
Abstract
The sol–gel method has been used for the synthesis of borosilicate gels from mixtures of methyltriethoxysilane (MTES) and dimethyldiethoxysilane (DMDES) and boric acid. The use of boric acid, B(OH)3 allows the hydrolysis and condensation of hybrid silicon alkoxides without further addition of water or catalyst. The use of difunctional silicon units, –(CH3)2SiO– promote the formation, during the sol–gel process, of linear oligomers which facilitate fiber drawing before gelation. Gel characterization performed by FT-IR, XRD, TG-DTA and DCS analysis indicates the formation of a mixed network with incorporation of the boron units via =B-O-Si≡ bridges. The formation of borosiloxane bonds seems favored by the presence of DMDES. SiBOC glasses were obtained after pyrolysis of the borosilicate gels in argon atmosphere at 1000 °C. TG-DTA study indicates that the ceramic yield decreases by increasing the amount of DMDES. Gel fibers were successfully prepared from convenient partially-aged solutions by hand drawing. Pyrolysis of the obtained gel fibers under argon atmosphere at 1000 °C open the possibility to produce SiBOC homogeneous glass fibers with diameter as low as 10 μm.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
The sol–gel method for making glasses and ceramics at low temperatures has been successfully developed in the last years. The obtained products, such as fibers, coatings and bulk glasses, have peculiar features and properties which make them suitable for highly specific applications (optical fibers, foams, catalysts, reactive powders, hollow spheres, glass-ceramic polymer composites, reflective and anti-reflective coatings, etc) [1–3].
Conventional methods for producing glass and ceramic fibers require high temperature melting of the raw materials, typically oxides or inorganic salts [4]. The sol–gel method is an useful technique for making glass and ceramic fibers because the simple chemistry and mild processing allow to easily produce fibers avoiding the difficulties of conventional methods [5, 6]. Gel fibers can be prepared by spinning a sol containing organic linear polymers (e.g. cellulose, algenate...) but they can also be obtained from partially hydrolyzed metal alkoxides solutions under acidic conditions, normally by using an amount of water smaller than that required for the complete hydrolysis of the reagents. Such a sol contains linear type of metalloxane polymers from which fibers can be drawn without the addition of spinning-aids and it is known as the “polymerization” method [7]. The gel fiber obtained at room temperature is converted into glass or ceramic fiber by the thermal treatment in air or inert atmosphere at temperatures above 800 °C [8].
When hybrid silicon alkoxides, such as methyl-triethoxysilane, are employed for synthesizing the gel fibers, the pyrolysis in inert atmosphere leads to SiCO fibers [9]. Silicon oxycarbides glasses display better properties, compared to silica glass, due to the presence in the amorphous network of Si–C bonds and graphene layers. In particular they show improved high temperature stability, better mechanical properties and durability against harsh environments [10, 11].
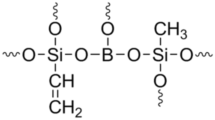
In this work we report on the synthesis and characterization of hybrid borosilicate gels and we show that they are promising candidates for the formation of SiBOC fibers. The motivations for this work are: (i) the introduction of B in SiCO glasses improves the high temperature stability leading to a more refractory material [12, 13], (ii) hybrid silicon alkoxides readily form, when reacted with boric acid, B(OH)3, a homogeneous borosilicate gel which is the ideal precursor for SiBOC glasses [14]. Indeed, the formation of a borosilicate gel from a silicon alkoxide and boric acid is based on the ability of B(OH)3 to hydrolize the Si–OR groups forming Si–OH moieties which can then condense with residual B–OH to form a stable borosiloxane bridge, =B–O–Si≡. Alternatively, B–OH can directly condense with a Si–OR group forming the borosiloxane bond and the corresponding alcohol molecule, ROH [14].
This work deals with the synthesis, using B(OH)3 as reactant, of borosilicate gels from mixtures of trifunctional methyltriethoxysilane (MTES) and di-functional dimethyldiethoxysilane (DMDES). The use of difunctional silicon units from DMDES, –(CH3)2SiO– promotes the formation, during the sol–gel process, of linear oligomers which should facilitate fiber drawing before gelation.
Two different B(OH)3/(MTES + DMDES) molar ratios were used in order to vary the amount of B in the final SiBOC materials and to control the crosslinking degree and the molecular weight of the oligomers formed in the solution. Indeed, in our process the boric acid has a two-fold role: it serves as a source of boron but also it promotes the condensation of the silicon alkoxides and the formation of the intermediates oligomers and the final gel network.
