
Abstract
The effect of powder particle size on non-isothermal crystallization kinetics of Ge11Ga11Te78 glass was examined by differential scanning calorimetry. Multivariate kinetic analysis was used to deconvolute the complex crystallization data. The overall process was found to be composed of initial tellurium precipitation followed by simultaneous formation of Ga2Te5 and GeTe phases. While the Te precipitation followed the nucleation growth Johnson–Mehl–Avrami kinetics with the growth originating mainly from surface and mechanically induced defects, the formation of gallium and germanium tellurides proceeded autocatalytically with a significantly lower involvement of the defects-based crystal growth. The model-based part of the kinetic behavior was found to be uninfluenced by the change in powder coarseness—both crystallization sub-processes were described in terms of similar models with similar kinetic exponents for all particle size fractions. On the other hand, the model-free kinetic parameters (activation energies, pre-exponential factors and crystallization enthalpies) were found to be significantly changing with increasing particle size; the observed trends were explained in terms of the changing amounts of the two involved types of crystallization centers.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The Ge–Ga–Te chalcogenide glasses belong among the most promising materials for far-infrared optics applications. Since the Ge–Ga–Te chalcogenide matrix is fully tellurous, the transmission window is extended to ~28 µm, which enables, for example, detection of H2O and CO2 signals [1]. Since the glass-forming region is in the Ge–Ga–Te system centered along the pseudo-binary GeTe4–GaTe3 line, most of the research articles aimed at the optical applications investigated compositions close to this line [2–8]. Worse glass stability and limiting conditions of the glass-processing procedures (molding, fiber-drawing, etc.) due to the impending crystal growth processes are the main drawbacks of the fully tellurous chalcogenide matrix. At the same time, these are also the main reasons for which the crystallization kinetics of these glasses is often studied, so that the crystallization behavior can be predicted for various experimental conditions. However, while majority of the applications of chalcogenide materials works with bulk glass, the crystallization kinetics is often studied for powders. Therefore, recently a great benefit of the particle size-dependent crystallization studies has been recognized [9–12]. Based on these studies, one is able to extrapolate the crystallization behavior reported for various glassy materials from the powdered to bulk material (reliable extrapolation can be of course made only for similar/related materials).
In the present study, the influence of particle size of the crystallization kinetics of the Ge11Ga11Te78 (50% of GeTe4 and 50% of GaTe3) glass will be studied in detail. This Ge–Ga–Te composition lies approximately in the middle of the glass-forming region, and hence, it is probably very close to the optimum stability. In the study, the major differences between the crystallization behavior of powders and bulk materials will be described and interpreted with respect to the material structure so that the conclusions can be extrapolated to the other Ge–Ga–Te glassy materials.
Experimental
The Ge11Ga11Te78 glass was prepared by the melt-quenching technique (annealing at 950 °C for 24 h, quenching in cold water). The prepared glass ingot was ground in an agate mortar and sieved (using a set of sieves with defined mesh sizes) so that the following powder fractions were prepared: 0–20, 20–50, 50–125, 125–180, 180–250, 250–300 and 300–500 μm. X-ray diffraction technique (XRD) was used to confirm the amorphous character of the prepared material; it was also verified that the grinding procedure did not cause any crystallization. The XRD analysis of amorphous and crystalline samples was performed using a Bruker AXS diffractometer D8 Advance equipped with a horizontal goniometer and scintillation counter utilizing CuKα radiation.
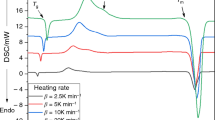
The crystallization experiments were performed using a Q2000 heat flow differential scanning calorimetry (DSC) (TA Instruments) equipped with an autosampler, RCS90 cooling accessory and T-zero technology. The instrument was calibrated using In, Zn, and H2O; dry N2 was used as purge gas at a flow rate of 50 cm3 min−1. Sample masses varied between 8 and 10 mg. The DSC measurements were realized as a simple heating scan from 150 to 350 °C; the following heating rates were applied for each particle size fraction: 0.5, 1, 2, 3, 5, 7, 10, 15, 20 and 30 °C min−1. Very good reproducibility of the experimental data was achieved. In order to achieve maximum precision during the consequent data acquisition, the tangential area-proportional baseline was used to subtract the thermokinetic background (see Fig. 1 for an example of the baseline construction). In Fig. 1, also two examples of DSC curves datasets with subtracted thermokinetic backgrounds are shown. Note that no other crystallization peaks/effects occur at higher temperatures (measured up to the melting).

