wang Y, McAnally J et al (2006) TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. J Clin Invest 116:3114–3126. doi:10.1172/JCI27702
Launay P, Fleig A, Perraud AL et al (2002) TRPM4 is a Ca2+-activated nonselective cation channel mediating cell membrane depolarization. Cell 109:397–407
Launay P, Cheng H, Srivatsan S et al (2004) TRPM4 regulates calcium oscillations after T cell activation. Science 306:1374–1377. doi:10.1126/science.1098845
Li R, Zheng W, Pi R et al (2007) Activation of peroxisome proliferator-activated receptor-alpha prevents glycogen synthase 3beta phosphorylation and inhibits cardiac hypertrophy. FEBS Lett 581:3311–3316. doi:10.1016/j.febslet.2007.06.017
Li Y, Baylie RL, Tavares MJ, Brayden JE (2014) TRPM4 channels couple purinergic receptor mechanoactivation and myogenic tone development in cerebral parenchymal arterioles. J Cereb Blood Flow Metab 34:1706–1714. doi:10.1038/jcbfm.2014.139
Liu H, El Zein L, Kruse M et al (2010) Gain-of-function mutations in TRPM4 cause autosomal dominant isolated cardiac conduction disease. Circ Cardiovasc Genet 3:374–385. doi:10.1161/CIRCGENETICS.109.930867
Liu H, Chatel S, Simard C et al (2013) Molecular genetics and functional anomalies in a series of 248 Brugada cases with 11 mutations in the TRPM4 channel. PLoS ONE 8:e54131. doi:10.1371/journal.pone.0054131
Makarewich CA, Zhang H, Davis J et al (2014) Transient receptor potential channels contribute to pathological structural and functional remodeling after myocardial infarction. Circ Res 115:567–580. doi:10.1161/CIRCRESAHA.115.303831
Mann N, Rosenzweig A (2012) Can exercise teach us how to treat heart disease? Circulation 126:2625–2635. doi:10.1161/CIRCULATIONAHA.111.060376
Mathar I, Kecskes M, Van der Mieren G et al (2014) Increased β-adrenergic inotropy in ventricular myocardium from Trpm4−/− mice. Circ Res 114:283–294. doi:10.1161/CIRCRESAHA.114.302835
Molkentin JD, Lu JR, Antos CL et al (1998) A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell 93:215–228
Nakayama H, Wilkin BJ, Bodi I, Molkentin JD (2006) Calcineurin-dependent cardiomyopathy is activated by TRPC in the adult mouse heart. FASEB J 20:1660–1670. doi:10.1096/fj.05-5560com
Nilius B, Prenen J, Droogmans G et al (2003) Voltage dependence of the Ca2+-activated cation channel TRPM4. J Biol Chem 278:30813–30820. doi:10.1074/jbc.M305127200
Nilius B, Prenen J, Janssens A et al (2005) The selectivity filter of the cation channel TRPM4. J Biol Chem 280:22899–22906. doi:10.1074/jbc.M501686200
Oliveira RSF, Ferreira JCB, Gomes ERM et al (2009) Cardiac anti-remodelling effect of aerobic training is associated with a reduction in the calcineurin/NFAT signalling pathway in heart failure mice. J Physiol 587:3899–3910. doi:10.1113/jphysiol.2009.173948
Onohara N, Nishida M, Inoue R et al (2006) TRPC3 and TRPC6 are essential for angiotensin II-induced cardiac hypertrophy. EMBO J 25:5305–5316. doi:10.1038/sj.emboj.7601417
Palkar R, Lippoldt EK, McKemy DD (2015) The molecular and cellular basis of thermosensation in mammals. Curr Opin Neurobiol 34:14–19. doi:10.1016/j.conb.2015.01.010
Park J-Y, Hwang EM, Yarishkin O et al (2008) TRPM4b channel suppresses store-operated Ca2+ entry by a novel protein-protein interaction with the TRPC3 channel. Biochem Biophys Res Commun 368:677–683. doi:10.1016/j.bbrc.2008.01.153
Qi J-Y, Xu M, Lu Z-Z, Zhang Y-Y (2010) 14-3-3 inhibits insulin-like growth factor-I-induced proliferation of cardiac fibroblasts via a phosphatidylinositol 3-kinase-dependent pathway. Clin Exp Pharmacol Physiol 37:296–302. doi:10.1111/j.1440-1681.2009.05282.x
