
Abstract
The trace impurities of a uranium ore concentrate (UOC) can be examined to determine mine source, methods of production, and quality. This study presents a method to determine the concentration of halides and main group elements, specifically P, S, Br and I, utilizing triple quadrupole inductively coupled plasma–mass spectrometry. These analytes were measured in a uranium matrix to simulate a UOC sample. The concentrations determined with this method showed agreement with known values. Solutions with and without uranium were compared. A UOC certified reference material, CUP-2, was analyzed to further demonstrate the effectiveness of the method.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Nuclear reactors supply over ten percent of the world’s energy needs [1]. Most nuclear reactors use uranium as the fissile material. One of the initial steps in the uranium nuclear fuel cycle is the production of a uranium ore concentrate (UOC) from various deposits (e.g. sandstone, lignite, and vein). Extraction of uranium from the mined material often results in high amounts of impurities within the final product, and analysis of these UOC impurities can be used in forensic and safeguard applications.
UOCs are produced by adding acids to mined and milled ore to leach out and concentrate uranium. The extraction method will vary depending on the type of mineral [2]. The impurities present in the UOC are dependent on both the production method and the geographic area from which the ore was mined [3]. The geological location of the mined ore is important as different locations may have varying degrees of impurities present. Quantification of these impurities can help identify the original source of the UOC. The specific chemical and elemental signatures found in nuclear materials can provide information regarding the production history of the material through region specific trace element patterns, including rare earth elements and anions [4,5,6]. Several studies have demonstrated that analysis of these can be used to identify a UOC to a specific origin [3, 7, 8].
No single signature has been shown to uniquely determine the source of a given material, so it is often necessary to analyze multiple signatures as part of complete forensic analysis [8]. The measurement of halides and main group elements, often present in UOCs as anions, can provide information regarding the process history [9]. For example, Badaut et al. found clear differences in anion concentrations when analyzing UOC samples from different geographical locations. The authors were able to determine which sample set originated in Beverley Mine (Australia) based on a low sulfate concentration, 15 µg mL−1 versus 100–900 µg mL−1 at other locations, as only one location in the study involved a process history that did not use large amounts of sulfate [5]. Anion content was determined using ion chromatography (IC) with conductivity detection.
Several techniques have been used to measure the composition of UOCs [10]. Handheld Raman spectrometers can be used as a screening method during the initial production. These systems determine the chemical form of uranium oxides as well as impurities with minimal sample preparation [11,12,13]. This method suffers in sensitivity. Fourier-transform infrared spectrometry (FTIR) has also been demonstrated for UOC analysis, identifying uranium oxides and some anionic species that fall in the mid IR range [14]. For example, sulfates and nitrates are often analyzed via FTIR due to their prominent bands at 1120 cm−1 and 1384 cm−1, respectively [2]. One drawback to FTIR is the reliance on a peak in the IR range which limits the applicability of the measurement. The most common methods for determining a multitude of trace elements in a UOC include inductively coupled plasma–mass spectrometry (ICP–MS) [15, 16] and inductively coupled plasma—optical emission spectroscopy [17, 18]. ICP–MS is a method of choice for many studies because it can analyze a wide range of elements at very low concentrations (ng mL−1) [3, 8, 15, 19,20,21]. Although halides are generally present in UOCs as salts or anions, they may be analyzed by ICP–MS if the ionization energy of the plasma is sufficient. In the case of sulphate and phosphate, the ICP–MS only detects S and P, but it is assumed that they originated as sulphate and phosphate ions.
The halides and some main group elements like F, Cl, P, S, Br, and I, typically present as anionic species, have unique properties that must be addressed for accurate or precise measurement when analyzing by ICP–MS. These elements are prone to volatility, spectral interferences, and sample carry-over effects. Some studies have minimized these issues by using a pyrohydrolysis method to extract the anions, IC to separate them, and conductivity detection for analysis [8, 22,23,24]. This method has been shown to be effective, but the detection limits are generally orders of magnitude lower when employing an ICP–MS. For example, Balcone-Boissard et al. found that Br and I concentrations fell below detection limits when analyzing by conductivity detection but were measurable by ICP–MS [22]. If utilizing this instrument, carry-over can be addressed by washing through a base such as tetra-methyl ammonium hydroxide (TMAH). TMAH can also address volatility issues by keeping the anion in solution [25, 26].