a Example of the subtraction of the thermokinetic background by means of the tangential area-proportionate baseline. b DSC curves obtained for the 50- to 125-μm powder (data for the lowest heating rates are omitted for better clarity). The thermokinetic background is already subtracted. c DSC curves obtained for the 250 to 300-μm powder (data for the lowest heating rates are omitted for better clarity). The thermokinetic background is already subtracted
Results and discussion
Deconvolution procedure
Figure 1 shows that the crystallization behavior of the Ge11Ga11Te78 glass is complex, exhibiting two overlapping peaks, where the degree of the overlap increases with heating rate (slightly) and particle size. In order to separate the two respective sub-processes, we have applied the kinetic deconvolution based on multivariate analysis [13]—main advantage of this approach lies in the fact that already the primary deconvolution takes into account the basic kinetic equation:
where Φ is the measured heat flow, ΔH is the crystallization enthalpy, A is the pre-exponential factor, E is the apparent activation energy of the process, R is the universal gas constant, T is temperature, and f(α) stands for an expression of a kinetic model with α being conversion. Note that most deconvolution procedures (like, for example, the original Fraser–Suzuki deconvolution [14]) are purely mathematical and only the consequent kinetic analysis can confirm their justness and accuracy.
Regarding the deconvolution methodology, in the first step the model-free methods, Ozawa [15] and Ozawa–Flynn–Wall methods [16], were used to estimate the apparent activation energy of crystallization E and pre-exponential factor A for each set of DSC curves measured for the given particle size. These methods are based on the following respective equations:
where q + is heating rate, T p is the temperature corresponding to the maximum of the peak, T α is the temperature corresponding to certain chosen values of conversion α.
The estimated E and A values are listed in Table 1. Secondly, the appropriate kinetic model was chosen—this step is based mostly on the researcher’s experience and amount of information provided by supplemental techniques like, for example, microscopy or XRD (preliminary mathematical deconvolution can also help to suggest the appropriate kinetic models). Note that the early/late correct choice of the crystallization model can only shorten/prolong the determination of the overall reaction mechanism; it does not have impact of the final quality of the results. Based on the asymmetry of the crystallization peaks (see Fig. 1), we have estimated the overall crystallization mechanism to be composed of one nucleation growth Johnson–Mehl–Avrami [17–20] process (the initial one) and one autocatalytic Šesták-Berggren [21] process (the following one). The JMA and AC models are expressed by the following equations, respectively:
where m, M and N are the kinetic parameters of the two models. The JMA parameter m is well known to reflect the dimensionality of the growing crystallites as well as nucleation conditions (the parameter usually varies between 1 and 4) [20, 21], whereas the M and N parameters of the AC model do not have direct physical meaning as they correspond to the shapes of the two respective sides of the kinetic peak (common values of these parameters range between 0 and 2). Regarding the relation between the two processes, in the first approximation we have set them to be independent. In the third step of the kinetic deconvolution, the multivariate analysis [13] was applied, based on the minimization of the sum of squared residuals RSS according to the following equations:
where n is number of measurements, j is index of the given measurement, First j is the index of the first point of the given curve, Last j is the index of the last point of the given curve, Yexpj,k is the experimental value of the point k of curve j, Ycal j,k is the calculated value of the point k of curve j, and w j,k is weighting factor for the point k of curve j. Note that the expression for w j,k (Eq. 7) is optimized for the DSC measurements, so that each measured curve is weighted despite the different number of measured points and increased errors due to the magnifying influence of heating rate on measured heat flow.
For each respective particle size fraction, the whole set of DSC curves obtained at different heating rates was optimized altogether. The correlation coefficients R 2 obtained for the overall fits of the sets of DSC curves (corresponding to the measurements taken for the respective particle size fractions) ranged between 0.992 and 0.997. All the kinetic parameters involved in the optimization (E 1, A 1, ΔH 1, m 1, E 2, A 2, ΔH 2, M 2 and N 2) were set as variable, i.e., the E and A values obtained via the model-free methods were used only as initial inputs and were further improved during the optimization. The suitability of the pre-selected models (JMA for the first process and AC for the second process) was confirmed by additional testing of all available different kinetic models (21 different models including those finally selected). Furthermore, the possibility of the mutual dependency of the two processes was tested by means of the multivariate analysis—the following interdependence mechanisms were ruled out: parallel, competing, switching, reversible reaction, independent inhibition, by-product inhibition. The only competitive correlation coefficient was obtained for the consecutive reaction mechanism (A → B → C); however, this mechanism can be ruled out based on the XRD results of the crystalline products. The temperature-resolved XRD data have shown that the first crystallization peak corresponds to the formation of hexagonal Te (P3121), while the second crystallization peak corresponds to the formation of hexagonal Ga2Te5 (P3m1) and rhombohedral GeTe (R3m). The corresponding diffraction patterns are shown in Fig. 2. This left the only option of the two crystallization processes being independent (as was suggested in the first place). Lastly, we have also tested how the correlation coefficients improve when the overall DSC curves are fitted using three processes—no consistent improvement was observed. The separated sets of DSC peaks were also evaluated by means of the single-process kinetic analysis, and none of the kinetic parameters (E, A, model parameters) exhibited any dependence on α or q +. Considering the already very good correlation coefficients, we can conclude that the currently presented combination of the JMA and AC processes represents the true formal kinetics of the complex process.

