
Abstract
Differential scanning calorimetry was used to study crystallization behavior in selenium glass under isothermal conditions. In the current work, which is the third in a sequence of articles dealing with the crystallization kinetics of complex processes, the isothermal crystallization kinetics was described in terms of the Johnson–Mehl–Avrami nucleation-growth model. The study was performed in dependence on particle size so that the advanced interpretation of characteristic kinetic functions could be employed. The complexity of the crystallization process was found to be represented by overlapping competing surface and bulk nucleation-growth mechanisms. Based on this information, the deconvolution in terms of the Johnson–Mehl–Avrami process was performed, separating the mechanisms involved. High consistency of the resulting kinetic parameters confirms the accuracy and physical meaningfulness of the deconvolution procedure. The resulting concept not only describes the isothermal crystallization process in glassy selenium very well, both qualitatively and quantitatively, but is also capable of explaining all accessible data from the literature on this topic. In addition, comparisons of the present and literature data clearly imply that the nucleation processes play a major role in the isothermal crystallization of amorphous selenium.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Recently, a thorough research of non-isothermal crystallization kinetics of pure selenium glass was performed by using state-of-the-art differential scanning calorimetry (DSC) [1, 2]. Careful examination of the precisely recorded DSC data revealed a high degree of complexity of the crystallization process, where surface and bulk/volume crystallization mechanisms simultaneously manifest. In the first paper [1], the overall crystallization response was studied in detail. The process complexity was described based on the “advanced interpretation of characteristic kinetic functions” [3], identifying the nature of the two mechanisms involved as well as determining the particular experimental conditions for their manifestation. In the second paper [2], these data are further deconvoluted using the “modified Fraser–Suzuki deconvolution” [4], so that the peaks corresponding to the two respective crystallization mechanisms are separated. The correctness and accuracy of the deconvolution procedure were confirmed by the high level of consistency of the deconvoluted data and their kinetic analysis. Furthermore, in the second paper, the accessible literature data were taken and explained in terms of the newly introduced concept of the overlapping manifestation of the surface and volume crystallization mechanisms. In this literature survey, we have also attempted to extrapolate the conclusions that we obtained for non-isothermal experiments toward the isothermal data.
In the present paper, the isothermal crystallization kinetics of glassy selenium will be described in detail based on a new, extensive series of isothermal measurements. The deconvolution procedures will be implemented in order to reveal and separate the occurring sub-processes driven by different crystallization mechanisms. Based on these findings we will update our previous extrapolated conclusions, revisit the isothermal literature studies, and specify the interpretations of these studies in greater detail.
The measurements of isothermal crystallization were performed by means of DSC. The kinetic equation of a DSC curve can be described [5] as follows:
where Φ is the measured heat flow, ΔH is the crystallization enthalpy, A is the pre-exponential factor, E is the apparent activation energy of the process, R is the universal gas constant, T is temperature, and f(α) stands for an expression of a kinetic model with α being the conversion. In the case of isothermal DSC data, the apparent activation energy E is calculated by isoconversional methods; in our study, that by Friedman [6] will be used. Within the framework of Friedman’s methodology, the activation energy is calculated for various chosen degrees of conversion and then averaged (usually for the interval of α = 0.3–0.7, where the influence of experimental conditions is minimized, in contrast to the peak tails). The Friedman equation may then be expressed as
where Φ α is the value of heat flow determined for the chosen degree of conversion α and a given temperature T α .
In the second step of kinetic analysis, an appropriate kinetic model is determined. In our work, the method developed by Málek [7, 8] will be used. It is based on a simple transformation of the DSC data into the so-called characteristic kinetic functions:
The data are usually normalized on both axes for better clarity when comparing a whole set of curves measured at different heating rates. In order to determine an appropriate kinetic model from the y(α) and z(α) functions, the conversion rates corresponding to their maxima, α max,y and α max,z , have to be found. These values are characteristic for each kinetic model; the algorithm mentioned previously, for example in [8], summarizes the characteristic maxima for several of the most typical kinetic models.
In this paper, the two most important kinetic models will be considered and tested. The nucleation-growth Johnson–Mehl–Avrami model (JMA(m)) [9–12] is expressed by
where m is the parameter reflecting the nucleation and crystal growth mechanisms as well as the crystal morphology. And the semi-empirical autocatalytic (AC(M, N)) model [5] is
where M and N are phenomenological parameters describing the curvatures of the ascending and descending halves of the described peak.