This paper demonstrates a method to determine the content of specific halide and main group elements in a uranium matrix using a triple quadrupole (TQ) ICP–MS. The TQ removes spectral interferences by employing a reaction gas in the second quadrupole and eliminating the interference in the third quadrupole. Using this methodology individual analyte separation, such as IC, is not needed before analysis. Additionally, a hazardous alkaline solution like TMAH was not required when using extended wash times to minimize carry-over effects.
Experimental
Materials and methods
All standards and samples were prepared in 2% nitric acid (HNO3), diluted from a concentrated stock of Fisher Optima grade (Pittsburgh, PA, USA) using ASTM Type I water (18.2 MΩ cm) produced by a Thermo Scientific Barnstead GenPure xCAD Plus Ultra-Pure water purification system (Waltham, MA, USA). Trace amounts of Fisher Optima grade hydrofluoric acid (HF) was used in the preparation of CUP-2 reference material.
Standards were prepared volumetrically by serial dilution from single element stocks (10 µg mL−1) purchased from High Purity Standards (Charleston, SC, USA). Solutions were prepared fresh in HDPE containers. Quality control (QC) solutions were also prepared by serial dilution from a single element stock of iodide (100 µg mL−1 I) and a multi-element stock containing P, S, and Br (100 µg mL−1 PO4, SO4, and Br) (Multi Element IC Std 1 Solution A) purchased from High Purity Standards. External calibration was utilized for quantification with a 4-point calibration curve. P, Br and I had concentrations of 0, 1, 5, and 10 ng mL−1 while S had concentrations of 0, 50, 75, and 100 ng mL−1. The QC solution contained concentrations of 5 ng mL−1 Br and I, 1.6 ng mL−1 P, and 50 ng mL−1 S. All solutions in the uranium matrix runs, including the blank, were analyzed in a uranium matrix of 50 µg mL−1 that was prepared from a stock solution of uranium (High Purity Standards). Extended wash times of at least 10 min were performed with 2% HNO3. Solutions were analyzed immediately after preparation to minimize losses due to volatility.
CUP-2 was prepared by weighing a 200 mg fraction of the dried UOC powder and adding 8 M HNO3—0.05 M HF. The sample was microwave digested using a CEM (Mathews, NC, USA) Discover SP-D microwave. The dissolved UOC was diluted gravimetrically to approximately 10 mg UOC g solution−1. The uranium concentration was determined using Davies-Gray titration, and the sample was subsequently diluted to a uranium concentration of 50 µg mL−1 in 2% HNO3—1 mM HF.
Instrumentation
A Thermo Scientific (Bremen, Germany) iCAP TQ ICP–MS was used for analyte measurement. The TQ has the ability to utilize ultrahigh purity (99.994%) gases within the reaction cell. Available reaction gases include oxygen, hydrogen, ammonia, and nitrogen (Airgas, Radnor, Pennsylvania). The optimized instrument parameters are listed in Table 1. A Peltier cooled quartz cyclonic spray chamber was used with a self-aspirating PFA 50 µL min−1 nebulizer (ESI, Omaha, Nebraska, USA). The cyclonic spray chamber removes larger droplets from the aerosol, and the Peltier cooling system reduces the amount of solvent that enters the plasma, leading to reduction of interferences such as oxides and hydroxides. The constant temperature also helps reduce signal drift associated with environmental temperature changes [27].
Results and discussion
Reaction gas determination and interferences
The analytes begin as anions in solution, but when they enter the plasma, the ionization energy is high enough to remove electrons converting them to cations despite their high electronegativities. Fluorine and chlorine were investigated, but their ionization potentials limited the formation of cations in the plasma. The difficulty in measuring these elements by ICP–MS with a sufficient detection limit has been demonstrated in another study [15]. No further analysis of F or Cl was performed. The TQ ICP–MS can be used for on-mass or off-mass analysis as shown in Fig. 1. On-mass analysis is when the mass detected is the same as the analyte mass set to pass through Q1, and the spectral interferences react with the collision cell gas so that Q3 will filter them out. For the case of off-mass analysis the analyte itself reacts with the gas in the collision cell so that the mass is shifted away from the spectral interferences, and Q3 filters out any mass other than that of the combined mass of the selected gas and the original analyte (Fig. 1). On-mass analysis was employed for 127I+ and off-mass analysis was employed for 31P16O+, 32S16O+, and 79Br16O+.

