
Abstract
An intercomparison was organized, with six laboratories tasked to quantify sixty-nine impurities in two uranium materials. The main technique employed for analysis was inductively coupled plasma mass spectrometry in combination with matrix-matched external calibration. The results presented highlight the current state-of-the-practice; lessons learned include previously unaccounted polyatomic interferences, issues related to sample dissolution, blank correction and calibration, and the challenge of estimating measurement uncertainties. The exercise yielded consensus values for the two analysed materials, suitable for use as laboratory standards to partially fill a gap in the availability of uranium reference materials characterized for impurities.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Analysis of trace elements (impurities) in uranium-bearing materials, from uranium ore to nuclear fuel, is performed in a variety of fields and for different purposes. The presence of certain elements affects the chemical, physical, and nuclear properties of materials involved in the nuclear fuel cycle; therefore industrial standards specify limits for a range of impurities [1, 2]. The information on trace elements is also utilized for attributing materials or for understanding variations through the uranium processing [3–8]. The International Atomic Energy Agency (IAEA) uses information on trace elements in uranium-bearing materials, such as uranium ore concentrate (UOC) and intermediate conversion products, to support verification of States’ declarations in terms of correctness and completeness. The information on impurity concentrations is used in several ways, in particular to judge material conformity to the nuclear-grade material specifications, to evaluate facility design information, to assess material origin and to detect possible substitution of material [9–12].
Impurities in uranium materials have been analysed over the past decades using an array of different analytical techniques and procedures [13] including atomic absorption spectrometry [14], atomic emission spectrometry [15–17], X-ray fluorescence spectrometry [3], ion chromatography [4], variations of inductively coupled plasma mass spectrometry (ICP-MS) including combination with laser ablation [18], quadrupole ICP-MS with or without collision/reaction cell [3, 19–21], time-of-flight ICP-MS [22], sector-field ICP-MS [6, 23, 24], and liquid chromatography ICP-MS [25]. The different analytical procedures exhibit specific merits and drawbacks hence they demonstrate diverse analytical performance and applicability.
To evaluate laboratory performance in the analysis of impurities in uranium materials, and to obtain a batch of well characterized material to serve as a laboratory standard for the analysis of impurities in uranium, in 1983–1984 the IAEA conducted two interlaboratory comparisons [26, 27], which involved the same U3O8 powder material.Footnote 1 The 19 participating laboratories applied different analysis techniques, predominantly variations of absorption and emission spectroscopy. As a result of the exercise, the U3O8 powder was certified as a reference material (RM), with concentrations of 19 impurity elements characterized [28].
Recently, CETAMA organized a similar performance testing, which involved 17 laboratories from the nuclear industry and the nuclear safeguards community [29]. The laboratories performed impurity measurements using predominately ICP-MS and, to some extent, ICP atomic emission spectrometry (ICP-AES). The comparison was carried out on one solution of uranyl nitrate, with only 12 elements requested for analysis, having concentration range of about three orders of magnitude. Generally, ICP-MS results showed better performance than ICP-AES. To the authors’ knowledge this was the only exercise on the subject performed within the past decade or more.
The interlaboratory comparison discussed herein was organized by the IAEA during 2011–2012. Six laboratories participated in the exercise: Canadian Nuclear Safety Commission Laboratory, Institute for Transuranium Elements (ITU) of the European Commission Joint Research Centre (EC JRC), the IAEA Safeguards Analytical Laboratory (SAL), and three laboratories of the US Department of Energy: Los Alamos National Laboratory, Lawrence Livermore National Laboratory, and Oak Ridge National Laboratory.
The laboratories were tasked to analyse trace elements in two powder materials of composition typical for UOC, which is a considerably more challenging matrix compared to a solution. The samples were to be chemically treated by the laboratories, including the sample dissolution as is employed in the analysis of real-world samples. Determination of 69 trace elements was requested, with the concentration range spanning seven orders of magnitude. This interlaboratory comparison also served the purpose of identifying the current state-of-the-practice as well as establishing consensus values for the two materials.
Experimental
Scope of the interlaboratory comparison
The goal of this exercise was to identify analytical capabilities of the participating laboratories and to assess the performance of the different analytical procedures in place to determine between-laboratory differences and their potential causes, to identify chemical elements that are problematic and/or difficult to analyse with established analytical techniques, to review the associated measurement uncertainties as reported by the laboratories, and, finally, to assign consensus values to the two materials for their future use for quality control (QC) in analysis of impurities in uranium materials.