The obtained gels and fibers were characterized by fourier transformed infrared spectroscopy (FT-IR), thermogravimetry and differential thermal analysis (TG-DTA), X-ray diffraction (XRD) and differential scanning calorimetry (DSC) and the glass fibers by FT-IR and scanning electron microscopy (SEM).
2 Experimental procedure
The starting hybrid silicon alkoxides, methyltriethoxysilane (MTES) and dimethyldiethoxysilane (DMDES) were purchased from ABCR, (Germany) while boric acid, B(OH)3, was purchased from Carlo Erba, Italy. All reagents were used as received, without further purification. Two sets of solutions with B/Si molar ratio of 0.33, labeled as L (LOW boron content), and 0.66 labeled as H (HIGH boron content) were prepared. The molar ratio between the silicon alkoxides (DMDES/MTES) was varied as follows: 0/3, 1/2, 2/1 and 3/0. The number after the letter H or L indicating the amount of boron, corresponds to the moles of DMDES used in the synthesis. For example, the sample labeled L2 refers to the composition obtained with B/Si = 0.33 and DMDES/MTES = 2.
Solutions were prepared at 70 °C by stirring the silicon alkoxides with the boric acid under reflux in nitrogen gas until complete dissolution. Clear solutions were obtained at this temperature for all samples except for the pure DMDES compositions, for which 90 °C were necessary for dissolving all the boric acid. After the solutions became transparent they were poured into polypropylene containers at room temperature. The tubes were left open for gelation. Table 1 lists the composition of the samples, the stirring time necessary for the complete dissolution of boric acid in the silicon alkoxide solutions and the gelling time.
The feasibility of fiber drawing was regularly checked during the aging of the sol–gel solutions. Accordingly, gel fibers were easily hand drawn with a spatula from all solutions. Typically, the production of the fibers was successful in partially aged solutions. Solutions were spinnable for several days before gelation. The gel fibers were pyrolyzed under 150 mL/min of flowing argon at 1000 °C to obtain the borosilicon oxycarbide fibers.
The samples (gel and fibers) were dried in an oven at 60 °C for 1 week. For the L3 and H3 compositions, which did not gel at room temperature, this mild heating process was enough to promote gelation.
FT-IR analyses were performed on the gels, on the gel fibers and on pyrolyzed products with a Magna Infrared Spectrometer (Nicolet Avatar 550 FT-IR from Thermo Electron Corporation, Waltham, MA). KBr pellets were prepared and 32 scans were recorded for each spectrum from 4000 to 400 cm−1 using a 2 cm−1 resolution and 1 cm−1 acquisition. Background was subtracted in all the cases.
Thermal analyses of the gels and pyrolyzed products were carried out by TG-DTA (NETZSCH, Geräteban, STA 409) from room temperature up to 1400°C under 100 mL/min of flowing argon using 10 °C/min of heating rate. The sample weight in each analysis was about 60 mg.
DSC was also performed on selected samples to get information on the glass transition temperature of the hybrid gels. The equipment used for this purpose was DSC 2010 from TA Instruments and the amount of material used for each analysis was about 12–20 mg.
Crystallinity of the gels and fibers was evaluated from the XRD patterns obtained in a Rigaku Dmax diffractometer, (Tokyo, Japan) in the Bragg-Brentano configuration using CuKα operating at 40 kV and 30 mA. The spectra were collected in the 10–60° 2θ range, with 5 s of time acquisition and 0.05° of step size.
Microstructure of the gel and pyrolyzed fibers was studied with a JEOL, JSM-5500 Scanning Electron Microscope.
3 Results and discussion
3.1 Characterization of gels
3.1.1 B(OH)3 dissolution and gelation time
Two sets of samples with different DMDES/MTES molar ratios and boric acid were synthesized by the sol–gel method. As it is shown in Table 1, the time necessary to dissolve the boric acid increases with both the boron content and the amount of DMDES. Moreover, when only DMDES was used as silicon precursor complete dissolution of the boric acid occurred only at higher temperatures (90°C). Dissolution of B(OH)3 in the alkoxide mixtures is facilitated by the presence of ethanol produced from the reaction of the Si-OEt groups with B-OH. Accordingly, the amount of ethanol formed in the presence of DMDES, which has only two hydrolizable Si-OEt moieties is smaller than that resulting from MTES, which has three Si-OEt groups per silicon atom, and this could explain the longer dissolution times for samples in which MTES is progressively substituted with DMDES. Another possibility could be a lower reactivity of the DMDES towards B(OH)3, even if, in general, difunctional silicon alkoxides are more reactive toward hydrolysis than ternary ones such as MTES [15].