A diagram demonstrating the difference between on-mass and off-mass analysis using the TQ ICP–MS. Depicts spectral interferences passing through Q1 but not Q3 while the desired ion passes through to the detector. This is a pictorial display of both modes of operation, these modes can be performed sequentially
The TQ ICP–MS utilized for this study incorporated four different reaction gases (hydrogen, helium, ammonia, and oxygen) employed as operating modes. Additionally, the kinetic energy discrimination (KED) mode uses a small amount of He in the collision cell to filter out unwanted spectral interferences based on their collision cross section. This requires a difference in size between the analyte and spectral interference rather than a difference in reactivity. The choice of collision cell gas for each ion depends on the thermochemistry and kinetics of the collisions that occur in the cell. For each ion, an ion exchange reaction with each gas can be proposed. For these reactions to occur, the enthalpy of formation of the reactants must be greater than that of the products. If this is not the case, then additional energy will be necessary for the reaction to proceed. This energy can originate from collisions that occur in the cell or by radio frequency [28]. If there is not enough energy to create a favorable reaction, then that reaction gas is not suitable for use with that ion. The best mode for each of the analytes was experimentally determined before method demonstration. Each species was analyzed via all modes, at 100 ng mL−1 to determine ion transmission with the reaction gas. Single element solutions of the interfering ions were then injected at µg mL−1 levels to determine spectral interference removal.
Phosphorus showed favorable response by either oxygen or KED mode. Both modes removed the spectral interferences listed in Table 2. The oxygen mode was chosen for P because it showed greater sensitivity (60x) than KED mode. For S, oxygen mode was the only mode that demonstrated the ability to resolve the spectral interferences by an off-mass shift (Table 2). Both oxygen and KED modes were evaluated for Br and showed favorable results. Upon analysis of single element interferences KED mode showed elevated Br counts due to an unresolved interference from Gd and Dy. For I, KED, oxygen, and helium were all investigated, all were able to eliminate spectral interferences (Table 2). The ion transmission was limited with KED having the least and helium having the greatest sensitivity. Oxygen provided half the sensitivity of helium for I analysis, but the number of counts were sufficient to make the measurements. Ultimately, oxygen mode was chosen for I as well. In this way all analytes studied utilized oxygen as the reaction gas in the collision cell. Using oxygen mode only kept analysis times down by limiting reaction gas equilibration times. These results were similar to the method developed by Balcaen et al. which used oxygen as a reaction gas for off-mass analysis in organic matrices containing P and S, but Br and I were analyzed using charge transfer reactions instead of on-mass or off-mass analysis [29]. Oxygen mode was used for the remainder of the experiment.
Method demonstration
For method demonstration the method was run on three separate days to determine accuracy and reproducibility. On each day the P, S, Br, and I were analyzed with and without a uranium matrix (50 µg mL−1), three times per day, to investigate the magnitude of uranium ion count suppression. The High Purity Standards IC solutions were used as a quality control to demonstrate the effectiveness of the method. All reported uncertainties are represented as 2σ as determined from the triplicate daily data points from each experiment. The reported uncertainties for the 3 day average concentrations are expanded uncertainties calculated using the sum of the square. The components in the uncertainty model were the slope of the calibration curve, the concentration of the stock standards, and the measurement variability. All RSDs were calculated by multiplying the standard deviation by 100 and dividing by the average concentration of the analyte.
It was necessary to use a weighted average of the matrix blank to correct for change in sensitivity across the run. The matrix blank was analyzed before and after a run, and a linear correction was applied to the sample. Signal sensitivity in the QCs decreased by an average of 20% during the run. After the corrections were applied, the daily average concentrations of P showed reproducibility across the 3 days of experiments, ~ 2.7% RSD without a uranium matrix and ~ 2.9% with a uranium matrix (Fig. 2). The average concentration of P among the 3 days was 1.74 ± 0.18 ng mL−1 when not in a uranium matrix and 1.64 ± 0.15 ng mL−1 in a uranium matrix. In both cases the uncertainties overlap with the known value of 1.630 ± 0.016 ng mL−1. The values for the 3-day average concentrations show agreement with each other.