Diffraction patterns obtained for the 20- to 50-µm powder heated at 5 °C min−1 to a 230 °C (to the maximum of the first crystallization peak) and b 270 °C. The insets visualize the two respective temperatures with respect to the given DSC curve (20–50 µm measured at 5 °C min−1). The symbols denote the particular crystalline phases formed during the crystallization
Selected examples of the deconvoluted peaks are shown in Fig. 3; note that the 0–20 and 20–50-µm particle size fractions did not have to be deconvoluted since the two peaks were well separated.

Examples of the deconvolution performed by means of the multivariate kinetic analysis—graphs a, b and c correspond to the data obtained at 20 °C min−1 for the 50–125, 180–250 and 300–500-µm particle size fractions
Interpretation of the kinetic parameters
In Fig. 4, the two respective activation energies E 1 and E 2 and pre-exponential factors A 1 and A 2 (corresponding to the two deconvoluted crystallization processes) are plotted in dependence on particle size—all lower indices in this section correspond to the denotation of the “first” and “second” process. The activation energies decrease with particle size, which is quite common for chalcogenide glasses [9, 10] and can be explained by decreasing influence of the crystal growth originating from surface and from mechanically induced defects (the defects-based crystallization was found to exhibit higher activation energies) [9, 10]. The course of the evolution of the pre-exponential factor A is mostly caused by the so-called compensation effect (mathematical correlation between the E and A values); however, this parameter is also responsible for the exact position of a given peak in temperature. Considering the evolution of both E and A, the significant difference between the values corresponding to the first and second peaks found for fine powders decreases with particle size—this is in agreement with the increasing degree of overlap observed for rising particle size (see Figs. 1, 3).

Apparent activation energies (E 1 and E 2) and pre-exponential factors (A 1 and A 2) determined by means of multivariate kinetic analysis for the first and second crystallization sub-processes and plotted in dependence on average particle size
Regarding the exact crystal growth mechanism, it was already mentioned in “Deconvolution procedure” section that the JMA and AC kinetics were attributed to the first and second peak, respectively. In Fig. 5, the kinetic exponents determined for these models (m 1 for the JMA model; M 2 and N 2 for the AC model) are plotted in dependence on average particle size. As can be seen, their values are more or less constant within the experimental errors, indicating that the very kinetic mechanisms are not influenced by changing ratio between the amount of the respective crystallization centers (surface and defects versus CNT volume-located nuclei). Incidentally, the indifference of the kinetic manifestation of the two respective processes also confirms their mutual independence.

JMA kinetic exponent (m 1) and AC kinetic exponents (M 2 and N 2) determined by means of multivariate kinetic analysis for the first and second crystallization sub-processes, respectively, and plotted in dependence on average particle size
Considering the obtained values of the JMA kinetic exponent m 1, they are in an excellent correspondence with our previous results on Ge20Te(80−x)Sex glasses [22–25], where also the first of the crystallization peaks corresponded to the precipitation of hexagonal Te, followed JMA kinetics and exhibited the m JMA values close to 1.4. Note that this value of the JMA exponent was found to correspond to the crystallization originating from mechanically induced defects [10, 22, 26]. It appears that the Te precipitation in the Ge–Te-based infrared glasses follows similar kinetics regardless of the subsequent crystallization mechanisms driven by the third added element (Ga or Se for our case), which further confirms the independent course of this process.