Gelation of the borosilicate solutions takes place over a time range of 1–4 weeks, approximately. We are aware that the gelation process for the studied samples is complicated by the concomitant events of solvent evaporation and moisture pick up from the environment. Nevertheless, few observations deserve mention: (i) gelation time is roughly independent from the MTES/DMDES ratio for the low boron composition (B/Si = 0.33) while it shows a clear trend in the case of using a B/Si ratio of 0.66. In general, the increase of gelation time when trifunctional MTES is substituted with the difunctional DMDES is due to the lower connectivity of the resulting siloxane network. Accordingly, the pure DMDES alkoxides, never reach the gelation point at room temperature, probably because it mainly forms linear chains or cyclic species.
3.1.2 Infrared study
FT-IR spectra of the dried gels show the following main bands (Fig. 1): νB-O in the range 1500–1300 cm−1, δSi-CH3 in the 1272–1262 cm−1, νSi–O at 1091 and 1040 cm−1, νSi-C at 860 cm−1, Si-CH3 at 804 cm−1 and δO–Si–O around 460 cm−1 [16–18]. The band at 890 cm−1, is the fingerprint of the borosiloxane bridges, =B–O–Si≡, formed in a hybrid silica matrix in which Si atoms form both Si–O and Si–C bonds at the same time [19]. The bands at 1197 and 547 cm−1, which are clearly visible in the DMDES rich samples (L2-3 and H2-3), suggest the separation of boric acid [19].

FT-IR spectra recorded on the borosilicate gels with low (L) and high (H) boron content. (*) Absorption bands related to B(OH)3
The evolution of the IR spectra with the amount of DMDES is similar for both of the investigated B/Si ratio. The spectral region between 1300 and 1550 cm−1, which characterizes the B–O bonds, broadens with the amount of DMDES. Moreover, several peaks can be distinguished inside this broad band (especially for the L3 and H3 compositions) which could be assigned to residual organic moieties such as Si–OEt or trapped ethanol. The presence of residual organic moieties for the DMDES-rich samples agrees with the lower reactivity of these gels toward B(OH)3 which has been deduced from the dissolution and gelling times previously reported. Another interesting trend shown by the FT-IR spectra is the following: in spite of the decreased reactivity of B(OH)3 in the DMDES rich-compositions, an increase of the absorption band of =B–O–Si≡ bridges at 890 cm−1 with the amount of difunctional alkoxide can be easily observed in both L and H samples.
3.1.3 X-ray diffraction (XRD)
For all the samples the XRD spectra are characterized by a broad halo centered at 22° associated with the formation of the siloxane network. XRD patterns of the DMDES-rich gels (L2-3, H2–3) show also the presence of crystalline B(OH)3. A typical XRD spectrum recorded on these DMDES-rich samples is shown in Fig. 2. Diffraction peaks at 2θ ≈ 14.6°, 28° and 40° are assigned to crystalline boric acid in the form of sassolite mineral [20]. For the samples rich in MTES the formation of crystalline boric acid was not observed. These results are in good agreement with those from FT-IR, in which the formation of B(OH)3 in the DMDES-rich compositions was suggested by the presence of the main band at 1197 cm−1 (see Fig. 1).

Typical XRD spectrum recorded on DMDES-rich gels: H2 composition
3.1.4 Thermal analysis (TG-DTA)
TG-DTA study was used to investigate the conversion process of the borosilicate gels into the SiBOC glasses. The DTA analysis did not show any relevant information and the patterns were characterized by broad endothermic effects corresponding to the various weight loss steps revealed in the TG study.
The thermogravimetrical evolution of the borosilicate gels in flowing argon is reported in Fig. 3. The pyrolysis process leads to a weight loss up to 800 °C; above this temperature the SiBOC glasses are thermally stable up to 1400 °C. The ceramic yield decreases by increasing the DMDES content in the starting gel. For the L3 and H3 gels, in which only difunctional –(CH3)2SiO– units are present in the network, the weight losses are higher than 60%.