Experimental
The selenium glass was prepared by a melt-quenching technique from the pure element (annealing at 350 °C)—the glass was cooled in water. The following glass powder fractions were prepared by grinding in agate mortar: 20–50, 50–125, 125–180, 180–250, 250–300, and 300–500 μm (amorphous character of the powders was confirmed by XRD). Crystallization processes in the prepared powder fractions were studied using a conventional Q2000 DSC (TA Instruments) equipped with cooling accessory, autolid, autosampler, and T-zero Technology. Dry nitrogen was used as the purge gas at a rate of 50 cm3 min−1. The calorimeter was calibrated through the use of melting temperatures of In, Zn, and H2O. The zero-line course was checked daily. The masses of the powder samples varied between 8 and 10 mg; hermetically sealed T-zero low-mass pans were used. Regarding the applied temperature program, the sample was first subjected to a 5-min isotherm at 45 °C and then heated at 100 °C min−1 to a selected temperature T i , where the sample was allowed to isothermally crystallize until the crystallization process was complete. The isothermal crystallization temperatures utilized in the case of each particle size fraction were 95, 100, 105, 110, 115, 120, 125, 130, 135, 140, 145, 150, 155, and 160 °C. In order to obtain a baseline for the isothermal measurement, each DSC pan with a crystalline sample of glass was kept in the DSC cell and the above-described temperature procedure was repeated (in this way the data subtracted from the isothermal crystallization signal truly surrogated the presence of an inert material with similar heat capacity, mass, grain size, and positioning in the DSC pan/cell). Perfect flatness and reproducibility of the baseline were confirmed. Prior to the measurements, the magnitude of the heating rate preceding the isothermal period was optimized: the rates of 10, 30, 50, and 100 °C min−1 were tested. It was found that q + = 100 °C min−1 best prevents the data loss during the pre-isothermal reaction time but still does not produce data-distortive measurement artifacts. We have also confirmed that the non-isothermal/isothermal transition and the consequent temperature stabilization generated negligible error compared to the actual experimental uncertainty and repeatability of the measurements themselves.
In the second part of the paper, the deconvolution procedure will be applied to the experimental data in order to separate the two overlapping crystallization processes. The deconvolution will be based on the JMA equation, expressed in the following form:
where K stands for the reaction rate constant, defined as
Thus, the deconvolution procedure utilizing the JMA model equation always provides physically meaningful solutions of the peaks separation. The Peakfit computer program (Systat Software Inc.) was used to perform the deconvolution; the parameters of the resulting JMA functions were obtained through a non-linear optimization method by using the Levenberg–Marquardt algorithm. The minimum of the residual sum of squares (RSS) was sought in order to obtain the best fit. The average correlation coefficient for all the performed deconvolution fits was 0.9991 ± 0.0014, which corresponds to a very high quality of the description.
Results and discussion
The isothermal crystallization behavior of selenium glass was studied in dependence on particle size. In the first part of the “Results and discussion” section, the overall crystallization response will be discussed in terms of basic kinetic analysis; secondly, the deconvolution of the sub-processes involved will be performed, and lastly the conclusions gained will be compared with data from the literature.
Basic kinetic analysis
The main task of the basic kinetic analysis is enumeration of Eq. 1. In this regard, the crystallization enthalpy ΔH is often neglected as its value is only needed to determine the pre-exponential factor A (in which not many scientists are interested). This is also why the determination of crystallization enthalpy is often simplified under the assumption that the crystallization enthalpy should be constant for all applied experimental conditions, and the ΔH value determined is presented in this way. However, as we have demonstrated for a number of materials and DSC instruments, the ΔH value determined from the crystallization peaks shows significant dependence on particle size, heating rate (for non-isothermal data), and temperature [1, 2, 13–17]. In Fig. 1a, the crystallization enthalpy ΔH is plotted in dependence on particle size and T i (average particle size d aver was calculated as the mid-point of the respective particle size intervals; results for T i = 95 °C are not displayed due to the very high scatter in the data). As can be seen, there is a significant dependence of ΔH on both variables.