The participating laboratories were requested to perform determination of concentrations (relative to uranium) for the following 69 chemical elements: Ag, Al, As, Au, B, Ba, Be, Bi, Br, Ca, Cd, Ce, Cl, Co, Cr, Cs, Cu, Dy, Er, Eu, Fe, Ga, Gd, Ge, Hf, Ho, In, Ir, K, La, Li, Lu, Mg, Mn, Mo, Na, Nb, Nd, Ni, P, Pb, Pd, Pr, Pt, Rb, Re, Rh, Ru, S, Sb, Sc, Se, Si, Sm, Sn, Sr, Ta, Tb, Te, Th, Ti, Tl, Tm, V, W, Y, Yb, Zn, Zr. The concentrations of these elements in the two materials selected for the exercise spanned more than seven orders of magnitude, ranging from below 1 ng/gU to higher than 1 mg/gU.Footnote 2
A total of five randomly selected aliquots of each material were sent to each participating laboratory, with a request to perform three replicate analyses per aliquot. Each replicate analysis included all steps of sample preparation and measurement, including sample dissolution and instrument calibration. Hence, a total of 3 × 5 = 15 data points per element and material were to be reported by each laboratory. In addition, for each result the laboratories were asked to report the associated measurement uncertainty (at 95 % level of confidence) and the critical level L c (detection decision at 99 % level of confidence, according to the widely accepted terminology introduced by Currie [30, 31]. Laboratories were requested to treat the materials in the same manner as they treat the majority of the samples routinely analyzed for impurities. A standardised template for the reporting of the measurement results was distributed to the participants, who were also requested to fill out a questionnaire aimed at gathering description of the analytical procedures and techniques employed.
The major limitation of this interlaboratory comparison was that the two materials used were not certified for the impurity concentrations; a few exceptions apply (see below). Hence, evaluating the accuracy of the reported data had its limitations; nonetheless between-lab differences were assessed. Furthermore, evaluating the reported measurement uncertainties as a statement of overall uncertainty against the ‘true value’ was not made; nonetheless an evaluation with respect to within-lab dispersion was performed to assess potential underestimation of the reported uncertainties.
The six laboratories participating in the exercise were assigned random codes, Lab 1 through Lab 6. Samples were distributed to the laboratories in autumn of 2011, with the deadline for reporting the final results to the IAEA set in February 2012. The pooled analysis results, their evaluation and findings were presented to the participants and discussed in May 2012.Footnote 3
Description of the two materials
Two isotopically natural uranium materials were selected for the interlaboratory comparison: an ammonium diuranate powder (ADU = (NH4)2U2O7, sample # 9064) and a triuranium octoxide (uranyl uranate) powder (U3O8, sample # 9449). The two materials were chosen based on several criteria, including availability of a sufficient amount of the material and its acceptable homogeneity. In addition, the U3O8 material was known to have a comparably low total amount of impurities allowing the limits of detection of the laboratories’ analytical procedures to be investigated, and the ADU material was known to have several impurities with comparatively high concentrations of 1 mg/gU or more.
Sample # 9064 was prepared at SAL from an ADU material obtained by the IAEA in 1994 from the Rössing uranium mine (Namibia). Uranium isotopic composition in this material was certified in 1995 by the EC JRC Institute for Reference Materials and Measurements (IRMM) as SMS 7267 [32]Footnote 4 and later measured at SAL using the method of modified total evaporation multi-collector thermal ionisation mass spectrometry (MTE MC-TIMS).Footnote 5 However this ADU was not certified for any impurities. A homogeneity study with respect to impurity distribution was performed at SAL in 2010 using ICP-MS, by analyzing eleven randomly selected aliquots of approximately 0.5 g each. For the current exercise, aliquots of 1.5–2.0 g were weighed into individual bottles and distributed to the participating laboratories. Polyethylene bottles were used deliberately, as it had earlier been demonstrated by dedicated experiments that storage and transportation of powder samples in glass bottles may result in a sample contamination with such common elements as Na and Si.
Sample # 9449 was prepared from the U3O8 material certified for impurities through the two IAEA interlaboratory comparisons conducted in 1980s [28]; however the original powder was later modified to increase the Fe concentration. A homogeneity study with respect to impurity distribution was performed at SAL in 2010 by analyzing eleven randomly selected aliquots of approximately 0.5 g each. Uranium isotopic composition in this material was also measured by SAL.Footnote 6 For the current exercise, aliquots of 1.5–2.0 g were weighed into individual polyethylene bottles and distributed to the participating laboratories.