TGA profiles recorded for samples with (a) low, L, and (b) high, H boron content heated in argon up to 1400 °C at a heating rate of 10°C/min
As a general trend, the pyrolysis in inert atmosphere of hybrid silica gels containing difunctional and trifunctional silicon units bearing Si-CH3 bonds can be divided in the following steps: (1) from room temperature to 300–400°C, the weight loss is associated to the evolution of water and ethanol molecules resulting from the residual Si-OH and Si-OEt groups present in the gels; (2) from 300–400°C up to 600°C, redistribution reactions between Si–O/Si–C bonds leads to the evolution of low molecular weight siloxane species [21] and (3) from 600 up to 800–900°C, the mineralization reactions take place with the homolytic cleavage of C–H and Si–C bonds and the formation of H2 and CH4 [22, 23]. In general, when difunctional silicon species, such as those derived from the DMDES or methyldiethoxysilane (MDES), are present in the starting gels the redistribution reactions leads to higher weight losses due to the easier formation of linear or cyclic volatile molecules [24]. The introduction of boron in borosilicate gels may lead to an increase of the low temperature weight loss step (up to 300–400°C) due to evolution of water from B–OH species [25].
We start discussing the TG curves from the high boron containing samples, the H series (Fig. 3b). It can be noticed that for the H0 and H1 samples, the weight is stable up to 400°C suggesting the formation of a well condensed gel network without residual ethoxy/hydroxy groups or B(OH)3. Above 400°C, the H0 and H1 gels show two well distinct weight loss steps in the following ranges: at 400–600°C assigned to the evolution of silanes/siloxanes formed by redistribution reactions, and between 600 and 700 °C due to the mineralization step with evolution of CH4 and H2. The magnitude of the weight loss is comparable for the two samples being slightly higher for the H1 composition. These results suggest that the DMDES units present in the H1 sample are homogeneously distributed in the siloxane network without the presence of DMDES long chains, which could easily led to the escape of cyclic siloxane and to the increase of the weight loss compared to the H0 samples. Contrary to the first two compositions, the gel H2 shows a weight loss from room temperature to 400 °C of 13 wt.% and a much more evident second step from 400 to 600 °C. The weight loss below 300 °C suggests the presence, in the H2 sample, of residual terminal moieties (–OEt and –OH) or even B(OH)3 as a separate phase, and supports our previous finding that the reactivity of B(OH)3 towards the silicon alkoxides decreases by increasing the amount of DMDES. For this sample, the weight loss step related to redistribution reactions becomes the most important one and accounts for a loss of 35 wt.% over a total loss of 55 wt.%. Such a high weight loss is certainly due to the presence of linear DMDES chains that can easily form during pyrolysis, either small oligomers or cyclic siloxanes such as (CH3)3Si–O–Si(CH3)3 and [(CH3)2SiO]4cy respectively. From 600 to 700 °C the mineralization step leads to a weight loss of ca. 5%, similar to the previous H0 and H1 samples. The H3 sample shows an almost complete decomposition above 400 °C with a weight loss above 60 wt.%.
The compositions with a lower amount of boron (L serie, Fig. 3a) display a similar but less defined trend. In this case even the two compositions with no or low amount of DMDES (L0 and L1) show a weight loss at low temperature. Moreover, the two high temperature weight loss steps observed for the H series are not well defined. We explain this result, and in particular the low temperature decomposition step, by assuming that the low amount of B(OH)3 leaves behind a higher amount of un-reacted Si–OEt groups which can, during the initial stages of pyrolysis, be responsible for the observed weight losses. We have to remind here that B(OH)3 serves, not only as a source of B but also promotes the hydrolysis and condensation of the silicon alkoxides and, therefore, it is reasonable to assume that the unreacted alkoxy moieties will increases by decreasing the amount of boric acid.
Even for this series of samples, L, the L2 gel shows a main weight loss in the range 300–600 °C due to redistribution reactions, and the L3 samples decomposes almost completely with a weight loss above 60 wt.% before 400 °C.
3.1.5 Differential scanning calorimetry (DSC)
The presence of linear PDMS chains in the siloxane structure has been inferred from the TG results, especially for the samples with higher amount of DMDES (L2-3 and H2-3 specimens). Among these samples, H2 shows the most clear and intense weight loss step in the range between 300 and 600 °C (besides the L3 and H3 compositions that decompose completely and therefore are not useful as ceramic precursors). Accordingly, a DSC analysis has been recorded on the H2 gel in order to confirm the formation of PDMS chains. Indeed, if linear –(CH3)2SiO– chains are present in the borosiloxane network they should give rise to a glass transition effect in the DSC pattern at a temperature dependant on the chain length and mobility in the network. The DSC curve recorded on the H2 gel is reported in Fig. 4. In the temperature range between −62 and −58 °C a clear endothermic effect has been observed and assigned to the glass transition effect of PDMS chains, result which is in agreement with our assumption. Glass transition temperature of infinite PDMS chains has been reported in the literature to be at −123 °C [26] while for PDMS oligomers crosslinked by various metal alkoxides, Tg occurs at higher temperature [27]. In particular, for PDMS oligomers with a molecular weight of 1500 uma, corresponding to chains with 22 repeating –(CH3)2SiO– units, crosslinked with titanium alkoxide, Tg shifts at higher temperature, 99 °C [26]. Accordingly, the glass transition temperature found in the H2 gel, corresponding to the inflexion point of the DSC curve, Tg = −60 °C, suggests the presence of shorter or less mobile oligomers in our material. This peculiar gel structure in which short linear oligomers are crosslinked by trifunctional ones could be the reason why the partially aged solutions are very well suited for fiber drawing, as it will be shown in the next paragraph.