To explain the trends shown in Fig. 1a, ΔH data for non-isothermal crystallization of a-Se published in [2] can be utilized; these data are displayed in Fig. 1b in dependence on d aver and heating rate q +. As can be seen in Fig. 1b, the surface map is relatively plain, confirming that there is no preliminary crystallization associated with preparation of the finest glassy powders and the consequent delay between the preparation and the very measurements; that is, the powders do not degrade within the first weeks after their preparation—their amorphous nature was confirmed by XRD. The mild decrease of ΔH with q + apparent in Fig. 1b was explained in [2] as a possible influence of energy dissipation occurring at slow heat evolution. A similar effect could contribute to the decrease of ΔH occurring at low T i s in the present study (see Fig. 1a), where the heat evolution is again extremely slow. Nevertheless, one more trend is apparent in Fig. 1a, namely, the marked decrease of ΔH for fine powders and high T is. This effect is caused by an extremely good heat transfer between the sample and the DSC pan, where even the heating at 100 °C min−1 preceding the isothermal period cannot fully prevent partial crystallization of the sample before the temperature T i is achieved. To quantify this effect, we have integrated the data from the initial non-isothermal step and calculated the corresponding enthalpy change (see Fig. 1c). It is apparent that for most measurements the portion of pre-crystallized material is negligible. However, in case of fine powders heated up to the highest applied T i a significant portion of the material crystallizes already during the initial non-isothermal heating step. If we sum the two ΔH contributions (isothermal and non-isothermal; i.e., Fig. 1a, c), we get the overall crystallization enthalpy depicted in Fig. 1d. In this graph, it can be seen that a relatively good resemblance can be found with the crystallization enthalpies evaluated from purely non-isothermal (Fig. 1c) measurements.
In the second step of kinetic analysis, the apparent activation energy E is determined. The ln(Φ α ) − T −1i dependences determined within the framework of the Friedman method were, however, curved, exhibiting two significantly different crystallization regions. An example of this evaluation is for the 125–180 μm particle size fraction shown in Fig. 2; symbol lines correspond to the particular data determined for each selected degree of conversion α. The main trend is similar for all α values: two clear temperature regions can be distinguished, each with significantly different values of apparent activation energy. The transient temperature region is then somewhere around 125–135 °C. Similar figures were obtained for all studied particle sizes. In Fig. 3a, the values of apparent activation energy E determined for the two crystallization regions are shown in dependence on d aver (the error bars are lower than the magnitude of points if not displayed otherwise). To complete the picture, the activation energy determined for the overall curved dependence is also displayed in Fig. 3a. However, this E overall value does not have a physical meaning in this case; it only demonstrates how the intermediate activation energy values can be reached by improper evaluation: either the too broad or the too narrow (for intermediary temperatures) temperature interval is considered. Similar issue of E Friedman being (or not) a representative activation energy value in case of complex processes was for non-isothermal processes discussed in [4, 18]. In [4, 18], it was shown that variation of E with experimental conditions indicates that only “apparent” activation energy was evaluated and that the process is almost certainly complex. In such case, the interpretation of E-α dependences needs to be handled with great care. In the present study, a similar conclusion was obtained for the isothermal conditions. The marked difference between the E values determined for the two borderline temperature intervals thus clearly suggests the presence and overlap of two crystallization mechanisms.

Evaluation of apparent activation energy according to Friedman; data for the 125–180 μm particle size fraction are shown. The curvature of the dependences corresponds to the transition between the dominant crystal growths of type A and type B spherulites

a Activation energy of crystallization determined from the Friedman plots (Fig. 2 and similar) in the three respective temperature regions. Data for all studied particle size fractions are depicted. b Kissinger kinetic plot obtained for non-isothermal data [1, 2]; the inset shows the activation energy of non-isothermal crystallization determined for low/high heating rates data in dependence on particle size [1, 2]
A similar conclusion was also reached for the non-isothermal crystallization experiments performed for a-Se [1, 2]. In Fig. 3b, the so-called Kissinger plot [19] is shown for the non-isothermal crystallization measurements; here the temperature T p corresponding to the maximum of the crystallization peak is utilized for evaluation of E. As can be seen, here the dependences also show curvature after reaching higher temperatures, again suggesting two different crystallization mechanisms. The transient temperature region corresponds well to the one observed during the isothermal measurements (this work, see Fig. 2). In fact, in the case of non-isothermal measurements, only the coarse powders exhibit the above-mentioned curvature because, even at the fastest heating rates, the crystallization of fine powders was finished before the critical transient temperature region was reached. The inset in Fig. 3b then shows the corresponding values of activation energies determined for the two respective temperature regions occurring in the Kissinger plot; the “high” and “low” heating rates data correspond to the higher and lower temperature regions, respectively. As can be seen, the activation energies determined for the two respective temperature regions are very similar for both isothermal and non-isothermal data, again indicating the combined manifestation of two distinctive crystallization mechanisms. It should be noted that the marked decrease of E evaluated for the high temperature region (140–160 °C) and the finest particle size (20–50 μm) is most probably caused by the high amount of pre-existing crystallites formed during the initial non-isothermal heating step.