Results and discussion
Summary of the questionnaires
The six participating laboratories were requested to return a completed questionnaire in order to provide details about their analytical procedure. A summary of the collected information is presented in Table 1.
Measurement techniques
The main technique employed by the laboratories was ICP-MS (mostly single-collector high-resolution sector-field instruments) in combination with matrix-matched external calibration. The majority of the participating laboratories developed their analytical procedures based on ASTM C1287-10 document [20], which represents a standard test method for the ICP-MS determination of 67 elements in nuclear grade uranium compounds without matrix separation. However, the analysts chose different options for sample dissolution and for the calibration of the ICP-MS instrument.
The calibration was performed using commercially available multi- and mono-element standards mixed with uranium matrix at concentration of about 10−4 g/gU of solution. Typically, 5–7 calibration points were used covering a range of 3–4 orders of magnitude of the analyte concentration; a linear fit was made. Lab 4 used a 2-point calibration based on the international norms [20, 33, 34], which require using a blank and one or two (matrix matched) standard solutions for the daily calibration.
Li, Sc, In, Bi, Rh, and/or 233U were added to samples as internal standards or, alternatively, ion intensities of impurities were normalized to isotope 235U after uranium isotopic composition was determined (see Table 1).
The measurement results were reported as relative concentrations of impurities normalized to the concentration of uranium in µg/gU. The uranium concentration was typically determined by Davies & Gray (D&G) titration and/or isotope dilution mass spectrometry (IDMS). In one case all intensities measured in samples and in matrix-matched standards with known uranium concentration were normalized to the 235U signal during ICP-MS measurement—an approach that does not require a determination of the uranium concentration in the samples.
Sample preparation
The general recommendations for dissolution of uranium materials can be found in ASTM C1347-08 [35] that is applicable to the test methods used for analysis of such materials for uranium elemental concentration, isotopic composition, and for impurities. According to the ASTM guidelines, the analyst may select appropriate dissolution procedures (e.g. heating with a steam bath or hot plate, or combination of acids and a high pressure microwave), reagents and labware to ensure safety and to avoid contamination of samples.
In this work the sample dissolutions were performed using nitric acid and heating, in some cases with a microwave oven, and/or supported by adding HCl or HF. The laboratories made an effort to perform measurements within one day after the sample dissolution, however holding times of as much as two to four weeks occurred in some cases.
Quality control
Uranium RM with certified or recommended values for impurity concentrations were utilized as QC materials, including NBL CRM 124, various CETAMA materials, and CANMET CUP-2. Results were monitored using QC charts.
Summary of the reported analytical results
The analytical results as reported by the laboratories were pooled, evaluated and plotted for visual inspection for each element and material separately.
For those impurity elements where an appropriate number of data points are available well above the critical level, it can be concluded that, overall, the between-laboratory relative differences are comparatively small. The between-laboratory relative standard deviation is usually within 30 % and for a large number of cases it does not exceed 15 %. However, several issues were identified within this interlaboratory comparison: potentially incomplete sample dissolution, unaccounted interferences, inaccurate blank correction, inaccurate calibrations, and significantly underestimated measurement uncertainties.
Effect of sample dissolution
It is known that the use of nitric acid alone might yield incomplete digestion of solid matrices [35]. Thus, significant differences in the reported results for Zr and Hf in material # 9449 were observed for those laboratories which used different acids for dissolution; see Fig. 1. One laboratory noted that a residue was observed after chemical dissolution of this material. The highest Zr and Hf concentrations were reported by those laboratories that employed HF, which likely indicates on incomplete sample dissolution or a precipitation if HF is not added. No correlation with other tetravalent elements including Ti, W, Mn, Sn, Ge, Re, Pb, or Mo is observed. On the other hand, the use of hydrofluoric acid may also generate insoluble fluorides of some elements, e.g. thorium fluoride [36], which complicates the analysis. As such there is a trade-off when using HF for sample dissolution. It highlights one of the challenges inherent to this type of analysis. The use of aqua regia resulted in better recoveries for some elements (e.g. tungsten); however the presence of HCl introduced additional interferences in the ICP-MS analysis.