DSC profile of gel H2. The mark indicates the glass transition temperature, Tg = −60 °C
3.2 Characterization of the fibers
Fibers up to near 5 m of length were easily obtained by immersing a spatula and pulling out material from solutions of appropriate viscosity. The fibers were very strong and therefore, they were easy-handled. It has been found that solution H2 was the most appropriate for an easy-production of fibers during periods as long as 2 weeks (after 1 week from its preparation).
The fibers obtained from every solution were pyrolyzed in an alumina tubular furnace under 150 mL/min of flowing argon up to 1000 °C to yield SiBOC glass fibers.
3.2.1 Fourier transform infrared spectroscopy (FT-IR)
The IR spectra of the gel and glassy SiBOC fibers obtained from H2 composition before and after pyrolysis are reported in Fig. 5. The IR spectrum of the gel fiber shows all the characteristic absorptions due to the presence of Si–CH3 (whose bending and stretching bands are located at 1261 and 802 cm−1, respectively) and Si–O stretching at 1074 cm−1. The small band at 1630 cm−1 is due to the presence of absorbed water. It is very interesting to notice the peaks associated to the B related IR bands: the B–O stretching band, located at 1500–1300 cm−1 has its maximum shifted toward lower wavenumbers, which is a typical feature of BO3 units well incorporated into the siloxane network [19]. The same indication arises from (i) the absence of the FT-IR peaks due to an additional boric acid phase and (ii) the presence of well defined and intense B–O–Si bands at 887 cm−1 and 680 cm−1. Finally, the absence of additional features in the broad B–O band points towards a gel fiber without residual alkoxy groups and trapped solvent molecules. Accordingly, compared to the bulk gel of the same composition, the H2 fibers shows a well hydrolyzed and condensed structure in which boron is well incorporated in the siloxane matrix via borosiloxane bridges without additional B(OH)3 phase. This difference can be related to the higher specific surface of the fibers compared to the bulk which makes easier the hydrolysis from the ambient moisture and solvent evaporation.

IR spectra of the H2 gel fiber and corresponding SiBOC ceramic fiber
In the spectrum of the pyrolyzed fiber all the bands related to the organic CH3 moieties have disappeared and the spectrum is typical of an inorganic SiBCO glass: B–O bonds at 1400 cm−1, Si–O at 1100 cm−1 and Si–C at 805 cm−1. The inorganic character is also indicated by the shift of the =B–O–Si≡ bridges from 887 cm−1 to 913 cm−1. The intense band at 1635 cm−1 due to absorbed water molecules may be related to the presence, in the pyrolyzed fibers of residual porosity which develops during pyrolysis and usually closes at T ≥ 800 °C [28].
3.2.2 Scanning electron microscopy (SEM)
Figure 6 shows some typical gel and ceramic SiOC fibers. As can be seen, fiber with a diameter as low as 10 μm (Fig. 6a) can be obtained from various solutions. However, it has to be reminded that the composition which allows the easiest fiber drawing is the H2. Fiber surface is usually smooth and defect free. Cross-sectional analysis indicated that the fibers are dense, pore-free and homogeneous. The pyrolysis process leads to ceramic fibers without damaging the fiber surface or introducing defects such as cracks or pores (Fig. 6b).

SEM of gel (a) and glass SiBOC fibers (b) obtained from solution H2. (The insert reports the cross-sectional view which shows the dense, pore free, structure of the pyrolyzed fiber)
4 Conclusions
Several borosilicate gels have been prepared from sol–gel solutions of MTES, DMDES and boric acid, with different molar ratios B/Si and MTES/DMDES. FT-IR study indicates that boron atoms are incorporated in the siloxane network via =B–O–Si≡ bridges. The presence of such borosiloxane bonds increases when the trifunctional MTES is substituted with the difunctional DMDES. For the DMDES-rich compositions a glass transition temperature of −67 °C has been observed from DSC and it has been assigned to the formation, in these gels, of linear polydimethylsiloxane, PDMS, chains.