The occurrence of the two distinguished crystallization temperature regions can be explained based on the studies of Ryschenkow, Faivre, and Bisault [20, 21], who performed an extensive research of Se crystals’ morphology by using various microscopic techniques. It was shown in [20, 21] that selenium may crystallize in three forms in this regard: single crystals, mode A spherulites, and mode B spherulites. The presence and quantity of the particular crystallite types are then driven by the temperature of crystallization and by the content of impurities that may act as nucleation centers. The single-crystal mode dominates at low undercooling near the melting temperature (~221 °C); spherulitic mode A is the intermediate temperature mode occurring in a wide interval around 160 °C, while the B type spherulites absolutely dominate for temperatures lower than 100 °C. In addition to the above-defined regions, a transition zone, extending roughly between 115 and 130 °C, exists, where both the spherulitic modes undergo a rapid change of their morphological features. The microscopic observations presented in [20, 21] are in very good agreement with the results derived from the development of E with T i presented in the current article. Hence, it can be assumed that the crystal morphology in the case of our isothermal “cold crystallization” measurements corresponds to the mixed A and B spherulitic modes, with eventual dominance of the respective modes occurring within the two mentioned crystallization regions. In [2], we have stated that the observations made in [20, 21] presumably correspond only to the “bulk” crystallization originating from the volume-located nuclei and that the exact morphology of the surface-located and/or bulk stress-induced crystallites remains unclear. Nevertheless, current research clearly shows (Fig. 2) that similar curvature occurs for all of the studied Se powders, regardless of their particle sizes. Thus, the same spherulitic crystal growth mechanisms seem to proceed in both bulk glass and very fine powders. This topic will be commented on further in “Deconvolution of isothermal crystallization data” section.
Deconvolution of isothermal crystallization data
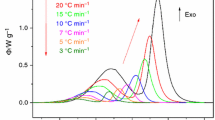
In the third step of the kinetic analysis, an appropriate kinetic model and its parameter(s) are usually determined. In the case of isothermal Se crystallization, however, no single-peak model was able to successfully describe the data. In Fig. 4, several selected DSC crystallization curves are displayed (and magnified); these particular data were obtained for the 125–180 μm particle size fraction, and the corresponding annealing temperatures T i are displayed next to the maxima of the respective isothermal peaks. As can be seen, all DSC curves show markedly complex behavior, for which not even the highly flexible semi-empirical AC model could account. Most crystallization curves clearly exhibit a sharp sub-peak on the low-α side in addition to the main peak, whose shape, however, indicates the presence of at least another two sub-processes. Similar information can be derived from the characteristic kinetic functions y(α) and z(α); see insets in Fig. 4. Both functions show significant scatter on the low-α side, corresponding to a change in the mutual ratios between the crystallization mechanisms involved. The presence of the multiple crystallization mechanisms is then apparent from the non-standard complex shape of the y(α) function. In addition, assuming that the crystallization processes in amorphous selenium should follow the physically meaningful JMA kinetics, the shift of the z(α) function maximum toward a value of ~0.7 indicates the presence of a secondary crystallization process preceding the main crystallization peak and so shifting its kinetics toward higher α values.