Average daily concentration of P with three analysis per day. Solid line denotes known concentration and the dashed lines indicate uncertainty in the QC
Runs with and without a uranium matrix proved reproducible over 3 days with an average concentration of S measured at 50.1 ± 2.5 ng mL−1 (~ 1.7% RSD) with no matrix and 47.4 ± 3.3 ng mL−1 (~ 1.6% RSD) in a uranium matrix (Fig. 3). Both measured values show good recovery of the known concentration of 50.00 ± 0.50 ng mL−1 within the uncertainty of the measurement at a 0.14% difference when not in a uranium matrix and a 5.4% difference in a matrix (Fig. 3). While there is a difference between solutions with and without a uranium matrix on the third day, this could be attributed to run to run variability of the method. The solutions show agreement with each other as the average concentrations fall within the uncertainties.

Average daily concentration of S with three analysis per day. Solid line denotes known concentration and the dashed lines indicate uncertainty in the QC
For Br, the method demonstrated run to run reproducibility for solutions both with (~ 2.2% RSD) and without (~ 2.6% RSD) a uranium matrix (Fig. 4). The 3 day average concentrations were 5.30 ± 0.30 and 5.15 ± 0.35 ng mL−1, respectively. These values showed agreement with the known concentration of 5.00 ± 0.10 ng mL−1 for both solutions not in a uranium matrix (3.0% different) and in a uranium matrix (5.7% different) as well as with each other. The agreement between the 3 day average concentrations indicates the method may be useful for the determination of Br in a uranium matrix.

Average daily concentration of Br with three analysis per day. Solid line denotes known concentration and the dotted lines indicate uncertainty in the QC
Iodine was more difficult to analyze than the other species due to the carry-over and volatility effects. Initial 3 min washout times were extended to 10 min to eliminate any carry-over effects from I. Issues with I carry-over has been previously reported [30]. While in an acid matrix it was necessary to run the sample immediately after preparation to avoid losing I to volatilization [31]. Since samples were analyzed within 10 min, more study is necessary to determine if I can be measured after microwave digestion. TMAH can be used to help keep iodine in solution, but at a pH above 8, uranium will precipitate out of solution [8, 32]. In the current method, the only added sample preparation for I is the addition of uranium to the blank and standards. This matrix matching step allows for the counts from the UO++ interference at 127 amu to be accounted for, leaving only I+ counts.
After performing a blank subtraction, the method showed agreement with the known value of 5.00 ± 0.10 ng mL−1 for average concentration of I both with (1.3% difference) and without (2.7% difference) a uranium matrix (Fig. 5). The method demonstrated run to run reproducibility with an RSD of ~ 2.2% when not in a uranium matrix and ~ 2.7% in a uranium matrix. An average 3 day concentration of 4.9 ± 1.1 ng mL−1 for solutions with no matrix and 4.9 ± 1.4 ng mL−1 with a uranium matrix was determined. The small difference between results with and without the matrix suggests that the method is suitable for analyzing I in UOCs with little sample preparation. Additionally, the method does not require the use of TMAH or other alkaline materials used in many other ICP–MS studies [33, 34].

Average daily concentration of I with three analysis per day. Solid line denotes known concentration and the dotted lines indicate uncertainty in the QC
Limit of detection
The limit of detection for each analyte was calculated as both an instrument detection limit (IDL) and a method detection limit (MDL). The IDL was used to compare the results of the method with and without a uranium matrix while the MDL shows the effectiveness of the method for each species. The IDL was calculated using Eq. (1) where \( \sigma_{B} \) is the standard deviation of the blank and m is the slope of the external calibration curve. The MDL was determined by propagating the IDL through the method preparation steps such that the uranium concentration was 50 µg mL−1.
The IDLs of P, S, and Br were nearly identical whether the uranium matrix was present or not (Table 3). This supports the assertion that these analytes can be analyzed in a uranium matrix with little impact on the limit of quantification. The IDL of I was higher in a uranium matrix which would be expected due to the presence of the UO++ interference (Table 2). While the IDL for I was higher than the other analytes, the method still demonstrated the ability to detect I in a uranium matrix at the concentrations utilized in this study (Fig. 5). The IDL for S was similar to that found in another study while the P and Br IDLs were two to three orders of magnitude lower than those found in the same study using IC and conductivity detection [5].