Gel fibers can be easily drawn from all the borosiloxane gels, especially form those compositions in which the formation of linear oligomers has been observed (DMDES-rich compositions). Pyrolysis of the gel fibers in inert atmosphere has shown the possibility of producing homogeneous, crack-free SiBOC fibers with a diameter as low as 10 μm.
References
Dislich H (1985) J Non-Cryst Solids 73:599
Kamiya K, Yoko T, Sano T, Tanaka K (1990) J Non-Cryst Solids 119:14
Alain C. Pierre (1998) Introduction to sol–gel processing. Kluwer Academic Publishers, Boston
Japan Patent, Nichias Corp., Jpn. Kokai Tokkyo Koho JP 60,167,924 [85,167,927]
Sakka S, Kamiya K, (1980) J Non-Cryst Solids 42(1–3):403
Brinker CJ, Scherer GW (1990) Sol–gel science: the physics and chemistry of sol–gel processing. Academic Press, San Diego
Sakka S, Kamiya K, Makita K, Yamamoto Y, (1984) J Non-Cryst Solids 63(1–2):223
Sakka S (1984) In: Brinker CJ, Clark DE, Ulrich DR (eds) Better ceramics through chemistry, Mat. Res. Soc. Symp. Proc. vol 32. North-Holland, New York, p 91
Kamiya K, Katayama A, Suzuki H, Nishida K, Hashimoto T, Matsuoka J, Nasu H (1999) J Sol–Gel Sci Technol 14:95
Renlund GM, Prochazka S, Doremus RH (1991) J Mater Res 6(12):2716
Soraru GD, Modena S, Guadagnino E, Colombo P, Egan J, Pantano C (2002) J Am Ceram Soc 85(6):1529
Klonczynski A, Schneider G, Riedel R, Theissmann R (2004) Adv Eng Mater 6:64
Gervais C, Babonneau F, Dallabona N, Soraru GD (2001) J Amer Ceram Soc 84[10]:2160
Soraru GD, Babonneau F, Gervais C, Dallabona N (2000) J Sol–Gel Sci Tech 18:11
Sorarù GD, D’Andrea G, Campostrini R, Babonneau F (1995) J Mater Chem 5:1363
Viart N, Rehspringer JL (1996) J Non-Cryst Solids 195:223
Peña-Alonso R, Rubio J, Rubio F, Oteo JL (2002) J Sol–Gel Sci Tech 25:255
Kasgoz A, Misono T, Abe Y (1999) J Non-Cryst Solids 243:168
Sorarù GD, Dallabona N, Gervais C, Babonneau F (1999) Chem Mater 11:910
JCPDS 30–199
Mutin PH (2002) J Am Ceram Soc, 85(5):1185
Trimmel G, Badheka R, Babonneau F, Latournerie J, Dempsey P, Bahloul-Houlier D, Parmentier J, Soraru GD (2003) J Sol–Gel Sci Tech 26:279
Pederiva L, Soraru GD, Latourneire J, Raj R, (2002) J Am Ceram Soc 85(9) 2181
Sorarù GD, D’Andrea G, Campostrini R, Babonneau F (1995) J Mater Chem 5:1363
Sorarù GD, Campostrini R, Maurina S, Babonneau F (1997) J Am Ceram Soc. 80:999
Clarson SJ (1993) In: Clarson SJ, Semlyen JA (eds) Siloxane polymers. PTR Prentice Hall, Englewood Cliff, p 216
Yamada N, Yoshinaga I, Katayama S (1997) J Mater Chem 7:1491
Schmidt H, Koch D, Grathwohl G, Colombo P (2001) J Am Ceram Soc 84[10], 2252
Acknowledgements
This work was partly supported by COFIN-2005 and by the European Community FP6 through MCRTN-019601, PolyCerNet. R. Peña-Alonso acknowledges the Ministerio de Educación y Ciencia from Spain for financial support under post-doctoral fellowship.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Peña-Alonso, R., Sorarù, G. Synthesis and characterization of hybrid borosiloxane gels as precursors for Si–B–O–C fibers. J Sol-Gel Sci Technol 43, 313–319 (2007). https://doi.org/10.1007/s10971-007-1558-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10971-007-1558-2