Several selected DSC curves corresponding to isothermal crystallization of the 125–180 μm particle size fraction. The respective T i values are displayed next to the maxima of the corresponding isothermal DSC peaks. The insets show characteristic kinetic functions z(α) and y(α) calculated for the isothermal data shown in the main graph
The complexity of crystallization processes can be described by means of deconvolution procedures. For non-isothermal data, the modified Fraser–Suzuki deconvolution [4] was found to be extremely advantageous, since it does not require specialized or expensive software equipment or large amounts of computer power and yet it provides accurate and physically meaningful kinetic results. In the case of isothermal crystallization, the calculations can be greatly simplified so that the very JMA model itself can be used to directly fit the experimental data. The procedure described in the “Experimental” section was used to deconvolute all the experimental data obtained within the framework of the present study. In Fig. 5, an example of such deconvolution is shown: data for the 125–180 μm particle size fraction and T i = 120 °C are displayed. As can be seen, by using three independent crystallization sub-processes a perfect description of the experimental data was achieved. Most of the deconvolution procedures were performed with similarly high correlation coefficients, which indicate not only precise deconvolution procedures but also a very high quality of the DSC data. In addition, we have also tested the possibility that the data can be described by only two independent processes; however, the quality and consistency of the description were not satisfactory.

Example of the JMA deconvolution procedure performed for the isothermal crystallization data (125–180 μm, T i = 120 °C). Dashed lines show the deconvoluted crystallization peaks. The assignment of the sequence order is demonstrated for the three deconvoluted crystallization processes
The occurrence of the second and third processes (see Fig. 5) is in good accordance with explanations provided in our earlier non-isothermal studies [1, 2, 17]. The second process most probably corresponds to surface crystallization, while the third process can be associated with crystallization originating in the volume of the glassy grains. Nevertheless, as pointed out before [1, 2, 17], the “surface” crystallization is something of a misnomer—a similar mechanism can also be associated with crystallization originating from mechanically and stress-induced defects within the glassy grain volume (the main characteristic of this process is that it does not follow the classical nucleation theory). The presence of the “first process” (see Fig. 5) was, on the other hand, not observed during our non-isothermal studies. This process represents a very fast crystallization of a relatively small amount of material. It can be reproduced well and, considering its changing kinetics, cannot be assigned to any DSC artifact; that is, it corresponds to a true crystallization process. The nature of this process does not seems to be clear, however. Since the rapid heating (performed at 100 °C min−1) realized prior to the isothermal crystallization step is the only difference compared to the non-isothermal experiment, we think that the origin of the “first process” can be associated with either local overheating of surface grain layers directly touching the bottom of the DSC pan or rapid transformation of energetically and sterically favorable crystallization centers (possibly the outer and most damaged surface layers of grains), where the higher kinetic barriers are overcome during the very fast preliminary heating.
Complete results of all the deconvolution procedures can be found in Online Appendix 1; obtained values of the parameters ΔH, K, and m corresponding to those occurring in Eq. 7 are listed for the three observed crystallization processes and all combinations of particle sizes and temperatures T i. In the following text, a brief survey of the most important results will be given. One of the main aspects of physically meaningful deconvolution is consistency of the magnitudes (or their trends) of the deconvoluted sub-processes. One of the most suitable ways to express these trends for the particular sub-processes is by the percentage of their ΔH relative to the crystallization enthalpy of the whole crystallization peak. Since the magnitude of the “first” process is negligible for most of the data (a few percent) compared to the magnitude of the other two listed processes, the ΔH trends are basically reduced to the ratio between the magnitudes of the two main competing mechanisms: “surface” crystallization originating from mechanically and stress-induced defects (the “second” process) and volume crystallization following the CNT (“third” process). In Fig. 6, the percentual portions of the overall crystallization enthalpy corresponding to the two main sub-processes (“surface” crystallization in Fig. 6a and volume crystallization in Fig. 6b) are shown. The two graphs are nearly complementary, confirming the negligible magnitude of the “first” process. As can be seen in Fig. 6a, the magnitude of the “surface” crystallization process falls within a range of approximately 15–35 %. Given that no iterative correction procedures were performed and the results displayed in Fig. 6 correspond to a solitary deconvolution step, the scatter in the data is actually quite low, confirming the high accuracy and consistency of the deconvolution. The basic ratio between the magnitudes of the two isothermal crystallization mechanisms is thus somewhere around 1:3 (surface versus volume).