Runnels LW (2011) TRPM6 and TRPM7: a Mul-TRP-PLIK-cation of channel functions. Curr Pharm Biotechnol 12:42–53
Runnels LW, Yue L, Clapham DE (2001) TRP-PLIK, a bifunctional protein with kinase and ion channel activities. Science 291:1043–1047. doi:10.1126/science.1058519
Sah R, Mesirca P, Mason X et al (2013a) Timing of myocardial trpm7 deletion during cardiogenesis variably disrupts adult ventricular function, conduction, and repolarization. Circulation 128:101–114. doi:10.1161/CIRCULATIONAHA.112.000768
Sah R, Mesirca P, Van den Boogert M et al (2013b) Ion channel-kinase TRPM7 is required for maintaining cardiac automaticity. Proc Natl Acad Sci USA 110:E3037–E3046. doi:10.1073/pnas.1311865110
Serafini N, Dahdah A, Barbet G et al (2012) The TRPM4 channel controls monocyte and macrophage, but not neutrophil, function for survival in sepsis. J Immunol 189:3689–3699. doi:10.4049/jimmunol.1102969
Shimizu I, Minamino T (2016) Physiological and pathological cardiac hypertrophy. J Mol Cell Cardiol 97:245–262. doi:10.1016/j.yjmcc.2016.06.001
Simard C, Sallé L, Rouet R, Guinamard R (2012) Transient receptor potential melastatin 4 inhibitor 9-phenanthrol abolishes arrhythmias induced by hypoxia and re-oxygenation in mouse ventricle. Br J Pharmacol 165:2354–2364. doi:10.1111/j.1476-5381.2011.01715.x
Simard C, Hof T, Keddache Z et al (2013) The TRPM4 non-selective cation channel contributes to the mammalian atrial action potential. J Mol Cell Cardiol 59:11–19. doi:10.1016/j.yjmcc.2013.01.019
Tao L, Bei Y, Zhang H et al (2015) Exercise for the heart: signaling pathways. Oncotarget 6:20773–20784. doi:10.18632/oncotarget.4770
Ullrich ND, Voets T, Prenen J et al (2005) Comparison of functional properties of the Ca2+-activated cation channels TRPM4 and TRPM5 from mice. Cell Calcium 37:267–278. doi:10.1016/j.ceca.2004.11.001
Umar S, Nadadur R, Iorga A et al (2012) Cardiac structural and hemodynamic changes associated with physiological heart hypertrophy of pregnancy are reversed postpartum. J Appl Physiol 113:1253–1259. doi:10.1152/japplphysiol.00549.2012
Vennekens R, Olausson J, Meissner M et al (2007) Increased IgE-dependent mast cell activation and anaphylactic responses in mice lacking the calcium-activated nonselective cation channel TRPM4. Nat Immunol 8:312–320. doi:10.1038/ni1441
Wilkins BJ, Molkentin JD (2004) Calcium-calcineurin signaling in the regulation of cardiac hypertrophy. Biochem Biophys Res Commun 322:1178–1191. doi:10.1016/j.bbrc.2004.07.121
Wilkins BJ, Dai Y-S, Bueno OF et al (2004) Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ Res 94:110–118. doi:10.1161/01.RES.0000109415.17511.18
Xie J, Cha S-K, An S-W et al (2012) Cardioprotection by Klotho through downregulation of TRPC6 channels in the mouse heart. Nat Commun 3:1238. doi:10.1038/ncomms2240
Zhang Y-J, Ma N, Su F et al (2015) Increased TRPM6 expression in atrial fibrillation patients contribute to atrial fibrosis. Exp Mol Pathol 98:486–490. doi:10.1016/j.yexmp.2015.03.025
Acknowledgements
This work was supported by École Doctorale Sciences du Mouvement Humain (SMH) ED463 and the “Agence Nationale pour Recherche” -Target channel (RPV09046F5A).
Author information
Authors and Affiliations
Corresponding author
Additional information
Mélanie Gueffier and Justin Zintz have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Gueffier, M., Zintz, J., Lambert, K. et al. The TRPM4 channel is functionally important for the beneficial cardiac remodeling induced by endurance training. J Muscle Res Cell Motil 38, 3–16 (2017). https://doi.org/10.1007/s10974-017-9466-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10974-017-9466-8