The MDLs for P, Br, and I were similar while S was about one order of magnitude higher in concentration (Table 3). Sulfur had a higher MDL than the other analytes due to the higher background (50 × counts) in the blank solutions. Sulfur values vary depending on the process in the formation of UOCs, and this element is a more valuable process indicator [35]. Thus a high S blank, with the associated higher MDL, does not preclude a useful measurement of this species in UOC materials. Also, the higher MDL for S should not be an issue in analysis due to the generally high values of SO42− in UOC [5]. This is consistent with the results of an interlaboratory study, primarily utilizing sector field ICP–MS, where the average level of reported S in a UOC was nearly 6000 µg gU−1 [15]. The MDLs for P and S in the present work were similar to reported values from the interlaboratory study [15]. No participants reported values for Br or Cl due to the difficulty in quantifying them by ICP–MS. This method was successful in quantifying Br and had a MDL that was the lowest of all analytes studied (Table 3).
CUP-2 analysis
A certified reference material (CRM), CUP-2, was analyzed for anion content and the results were compared to the recommended values. CUP-2 is a UOC CRM that was jointly produced by the Canadian Certified Reference Materials Project and the Canadian Uranium Producers Metallurgical Committee. The CRM solution was diluted to a concentration of 50 µg U mL−1, consistent with the uranium content used during method demonstration. The calibration standards were matrix matched in the same manner as was performed previously. CUP-2 was evaluated three times on three separate days so an average concentration value could be reported, and the reproducibility of the method for the CRM could be investigated. The CRM was analyzed for P, S, Br, and I, although there is no recommended value for Br or I.
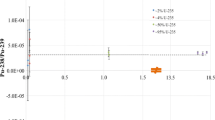
The average P and S concentrations determined using the method described in this study show agreement with the provisionally recommended values for the CUP-2 CRM (Fig. 6). The average 3 day P concentration (410 ± 45 µg g U−1) was within 3.1% of the recommended value (397 µg g U−1), and the S concentration (10,700 ± 900 µg g U−1) was within 0.89% of the recommended value (10,607 µg g U−1) (Fig. 6). This agreement suggests that the TQ ICP–MS method is suitable for detection of P and S in UOC materials. As mentioned previously, Br and I values are not available for CUP-2, because of the challenge of measuring the halides in material such as UOCs, but they were analyzed using the method developed in this study. No I was detected in CUP-2, nor has it been previously reported. This may be due several factors (e.g. extended storage time, volatility, and sample preparation). Further studies are required to determine if I could be detected in CUP-2 immediately after the sample was prepared. Bromine was detected utilizing this method with a 3 day average concentration of 21.7 ± 2.6 µg g U−1. While there is no recommended value to compare this result to, the determined concentration is above the MDL of this study (Table 3). The use of the TQ and the method developed here may open the door for more routine analysis of these analytes in the future, allowing an evaluation of their value as signatures of the UOC source.

Cup-2 data for P, S, and Br with a line indicating the recommended value
Conclusions
A method for analyzing P, S, Br, and I in a uranium matrix using a TQ ICP–MS is presented here. The instrument removed interferences that are common for the analytes in this study, and the results showed agreement between runs with and without a 50 μg mL−1 uranium matrix for P, S, Br, and I. While the method may sacrifice precision on a multiple day analysis and stability issues in the acid matrix were observed for I, the average concentrations for all analytes agreed with known values. When analyzing the data for possible matrix effects, a comparison of the results with and without uranium shows that this method is suitable for accurate quantification of P, S, Br, and I when using matrix matching standards. When analyzing CUP-2, the experimentally determined concentrations for P and S agreed with the recommended values. This suggests that the method may be useful for those species and allows the possibility of Br and I analysis which have not traditionally been undertaken by ICP–MS in UOC materials in the past. The method allows for faster analysis times with less sample preparation, bypassing the use of chemical separations and alkaline washes.