Percentage of the overall crystallization enthalpy corresponding to the particular deconvoluted peaks plotted in dependence on temperature T i (each dependence corresponds to one studied particle size fraction). Graph A shows data for the second deconvoluted peak (“surface” crystallization); graph B shows data for the third deconvoluted peak (volume crystallization)
As can be further seen in Fig. 6, the “surface” crystallization mechanism starts to dominate after reaching some critical temperature. In the case of the finest powders, this switch between the two crystallization mechanisms already occurs at relatively low temperatures (~120 °C for the 20–50 μm particle size fraction). With increasing particle size, this critical temperature also increases, while for the coarsest powders the volume crystallization mechanism dominates throughout the whole investigated temperature region (up to 160 °C). There are two possible explanations for this phenomenon: First, it can be associated with the overall rapidity of the two processes (expressed by the pre-exponential factor A). The decrease of the critical temperature with decreasing particle size may then be caused by the combined effects of an increased number of crystallization centers (defects) and further lowered activation energy of the “surface” mechanism resulting from more severely damaged glassy grains. Second, in the authors’ opinion more plausible explanation then employs the presence of the pre-existing crystallites formed during the initial non-isothermal heating step. These crystallites can act as crystallization centers in a similar way as mechanically induced defects and the increased amount of these crystallites may lead to their preferred growth, effectively suppressing the complementary crystal growth mechanism.
Regarding the activation energy of the particular processes, it can be evaluated from the temperature dependences of the respective deconvoluted parameters K. Scatter of the data enables only estimating of the activation energy values; however, it is clear that in case of all three processes these dependences exhibit similar curvature as those depicted in Fig. 2. This then also leads to similar values of activation energies as depicted in Fig. 3a. Similarity of the estimated activation energies confirms the above-suggested importance of the pre-exponential factor in the crystallization kinetics of the two main discussed sub-processes.
In Fig. 7, averaged values of the JMA kinetic exponent m are shown for the deconvoluted data of all studied particle sizes: data for the “second” process are slightly shifted toward higher d aver so that the error bars are clearly visible and distinguishable for all data. Again, the consistency of the results with respect to changing the particle size confirms the accuracy of the deconvolution procedure. Compared to the non-isothermal data [1, 2], the m values determined for the isothermal crystallization are significantly lower. This may be a consequence of the fact that the nucleation period is greatly reduced (compared to the non-isothermal measurements), which would force the crystallization processes to increase the utilization of the mechanically induced defects during the secondary nucleation/crystal growth of the a-Se spherulites (and hence decrease the apparent kinetic exponent m). Despite the generally lower m values in the case of isothermal crystallization, there is a significant and consistent difference in m values for the two main crystallization mechanisms, confirming their presence and the physical meaningfulness of the deconvolution procedure.

Values of deconvoluted JMA kinetic exponents m averaged for each particle size fraction
Comparison with literature data
Comparisons with our previous non-isothermal particle-size-based studies [1, 2] were made throughout the previous texts. To conclude, values of activation energy E and crystallization enthalpy ΔH were in a good agreement for both isothermal and non-isothermal data. The JMA kinetic exponents m determined from the isothermal data were somewhat lower than we expected based on the trends found for the lowest heating rates during the non-isothermal measurements. This is most probably a consequence of the rapid pre-isothermal heating (performed at 100 °C min−1), which markedly shortened the period of time for which the sample was allowed to nucleate compared to the non-isothermal experiments performed at low heating rates, where the sample was slowly heated from laboratory temperature, that is, through the whole nucleation temperature range. The small amount of volume nuclei formed during the rapid pre-isothermal heating then forced the crystallization processes to increase utilization of the mechanically induced defects during the secondary nucleation/crystal growth of the a-Se spherulites (and hence decreased the apparent kinetic exponent m).