Pooled results for elements Hf (upper plot) and Zr (lower plot) for material # 9449. Blue dots with uncertainty bars are the reported results; red lines are the respective L c values; blue lines show average (thick line) and average ±3 standard deviations (thin lines) values for each laboratory. Lab 1 and Lab 3 employed the help of hydrofluoric acid in the sample digestion. These two laboratories reported the highest Hf and Zr concentrations, and Hf results correlate with Zr results. The pattern observed for Zr of Lab 6 results hint on incomplete dissolution as well. (Color figure online)
Effect of interferences
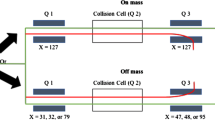
Some laboratories reported results that exhibited a positive bias for concentrations of elements Ag, La, or Sc. It was confirmed that these biases resulted from unaccounted interferences of ZrO+, UAr2+, and Zr2+ ions (Fig. 2). Other interferences relevant to the ICP-MS analytical procedures were accounted for by the laboratories to some degree or another; they are summarized in Table 2. These interferences are mostly double-charged species and/or oxides species, in addition to some recognized species produced by the uranium matrix (e.g., UAr2+). As a matrix-matched approach is used by the laboratories, interferences stemming from uranium species are to be expected. Since the formation of interferences is influenced by the elemental composition of the impurities in the uranium material, many more interferences may need to be accounted for.

Pooled results for elements La (upper plot) and Sc (lower plot) for material # 9064. Blue dots with uncertainty bars are the reported results; red lines are respective L c values; blue lines show average (thick line) and average ±3 standard deviations (thin lines) values for each laboratory; dotted green line shows average value for all laboratories. The results reported by a few laboratories were biased or associated with larger uncertainties due to inaccurate correction of interferences that affected these analytes (Lab 2, Lab 3 for La; Lab 3 for Sc). Lab 1 did not report Sc concentrations as this element was used for calibration as an internal standard in the analytical procedure. (Color figure online)
Blanks and cross-contamination
A disproportionally large number of positive outliers (deviating by as much as 4 standard deviations) are found in the results for concentrations of Ba, Ca, Mg, Na, K, Pb, and Zn. Concentrations of these elements in the analyzed sample solutions as well in the process blank solutions can be potentially subject to significant non-controlled changes during sample preparation and analysis because of a high risk of sample contamination with ubiquitous elements present in the laboratory environment. Positive biases in selected Rb and B data were also observed, potentially due to blank issues.
Calibration
Trends or biases in reported results are apparent for selected elements or laboratories. These were likely caused by calibration issues, e.g., significant day-to-day variations or negative bias due to issues related to the matrix-matching or to the determination of the uranium content.
Measurement uncertainty
The participating laboratories made an effort to identify and propagate uncertainty sources, in some cases applying the GUM methodology [37]. Nonetheless, in a large number of cases the reported uncertainties appear to be significantly underestimated with respect to the observed within-lab dispersion. Considering that the between-lab dispersion is usually larger than the within-lab dispersion, the reported uncertainties are potentially underestimated in even more cases. The laboratories tend to assess the measurement repeatability using internal precision which is prone to result in significant underestimations. Other significant sources of uncertainty, including calibration, interferences, and blank corrections were either not included or appear to be underestimated.
Challenging elements
Several laboratories did not report data for phosphorus, sulphur, and silicon, because these elements are difficult to quantify by ICP-MS. However, P, S, and Si results were reported by selected laboratories which used sector-field ICP-MS instruments. On Fig. 3, these sector-field ICP-MS results are compared to the X-ray fluorescence (XRF) results for sulphur submitted by one laboratory; these results are judged to be in a reasonable agreement.

Pooled results for elements S (material # 9064), Si (material # 9449), and P (material # 9449). Blue dots with uncertainty bars are the reported results; red lines are respective L c values; blue lines show average (thick line) and average ±3 standard deviations (thin lines) values for each laboratory. One laboratory employed XRF for analysis of sulphur; all other results for the three elements were produced using ICP-MS. (Color figure online)
None of the participants reported results for Cl and Br as these elements are a challenge for quantification by ICP-MS. Several laboratories did not report data for Au and In. Indium is frequently used as internal marker for the ICP-MS signal normalization, and in such cases it cannot be quantified.
Detection decision
The values for the detection decision threshold (critical level, L c ) reported by the participants for the lanthanides are below 1 × 10−9 g/gU, while such elements as Ca, Fe, K, Li, Mg, Na, Ni, P, S, and Si have comparatively high L c —on the order of 10−4 g/gU. As illustrated on Fig. 4, this mirrors low versus high instrumental and chemical blanks, as expected for the respective elements. Reported L c values appear to be overestimated in a few cases.