References
Chu S, Majumdar A (2012) Opportunities and challenges for a sustainable energy future. Nature 488(7411):294–303
Mayer K, Wallenius M, Varga Z (2015) Sample characteristics and nuclear forensic signatures. In: Fedchenko V (ed) The new nuclear forensicsanalysis of nuclear materials for security purposes. Oxford University Press, Oxford, pp 93–127
Keegan E, Richter S, Kelly I, Wong H, Gadd P, Kuehn H, Alonso-Munoz A (2008) The provenance of Australian uranium ore concentrates by elemental and isotopic analysis. Appl Geochem 23(4):765–777
Moody KJ, Grant PM, Hutcheon ID (2015) Nuclear forensic analysis, 2nd edn. CRC Press Taylor & Francis Group, Boca Raton
Badaut V, Wallenius M, Mayer K (2009) Anion analysis in uranium ore concentrates by ion chromatography. J Radioanal Nucl Chem 280(1):57–61
Lin M, Zhao YG, Zhao LF, Li LL, Wang F, Zhu LC, Hu XN, Ning W (2015) Tracing origins of uranium ore concentrates (UOCs) by multidimensional statistical analysis of rare-earth impurities. J Anal At Spectrom 30(2):396–402
Svedkauskaite-LeGore J, Rasmussen G, Abousahl S, van Belle P (2008) Investigation of the sample characteristics needed for the determination of the origin of uranium-bearing materials. J Radioanal Nucl Chem 278(1):201–209
Keegan E, Wallenius M, Mayer K, Varga Z, Rasmussen G (2012) Attribution of uranium ore concentrates using elemental and anionic data. Appl Geochem 27(8):1600–1609
Significance of Mineralogy in the Developments of Flowsheets flow sheets for Processing Uranium Ores (1980) Technical Reports Series NO. 196. International Atomic Energy Agency, Vienna
Kristo MJ, Gaffney AM, Marks N, Knight K, Cassata WS, Hutcheon ID (2016) Nuclear forensic science: analysis of nuclear material out of regulatory control. In: Jeanloz R, Freeman KH (eds) Annual review of earth and planetary sciences, vol 44. Annual Reviews, Palo Alto, pp 555–579. https://doi.org/10.1146/annurev-earth-060115-012309
Su YF, Tonkyn RG, Sweet LE, Corbey JF, Bryan SA, Johnson TJ (2018) Characterization of uranium ore concentrate chemical composition via Raman spectroscopy. In: 19th meeting of the chemical, biological, radiological, nuclear, and explosives (CBRNE) Sensing as part of the SPIE defense and commercial sensing (DCS) symposium, Orlando, FL, Apr 16-18 2018. Proceedings of SPIE. Spie-Int Soc Optical Engineering, BELLINGHAM. https://doi.org/10.1117/12.2304304
Pointurier F, Marie O (2013) Use of micro-Raman spectrometry coupled with scanning electron microscopy to determine the chemical form of uranium compounds in micrometer-size particles. J Raman Spectrosc 44(12):1753–1759
Lin DHM, Manara D, Varga Z, Berlizov A, Fanghanel T, Mayer K (2013) Applicability of Raman spectroscopy as a tool in nuclear forensics for analysis of uranium ore concentrates. Radiochim Acta 101(12):779–784
Varga Z, Ozturk B, Meppen M, Mayer K, Wallenius M, Apostolidis C (2011) Characterization and classification of uranium ore concentrates (yellow cakes) using infrared spectrometry. Radiochim Acta 99(12):807–813
Burger S, Boulyga SF, Penkin MV, Bostick D, Jovanovic S, Lindvall R, Rasmussen G, Riciputi L (2014) Quantifying multiple trace elements in uranium ore concentrates: an interlaboratory comparison. J Radioanal Nucl Chem 301(3):711–729
Rogers KT, Giaquinto J, Essex RM, Metzger SC, Ticknor BW, Hexel CR (2018) Trace impurity analysis in uranium oxide via hybrid quantification techniques-gravimetric standard addition and isotope dilution mass spectrometry. J Radioanal Nucl Chem 318(1):685–694
Manard BT, Metzger SC, Quarles CD, Rogers KT, Ticknor BW, Bostick DA, McBay EH, Hexel CR (2019) Evaluation and specifications for in-line uranium separations using inductively coupled plasma optical emission spectroscopy (ICP-OES) detection for trace elemental analysis. Appl Spectrosc 73(8):927–935
Wylie EM, Manard BT, Quarles CD, Meyers LA, Xu N (2018) An automated micro-separation system for the chromatographic removal of uranium matrix for trace element analysis by ICP-OES. Talanta 189:24–30
Khumalo N, Mathuthu M (2018) Determination of trace elements and lanthanide (REE) signatures in uranium mine products in South Africa by means of inductively coupled plasma mass spectrometry. J Geochem Explor 186:235–242