In the work by Holubová et al. [22], isothermal measurements were performed for bulk samples in the 90–210 °C temperature range, where selenium glass was prepared by slow cooling in air. For isothermal experiments, the following values were obtained: ΔH = 65.5 ± 2.9 J g−1, E = 83 ± 2 kJ mol−1, and m ~3.5. The value of crystallization enthalpy is in good agreement with our data obtained for the coarsest powder. As shown in Figs. 2 and 3, the value of activation energy is highly dependent on the considered temperature range and evaluation methodology. As the actual data are not shown in [22], no detailed conclusions can be derived regarding the value of E except that it falls within the generally expected range and may have been affected (lowered) by the higher amount of volume nuclei formed during the slower glass-formation. The value of the JMA kinetic exponent m = 3.5 reported in [22], on the other hand, clearly suggests dominant involvement of the volume crystallization process based on CNT. It was probably the combined effect of the slow glass-formation and slower pre-isothermal heating (about which, unfortunately, no information is provided in [22]), which accounted for the significantly higher number of volume-located nuclei, leading to the 3D crystal growth observed in this work. In addition, samples in the form of bulk probably contained a very low amount of stress or mechanically induced defects, which suppressed the manifestation of surface crystallization. Unfortunately, the severe lack of information about the experimental setup and measurement procedures applied in [22] does not allow for definite explanation of the results published there, reducing their scientific value to a large extent.
In the next work discussed [23], Afify studied crystallization kinetics for a-Se prepared by melt-quenching, both isothermally and non-isothermally. Unfortunately, the exact form of the sample is not specified. Nevertheless, taking into account the published [23] values of E = 79.4 kJ mol−1 and m = 1.75 (non-isothermally, heating rates of 2.5–40 °C min−1) and comparing them with our previous non-isothermal results [1, 2], it can be assumed that the sample was in the form of coarse powder with a higher proportion of damaged surface. The isothermal kinetic parameters determined in [23] were the following: E = 75.3 kJ mol−1 and m = 1.85. Considering the assumed form of the sample and the investigated temperature interval (80–90 °C), these results are in very good agreement with those obtained within the framework of the present study.
Third, in their work [24], Abu-Sehly et al. studied isothermal kinetics of melt-quenched a-Se glass in a fine powder form. Strong dependence of the apparent activation energy E on the degree of conversion α and variation of the Avrami exponent m with the annealing temperature (values from 2 to 4 in the 80–100 °C range) suggests complex crystallization behavior with a dominating volume-located CNT crystal growth mechanism. This is also confirmed by the correspondence of low E and high m occurring at low degrees of conversion α, gradually shifting toward higher E and lower m with increasing degree of crystallinity. The marked dominance of the volume crystallization mechanism is certainly a consequence of slow pre-isothermal heating performed at 20 °C min−1. The rapidity of the crystallization process corresponding to the large number of volume-located nuclei can be confirmed by simple comparison with our own data: in [24], it took ~3 min to finish the crystallization process at 95 °C, while in our present work the crystallization proceeded for 50 min under the same conditions.
From the above-mentioned comparisons, it can be concluded that it is the nucleation processes that play the major role in the isothermal crystallization of amorphous selenium. In particular, the amount of volume-located nuclei originating either from the glass-formation or the pre-isothermal procedural steps (nucleation periods, rate of heating …) has an immense influence on the course of the crystallization process. Furthermore, based on the above-mentioned comparisons, it seems that only in the case of low amounts of volume-located nuclei do the surface and mechanically/stress-induced defects dominate and take control of the crystallization process. As was shown, variation of these aspects may produce markedly different results and seems to be the main source of inconsistencies found in the literature dealing with the crystallization kinetics of glassy materials. On the other hand, taking all these aspects into account makes it possible to obtain the intrinsic consistency and at least a qualitative explanation of all the accessible data. Thus, a detailed experimental part is one of the most important sections of every article dealing with a similar topic.
Conclusions
Crystallization kinetics in amorphous selenium was studied under isothermal conditions by using DSC. The study was performed in dependence on particle size. A type of complexity similar to that manifested for non-isothermal experiments (competition between the volume crystallization mechanism following the classic nucleation theory and surface crystallization originating from mechanically induced defects) was also confirmed in the case of the isothermal data. It was shown that by a combination of the JMA deconvolution procedure and advanced interpretation of characteristic kinetic functions z(α) and y(α), a complete picture regarding crystallization kinetics in a given material can be attained. The high level of consistency of the deconvolution results confirmed both the quality of the DSC data and the accuracy of the deconvolution procedure.