Minimum, median, and maximum of the reported L c values for the measured elements, combined for six laboratories and the two analysed materials
The effect of data ‘censoring’ by apparently overestimated L c values is demonstrated in Fig. 5, which presents relative concentrations of 14 rare earth elements (REE) normalized to the sum of lanthanides. Although the concentrations of six REE were reported by Lab 5 as ‘below L c ’ (Fig. 5a), the comparison of ‘uncensored’ data obtained from Lab 5 (Fig. 5b) with the results reported by Labs 1, 4 and 6 (Fig. 5c) demonstrates a very good agreement of relative REE concentrations determined by different laboratories. In practice, the use of the ‘censored’ data presented on Fig. 5a would reduce the ability of data evaluators to attribute samples by analyzing REE patterns. We conclude therefore, that a proper estimation of detection decision is essential to avoid a loss of useful information about investigated samples.

Relative concentrations of 14 rare earth elements: (a) reported by Lab 5; (b) Lab 5, including measurement results reported as ‘below L c ’; (c) reported by Labs 1, 4 and 6
Assigning consensus values to the materials
One of the goals of this interlaboratory comparison was to assign a consensus value with associated measurement uncertainty for the concentration of each impurity element of interest (in units of microgram per gram of uranium) in the two materials. The materials can then be used, for example, as laboratory standards for QC purposes. It is noted that they shall not be used as calibration standards, since the consensus values were not established by a rigorous certification campaign. Although the laboratories employed traceable standards for the analysis calibration, traceability of the consensus values to the SI is not claimed here, because of potentially unrecognized interferences or matrix-effects.
After initial submission of the analysis data, some laboratories later revised selected results, in order to address preliminary findings of the exercise. This included revisions to blank subtractions, interference corrections, and rejection of selected results demonstrated to suffer from calibration or other issues. These revised data were used in the calculation of the consensus values.
The methodology used to assign consensus values is based on ISO Guide [38] and was used earlier in the re-certification of NBL CRM 124 [39]. The methodology considers four cases:
-
1.
No consensus value is assigned if only n = 2 laboratories or less have reported data, which is indicated with ‘n/a’ for ‘not applicable’. In both materials, this applies to elements Br, Cl (n = 0) and Au, In (n = 2). It also applies to S in material # 9449 (n = 2; all data points are below the respective L c values). As elements P and Si (n = 2, both materials) are of particular interest in most applications of the impurity data, no consensus values are assigned, but the range is stated (minimum result to maximum result) for informational purpose only.
-
2.
No consensus value is assigned to Hf and Zr in material # 9449 (n > 4) due to potential issues with incomplete sample dissolution as discussed above. The range is stated (minimum result to maximum result) for informational purpose only.
-
3.
An upper limit is assigned as consensus value for an element concentration in a given material if at least n = 3 laboratories reported data with the majority of the data points below or close to the corresponding L c values. The upper limit is calculated as 95 % percentile (no uncertainty assigned). The 95 % percentile is calculated using all reported measurement values by all the laboratories. In case where a reported measurement value is below the corresponding L c , the measurement value is ignored, and the L c value is used instead. A selected number of data points were rejected for the calculation of the consensus values as these data points are likely affected by signal spikes (typically more than three times higher than the within-lab standard deviation) mostly due to issues with contamination (high blank result) and/or significantly larger uncertainty. The data points that were rejected amount to less than 0.5 % of the entire data set.
-
4.