Lastly, the evolution of the crystallization enthalpies of the two processes needs to be considered. In Fig. 6, the overall crystallization enthalpy ΔH determined is a sum of both overlapping peaks (empty points). In addition, the relative portion of the overall enthalpy corresponding to the first peak ΔH 1/ΔH is also displayed in Fig. 6 (filled points). As can be seen, the value of the overall crystallization enthalpy ΔH first increases with particle size, and then (at approx. 125- to 180-µm powder fraction), it stabilizes at the maximum value of ~42 J g−1. This effect was observed also for our earlier studies [9, 10] and can be explained by processing of the C p-T dependences in terms of the Kirchhoff’s law (note that the crystallization effects shift to higher temperatures with increasing d aver). The possibility of the enthalpy increase being caused by the change in crystallization products can be ruled out since for the fine powders, the ratio between the enthalpies of the two sub-processes is constant (filled points in Fig. 6). The dependence of ΔH 1/ΔH on d aver, however, exhibits a significant decrease for coarse powders. This effect can be explained by the fact that the Te precipitation (the first crystallization process) originates mainly from surface and mechanically induced defects—as was confirmed by the kinetic analysis. Since in case of coarser powders, the amount of these crystallization centers dramatically decreases, the number of correspondingly formed Te crystallites also goes down and the remaining “surplus” amorphous matrix is transformed in the second crystallization step.

Overall crystallization enthalpy ΔH (empty points) and the relative portion of the overall enthalpy corresponding to the first sub-process (filled points) plotted in dependence on average particle size
Lastly, we can use the actual position of the crystallization peaks in temperature (and its evolution with respect to particle size) to obtain information about the physicochemical background of the crystallization process: It is apparent in Fig. 3 that at lower d aver the surface crystallization of Te shifts to lower temperatures due to the increasing amount of mechanical defects that act as nucleation centers—proportions of the peak (the characteristic JMA asymmetry) confirm that the process is driven by the surface nucleation growth mechanism. On the other hand, the position of the second crystallization peak in temperature is almost unaffected by d aver (with the crystallization onset being ~260 °C), which indicates that formation of GeTe and Ga2Te5 is independent from the Te precipitation and that mechanical defects most probably do not act as direct crystallization centers. Instead, it may be formation of the Te-depleted amorphous regions (due to the diffusion/relocation of the Te atoms into Te crystallites associated with the formation of the Ge- and Ga-richer zones) that initiates the crystallization of GeTe and Ga2Te5 phases. This assumption is supported by evolution of the crystallization rapidity of these phases (see the shape and width of the respective crystallization peaks in Fig. 3). In case of fully finished Te crystallization (Fig. 3a), the consequent formation of the GeTe and Ga2Te5 phases proceeds more rapidly compared to the coarse powders (Fig. 3b), where the significantly lower amount of Te-depleted regions is created both overall and at the moment of start of GeTe and Ga2Te5 formation.
Conclusions
The crystallization kinetics of the Ge11Ga11Te78 glass was studied by means of DSC in dependence on particle size. The complex crystallization process was deconvoluted by means of multivariate kinetic analysis, and the following crystal growth mechanism was identified: The crystallization starts with the precipitation of hexagonal tellurium, following the nucleation growth JMA kinetics (the crystal growth was found to originate from mechanically induced defects); the precipitation of Te is then followed by formation of Ga2Te5 and GeTe, which follows the autocatalytic kinetics. With regard to the influence of particle size on the crystallization kinetics, expectedly it did not affect most intrinsic aspects of the overall kinetic behavior (models, their parameters and mutual correlation between the two processes). The only aspects influenced by the change in d aver were apparent activation energy and the ratio between the magnitudes of manifestation of the two particular sub-processes. The apparent activation energy was found to decrease with particle size for both sub-processes, which indicates that also the second (AC) crystallization sub-process involves growth from mechanically induced defects. In addition, the intensity of the Te precipitation was found to decrease for the coarsest powders, which corresponds to the significant decrease in the amount of respective crystallization centers (defects and surface imperfections).
As results from the present study, one needs to be cautious when using DSC crystallization data for determination of glass stability of the Te-based chalcogenide glasses due to the large variability of the “crystallization temperatures” dependent on the applied heating rate and sample form. In addition, since the primary crystallization process was found to be surface Te precipitation, the Ge–Ga–Te materials seem not to be suitable for development of glass–ceramics. However, the present results indicate that the intensity of Te precipitation decreases with d aver, which may favorably influence controlled crystallization of defects-free (bulk) samples.
References
Danto S, Houizot P, Boussard-Pledel C, Zhang XH, Smektala F, Lucas J. A family of far-infrared-transmitting glasses in the Ga–Ge–Te system for space applications. Adv Funct Mater. 2006;16:1847–52.
Wang G, Nie Q, Barj M, Wang X, Dai S, Shen X, Xu T, Zhang X. Compositional dependence of the optical properties of nvel Ge–Ga–Te-CsI far infrared transmitting chalcohalide glasses system. J Phys Chem Sol. 2011;72:5–9.
Danto S, Ruff Z, Wang Z, Joannopoulos JD, Fink Y. Ovonic memory switching in multimaterial fibers. Adv Funct Mater. 2011;21:1095–101.