Boulyga SF, Cunningham JA, Macsik Z, Hiess J, Penkin MV, Walsh SJ (2017) Development, validation and verification of an ICP-MS procedure for a multi-element analysis of uranium ore concentrates. J Anal At Spectrom 32(11):2226–2237
Varga Z, Katona R, Stefanka Z, Wallenius M, Mayer K, Nicholl A (2010) Determination of rare-earth elements in uranium-bearing materials by inductively coupled plasma mass spectrometry. Talanta 80(5):1744–1749
Balcone-Boissard H, Michel A, Villemant B (2009) Simultaneous determination of fluorine, chlorine, bromine and iodine in six geochemical reference materials using pyrohydrolysis, ion chromatography and inductively coupled plasma-mass spectrometry. Geostand Geoanal Res 33(4):477–485
Jeyakumar S, Mishra VG, Das MK, Raut VV, Sawant RM, Ramakumar KL (2014) Study on the identification of organic and common anions in the pyrohydrolysis distillate of mixed uranium-plutonium carbide for the interference free determination of chlorine and fluorine by ion chromatography. Radiochim Acta 102(4):291–302
Shimizu K, Suzuki K, Saitoh M, Konno U, Kawagucci S, Ueno Y (2015) Simultaneous determinations of fluorine, chlorine, and sulfur in rock samples by ion chromatography combined with pyrohydrolysis. Geochem J 49(1):113–124
Chai JY, Muramatsu Y (2007) Determination of bromine and iodine in twenty-three geochemical reference materials by ICP-MS. Geostand Geoanal Res 31(2):143–150
Oliveira AA, Trevizan LC, Nobrega JA (2010) REVIEW: iodine determination by inductively coupled plasma spectrometry. Appl Spectrosc Rev 45(6):447–473
Thomas R (2013) Practical guide to ICP-MS a tutorial for beginners, 3rd edn. CRC Press Taylor & Francis Group, Boca Raton
Tanner SD, Baranov VI, Bandura DR (2002) Reaction cells and collision cells for ICP-MS: a tutorial review. Spectroc Acta Pt B-Atom Spectr 57(9):1361–1452
Balcaen L, Bolea-Fernandez E, Resano M, Vanhaecke F (2015) Inductively coupled plasma–Tandem mass spectrometry (ICP-MS/MS): a powerful and universal tool for the interference-free determination of (ultra) trace elements—a tutorial review. Anal Chim Acta 894:7–19
Haldimann M, Eastgate A, Zimmerli B (2000) Improved measurement of iodine in food samples using inductively coupled plasma isotope dilution mass spectrometry. Analyst 125(11):1977–1982
Todorov TI, Gray PJ (2016) Analysis of iodine in food samples by inductively coupled plasma-mass spectrometry. Food Addit Contam Part A-Chem 33(2):282–290
Schnetger B, Muramatsu Y, Yoshida S (1998) Iodine (and other halogens) in twenty six geological reference materials by ICP-MS and ion chromatography. Geostand Newsl 22(2):181–186
Fecher PA, Goldmann I, Nagengast A (1998) Determination of iodine in food samples by inductively coupled plasma mass spectrometry after alkaline extraction. J Anal At Spectrom 13(9):977–982
Shah M, Wuilloud RG, Kannamkumaratha SS, Caruso JA (2005) Iodine speciation studies in commercially available seaweed by coupling different chromatographic techniques with UV and ICP-MS detection. J Anal At Spectrom 20(3):176–182
Krajko J, Varga Z, Wallenius M, Mayer K, Konings R (2016) Investigation of sulphur isotope variation due to different processes applied during uranium ore concentrate production. J Radioanal Nucl Chem 309(3):1113–1121
Acknowledgements
This work was supported by the Department of Energy’s National Nuclear Security Administration under contract DE-AC05-00OR22725 with UT-Battelle, LLC. Oak Ridge National Laboratory is managed by UT-Battelle for the Department of Energy under Contract DE-AC05-000R22725. The US government retains and the publisher, by accepting the article for publication, acknowledges that the US government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this manuscript, or allow others to do so, for US government purposes. DOE will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (http://energy.gov/downloads/doe-public-access-plan).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare they have no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Fletcher, N.D., Manard, B.T., Metzger, S.C. et al. Determining P, S, Br, and I content in uranium by triple quadrupole inductively coupled plasma mass spectrometry. J Radioanal Nucl Chem 324, 395–402 (2020). https://doi.org/10.1007/s10967-020-07057-0
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-020-07057-0