A consensus value is assigned to an element concentration in a material if at least n = 3 laboratories have reported data and the majority are above the corresponding L c . The consensus value is calculated as the mean of laboratory means. Data points below L c are ignored, however the number of data point thus ignored was small. An expanded uncertainty U(95.5 %) using respective coverage factor k is assigned using GUM methodology [33] by incorporating and estimating all recognized sources of uncertainty. These include:
-
Between-laboratory variation: standard deviation of laboratory means;
-
Uranium concentration determination: IDMS, D&G, or normalization to 235U signal is used, hence conservative U(k = 2) = 1 % rel. is assigned;
-
Weighing and dilutions: usually gravimetrical preparation, conservative U(k = 2) = 1 % rel. is assigned;
-
Uncertainties of certified values of mono- or multi-element calibration standards employed to establish metrological traceability: typically U(k = 2) of the certified value is smaller than 0.5 %, hence a conservative U(k = 2) = 0.5 % rel. is assigned;
-
Regression of calibration curve, uranium matrix effect, internal maker normalization: these uncertainty components are difficult to estimate but an inappropriate matrix-matching was observed to produce as much as 20 % bias, hence U(k = 2) = 20 % rel. is assigned;
-
Detector non-linearity including dead-time correction: assumed to be not more than U(k = 2) = 2 % rel.;
-
Purity of uranium material used for matrix-matching: no uncertainty is estimated for this component, as different materials were used by the laboratories and hence it is difficult to assess; it is assumed that this component of uncertainty will be reflected to some extent in the between-laboratory variation;
-
Blank correction, including process blank and memory effects: results for several elements including Mg, Ba, Ca, Na, K, Pb, and Zn show a significant number of signal spikes indicating memory effects, cross-contamination, and/or increased blanks; U(k = 2) = 10 % rel. is assigned to these elements;
-
Interference correction including isobars, oxides, double-charged ions, hydrides, and peak-tailing: difficult to estimate as it depends on the ratio of analyte to interfering element and on the formation of interferences as a function of the plasma settings, mass resolution applied, etc. At a minimum, elements Sc, Ag, Sn, and La were identified, for which significant interference corrections had to be performed. An U(k = 2) = 10 % rel. is assigned to these elements;
-
Sample inhomogeneity: no significant degree of inhomogeneity at the 0.5 g minimum sample size was observed, based on triplicate measurements of each element on five independent dissolutions of each material performed by each laboratory; hence the uncertainty component is assumed to be negligible, and not included.
-
Other sources of uncertainty are assumed to be insignificant. A revision of the uncertainty calculation will be performed in the future if other significant sources of uncertainty are identified or if the estimation of those listed above need to be improved. A revision of the consensus values will be performed in the future if additional data become available.
The consensus values established using the approach described above are listed in Tables 3 and 4 for materials # 9064 and # 9449, respectively. Typical (relative) expanded uncertainties (at 95.5 %) for material # 9064 are in the range of 10–50 %. For material # 9449 the range is significantly larger, from 20 to 80 %, mirroring the fact that the concentrations of many elements in this sample are significantly lower compared to sample # 9064 with several impurities approaching the corresponding critical levels. For material # 9449, certification of the concentrations of eight elements (Al, Co, Cr, Cu, Fe, Mn, Mo, and Ni) was performed in 1985 [28] using data from two interlaboratory comparisons [26, 27]. These certified values are listed in Table 4 as well. The consensus values established in this paper agree with the certified values from 1985. The only exception is the Fe impurity; this was expected since the original certified material was later modified by increasing the concentration of iron (by an amount not known to the authors). Relative differences of the participants’ results to the consensus values often not exceed 15 % for those elements where the concentrations are well above the detection decision.
Available uranium RMs for determination of impurities
It is a good practice to utilize RM for the purpose of validation and monitoring of the performance of the analytical procedure from sample preparation to data reporting. A survey of RM suitable for this purpose in the quantification of impurities in uranium materials is presented in Table 5. Several of these RMs were indeed used by the participating laboratories; refer to Table 1.
In general, the available uranium-matrix RM do not cover the complete range of elements that are of interest in the analysis of uranium-bearing samples. In addition, the chemical composition of the available uranium matrix RM does not always correspond to the composition of real-world samples. Analysis of materials with high amounts of elements such as Mg, Na, Zr, and Mo (e.g. low-quality UOC) might require different procedures for sample treatment and analysis, and specific approaches for correction of interferences. Laboratories are encouraged to share their experience on the use of RMs for QC purposes with the aim to select the most suitable RMs for potential future re-certification.
REE patterns are known to be powerful provenance indicators and have been utilized in the earth science community for decades [40]; its applicability for attribution of uranium materials has been recognized [7, 41–43]. Therefore, it is desirable to certify REE concentrations in at least one RM with a matrix and element composition that is typical for UOC. At present, there appears to be no uranium matrix RM available with all lanthanides certified for elemental concentration. Potential candidates among those listed in Table 5 are CETAMA materials Beryl and Feldspath.