Nie Q, Wang G, Wang X, Dai S, Deng S, Xu T, Shen X. Glass formation and properties of GeTe4–Ga2Te3–AgX (X = I/Br/Cl) far infrared transmitting chalcohalide glasses. Opt Commun. 2010;283:4004–7.
Petkov P, Ilcheva V, Wamwangi D, Wuttig M, Ilchev P, Petkova T. Phase transition of gallium containing telluride thin films. J Optoelectron Adv M. 2009;11:1261–4.
Wang X, Nie Q, Wang G, Sun J, Song B, Dai S, Zhang X, Bureau B, Boussard C, Conseil C, Ma H. Investigations of Ge–Te–AgI chalcogenide glass for far-infrared application. Spectrochim Acta A. 2012;86:586–9.
Bureau B, Danto S, Ma HL, Boussard-Plédel C, Zhang XH, Lucas J. Tellurium based glasses: a ruthless glass to crystal competition. Solid State Sci. 2008;10:427–33.
Zhang XH, Calvez L, Seznec V, Ma HL, Danto S, Houizot P, Boussard-Plédel C, Lucas J. Infrared transmitting glasses and glass-ceramics. J Non Cryst Sol. 2006;352:2411–5.
Svoboda R, Málek J. Thermal behavior in Se–Te chalcogenide system: interplay of thermodynamics and kinetics. J Chem Phys. 2014;141:224507-1-10.
Svoboda R, Málek J. Thermal behavior of Se-rich Ge2Sb2Se(5-y)Tey chalcogenide system. J Alloys Compd. 2015;627:287–98.
Svoboda R, Málek J. Thermal behavior of Se-rich GeSb2Se(4-y)Tey (glassy) system. J Alloys Compd. 2016;670:222–8.
Svoboda R, Málek J. Non-isothermal crystallization kinetics of As2Se3 glass studied by DSC. Thermochim Acta. 2014;579:56–63.
Opfermann J. Kinetic analysis using multivariate non-linear regression. J Therm Anal Calorim. 2000;60:641–58.
Perejón A, Sánchéz-Jiménez PE, Criado JM, Pérez-Maqueda LA. Kinetic analysis of complex solid-state reactions—a new deconvolution procedure. J Phys Chem B. 2011;115:1780–91.
Ozawa T. Kinetic analysis of derivative curves in thermal analysis. J Therm Anal. 1970;2:301–24.
Flynn JH, Wall LA. A quick, direct method for the determination of activation energy from thermogravimetric data. J Polym Sci B Polym Lett. 1966;4:323–8.
Avrami M. Kinetics of phase change I—general theory. J Chem Phys. 1939;7:1103–12.
Avrami M. Kinetics of phase change. II—Transformation-time relations for random distribution of nuclei. J Chem Phys. 1940;7:212–24.
Avrami M. Granulation, phase change, and microstructure—kinetics of phase change III. J Chem Phys. 1941;7:177–84.
Johnson WA, Mehl KF. Reaction kinetics in processes of nucleation and growth. Trans Am Inst Min (Metall) Eng. 1939;135:416–42.
Šesták J. Thermophysical properties of solids, their measurements and theoretical analysis. Amsterdam: Elsevier; 1984.
Svoboda R, Brandová D, Málek J. Non-isothermal crystallization kinetics of GeTe4 infrared glass. J Therm Anal Calorim. 2016;123:195–204.
Svoboda R, Brandová D, Beneš L, Málek J. The effect of Se↔Te substitution on crystallization micro-mechanisms evincing in GeTe4 glass. J Therm Anal Calorim. 2016;123:205–19.
Svoboda R, Brandová D, Málek J. Combined dilatometric and calorimetric study of kinetic processes occurring in Ge20Te76Se4 infrared bulk glass. J Non-Cryst Sol. 2016;432:493–8.
Svoboda R, Brandová D, Málek J. Thermal behavior of Ge20SeyTe80-y infrared glasses (for y up to 8 at.%). J Alloys Compd. 2016;680:427–35.
Svoboda R, Brandová D. Crystal growth from mechanically induced defects: a phenomenon observed for glassy materials. J Therm Anal Calorim. 2017;127:799–808.
Acknowledgements
This work has been supported by the Czech Science Foundation under project No. 17-11753S.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Svoboda, R., Brandová, D. The effect of powder coarseness on crystallization kinetics of Ge11Ga11Te78 infrared glass. J Therm Anal Calorim 129, 593–599 (2017). https://doi.org/10.1007/s10973-017-6148-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-017-6148-0