Furthermore, from comparison of the accessible literature data on isothermal crystallization of a-Se, it was found that it is in fact the nucleation process that completely determines the crystallization kinetics of this material and causes the large (virtual) discrepancies between the results. Taking into account the suggested crystallization complexity concept together with the dominant influence of volume nucleation, all the accessible literature data can be explained very well.
References
Svoboda R, Málek J. Crystallization kinetics of amorphous Se, part 1—interpretation of kinetic functions. J Therm Anal Calorim. 2013;114:473–82.
Svoboda R, Málek J. Crystallization kinetics of a-Se, part 2—Deconvolution of a complex process: the final answer. J Therm Anal Calorim. 2014;115:81–91.
Svoboda R, Málek J. Interpretation of crystallization kinetics results provided by DSC. Thermochim Acta. 2011;526:237–51.
Svoboda R, Málek J. Applicability of Fraser–Suzuki function in kinetic analysis of complex crystallization processes. J Therm Anal Calorim. 2013;111:1045–56.
Šesták J. Thermophysical properties of solids, their measurements and theoretical analysis. Amsterdam: Elsevier; 1984.
Friedman HL. Kinetics of thermal degradation of char-forming plastics from thermogravimetry. Application to a phenolic plastic. New York: Wiley Subscription Services; 1964.
Málek J. Kinetic analysis of crystallization processes in amorphous materials. Thermochim Acta. 2000;355:239–53.
Málek J. The kinetic analysis of non-isothermal data. Thermochim Acta. 1992;200:257–69.
Avrami M. Kinetics of phase change I—general theory. J Chem Phys. 1939;7:1103–12.
Avrami M. Kinetics of phase change. II—transformation-time relations for random distribution of nuclei. J Chem Phys. 1940;7:212–24.
Avrami M. Granulation, phase change, and microstructure—kinetics of phase change III. J Chem Phys. 1941;7:177–84.
Johnson WA, Mehl KF. Reaction kinetics in processes of nucleation and growth. Trans Am Inst Min (Metall) Eng. 1939;135:416–42.
Svoboda R, Brandová D, Málek J. Crystallization behavior of GeSb2Se4 chalcogenide glass. J Non-Cryst Solids. 2014;388:46–54.
Svoboda R, Málek J. Non-isothermal crystallization kinetics of As2Se3 glass studied by DSC. Thermochim Acta. 2014;579:56–63.
Svoboda R, Málek J. Amorphous-to-crystalline transition in Te doped Ge2Sb2Se5 glass. J Therm Anal Calorim. 2014;117:1073—83.
Svoboda R, Málek J. Nucleation in As2Se3 glass studied by DSC. Thermochim Acta. 2014;593:16–21.
Svoboda R, Málek J. Crystallization mechanisms occurring in Se–Te glassy system. J Therm Anal Calorim. 2014. doi:10.1007/s10973-014-4199-z.
Criado JM, Sánchez-Jiménez PE, Pérez-Maqueda LA. Critical study of the isoconversional methods of kinetic analysis. J Therm Anal Calorim. 2008;92:199–203.
Kissinger HE. Reaction kinetics in differential thermal analysis. Anal Chem. 1957;29:1702–6.
Ryschenkow G, Faivre G. Bulk crystallization of liquid selenium. J Cryst Growth. 1988;87:221–35.
Bisault J, Ryschenkow G, Faivre G. Spherulitic branching in the crystallization of liquid selenium. J Cryst Growth. 1991;110:889–909.
Holubová J, Černošek Z, Černošková E, Černá A. Crystallization of supercooled liquid of selenium: The comparison of kinetic analysis of both isothermal and non-isothermal DSC data. Mater Lett. 2006;60:2429–32.
Afify N. A new method to study the crystallization or chemical reaction kinetics using thermal analysis technique. J Phys Chem Solids. 2008;69:1691–7.
Abu-Sehly AA, Alamri SN, Joraid AA. Measurements of DSC isothermal crystallization kinetics in amorphous selenium bulk samples. J Alloys Compd. 2009;476:348–51.
Acknowledgements
This work has been supported by the Czech Science Foundation under Project No. P106/11/1152.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Svoboda, R., Málek, J. Crystallization kinetics of a-Se. J Therm Anal Calorim 119, 1363–1372 (2015). https://doi.org/10.1007/s10973-014-4201-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-014-4201-9