Conclusions
The current state-of-the-practice in elemental impurity analysis of uranium materials, as it applies to the majority of laboratories that participated in this interlaboratory comparison, is to use an ICP-MS in combination with matrix-matched external calibration. This technique permits to make a multi-element analysis of almost all element of interest within the same measurement. For digestion (dissolution) of uranium-containing samples, the participating laboratories used hot plate and microwave, nitric acid alone as well as mixtures of nitric acid with hydrochloric acid and nitric acid with hydrofluoric acid. Adding HF was observed to affect the sample dissolution significantly for Zr and Hf. In general, no universal procedure exists for the sample dissolution, which would make possible analysis of all investigated elements with the same level of quality. As such there is a trade-off when using different approaches for sample dissolution. It highlights one of the challenges inherent to this type of analysis. The quality of results as well uncertainty budgets also critically depend on the judicial choice of RM used for calibration, and on optimization of the ICP-MS conditions, such as sample introduction, interference removal and/or correction, and calibration. The analysis of procedural blanks was deemed useful for determining the detection limits. The quality of the sample digestion and overall analysis depend on the labware cleanliness and on the carryover from the previous sample in the ICP-MS.
Uranium RMs with certified or recommended values for impurity element concentrations were utilized by all laboratories for QC purposes, and results were monitored using QC charts. However, the available RMs did not cover the complete range of the analyzed elements. It is necessary for proper QC to subject a well characterized material (preferably standard RM) to the same sample preparation procedure as the unknown samples. There is a general need to have a RM for uranium ore concentrate with well characterized and certified concentrations of impurities, especially REE.
For those impurity elements, where an appropriate number of data points were available and the concentration was well above the detection threshold, the overall between-laboratory relative standard deviations were usually within 30 % and often not exceeding 15 %. Impurity concentrations were determined spanning seven orders or magnitude; a reliable quantification of the REE at levels of part per trillion was demonstrated. This was achieved in a multi-element analysis without separation or pre-concentration of the analytes. Within the community of analytical laboratories this is regarded as satisfactory performance. Of those 69 elements requested, elements Cl and Br are not readily analysed with the established ICP-MS techniques; elements P, S, and Si are challenging. Issues in the reported results were identified due to the sample dissolution, interference and blank corrections, and calibration. In selected cases, systematic trends are observed in replicate data points indicating potential day-to-day variations in calibration, instrument drifts, and memory effects.
Consensus values with associated measurement uncertainties for both materials were established based on the results of this interlaboratory comparison. Eight impurity elements in the U3O8 material (# 9449) were certified previously. The consensus values established herein for these eight elements are in agreement with the certified values. Relative differences of the participants’ results to the consensus values often did not exceed 15 % for those elements where the concentrations are well above the corresponding detection threshold.
Both materials are suitable for future use as laboratory standards; they fill gaps in the availability of uranium matrix RM characterized for impurities. It is noted that both materials shall not be used as calibration standards since the consensus values were not certified by a rigorous certification campaign. Although the participating laboratories employed traceable mono- or multi-element standards for calibration, traceability of the consensus values to the SI is not claimed because of potentially unrecognized interferences or matrix effects.
One of the advantages of using ICP-MS is the possibility to achieve comparatively low detection thresholds even when using a matrix-matched approach. This is reflected in the reported L c values which for the lanthanide elements are as low as sub-nanogram per gram uranium. With respect to the lanthanides, low L c values and reasonable accuracy of the element concentrations even at very low levels are indeed desirable as these results are featured in the REE patterns. However, for elements including Ca, Fe, K, Li, Mg, Na, Ni, P, S, and Si the reported L c values are as high as several hundred microgram per gram uranium (see Fig. 4). This observation is expected (as it mirrors high instrumental and chemical blanks for the respective elements) and highlights one of the analytical challenges. It is noted that the maximum reported detection threshold values for such impurities as B, Ca, Fe, Mg, Ni, P, and Si approach the corresponding upper limits specified for nuclear-grade material [1], therefore reliable quantification of these elements may be an issue when utilising the impurity data to assess sample conformity to the nuclear-grade material requirements.
In some cases, L c values appear to be too conservative (overestimated). In practice, overestimation of detection thresholds can lead to a loss of valuable information if measurement results are replaced with overestimated ‘below L c values’ in reporting. That can particularly affect the use of REE patterns. Such loss of information should be avoided by a proper and realistic estimation of the L c values. Therefore laboratories are encouraged to report analysis results even if the measured concentrations are below the estimated L c values. This information can be used for assessing the current capabilities of the analytical laboratories for impurity measurements in the low concentration range. It is furthermore desirable to develop unified approaches for the calculation and expression of L c .
With respect to measurement uncertainties, the laboratories made an effort to identify and propagate uncertainty sources, in some cases using GUM methodology. Nonetheless, in a large number of cases the reported uncertainties were significantly underestimated. The laboratories should try to identify those sources of uncertainty that were not (properly) accounted for to improve the estimation of associated measurement uncertainties in the future.
Notes
The preparation of the U3O8 material was arranged by the Laboratoire Central d’Analyse et de Controle in Grenoble. A large uranium metal billet of industrial origin was molten together with a small uranium ingot carrying the trace elements. It was then converted to U3O8 by ignition under a stream of oxygen. The resulting U3O8 itself was milled, ground, and sieved in order to obtain a 100 mesh grain size powder. The whole batch was then homogenized, and the homogeneity was verified by six laboratories. In 1982 the French Government donated a batch of the resulting material to the IAEA.
Concentrations of Br and Cl impurities, not measured by any of the participating laboratories, remain undetermined.
Technical Meeting on Analysis of Elemental Impurities in Uranium Samples. IAEA, Vienna, 30 May–1 June 2012.
The certified atom (molar) ratios with respective expanded uncertainties (k = 2) are: 234U/238U = 0.00005472(76), 235U/238U = 0.0072568(36). Certified atom ratio 236U/238U is < 2 × 10−8.
Recent measurements at SAL using MTE MC-TIMS yielded the following atom (molar) ratios, traceable to CRM IRMM-184, with respective expanded uncertainties (k = 2): 234U/238U = 0.00005471(21), 235U/238U = 0.0072544(29), and 236U/238U < 5 × 10–9.
Recent measurements at SAL using MTE MC-TIMS yielded atom (molar) ratios traceable to CRM IRMM-184, with respective expanded uncertainties (k = 2): 234U/238U = 0.00005449(23), 235U/238U = 0.0072561(30), and 236U/238U < 1 × 10–8.
References
ASTM C753-04 (2009) Standard specification for nuclear-grade sinterable uranium dioxide powder
ASTM C967-13 (2013) Standard specification for uranium ore concentrate
Keegan E, Richter S, Kelly I, Wong H, Gadd P, Kuehn H, Alonso-Munoz A (2008) The provenance of Australian uranium ore concentrates by elemental and isotopic analysis. Appl Geochem 23:765–777
Badaut V, Wallenius M, Mayer K (2009) Anion analysis in uranium ore concentrates by ion chromatography. J Radioanal Nucl Chem 280(1):57–61
Varga Z, Wallenius M, Mayer K, Meppen M (2011) Analysis of uranium ore concentrates for origin assessment. Proc Radiochim Acta 1:1–4
Keegan E, Wallenius M, Mayer K, Varga Z, Rasmussen G (2012) Attribution of uranium ore concentrates using elemental and anionic data. Appl Geochem 27:1600–1609
Peńkin MV, Zhao K, Fischer DM, Boulyga SF (2012) Use of rare-earth elemental impurity patterns for origin assessment of uranium materials. In: 9th international conference on methods and applications of radioanalytical chemistry (MARC IX), Kailua-Kona
Button P, Healey G, Chipley D (2013) Change in impurities observed during the refining and conversion processes. ESARDA Symposium, Bruges
Mayer K, Tushingham J, Boulyga S, Aregbe Y (2009) Report on the workshop on measurements of impurities in uranium. ESARDA Bull 43:57–64
Fischer DM (2010) The evolution of environmental sampling for safeguards. In: IAEA symposium on international safeguards, Vienna. IAEA-CN-184/138
Balsley S (2010) Destructive nuclear material analysis for safeguards: importance and future trends. In: IAEA symposium on international safeguards, Vienna. IAEA-CN-184/278
Peńkin M (2012) Safeguards needs for characterization of uranium compounds. In: IAEA technical meet on analysis of elemental impurities in uranium samples, Vienna
De Souza AL, Cotrim MEB, Pires MAF (2013) An overview of spectrometric techniques and sample preparation for the determination of impurities in uranium nuclear fuel grade. Microchem J 106:194–201
Goyal N, Purohit PJ, Dhobale AR, Patel BM, Page AG, Sastry MD (1987) Electrothermal atomisation atomic absorption spectrometric determination of silver, beryllium, calcium, iron, lead and tin in uranium without preliminary separation. J Anal At Spectrom 2:459–461
Santoliquido PM (1988) Determination of trace-elements in uranium oxide (U3O8) by inductively coupled plasma emission-spectrometry and graphite furnace atomic-absorption spectrometry. J Res Natl Bur Stand 93:452–454
Premadas A, Srivastava K (2002) Inductively coupled plasma atomic emission spectrometric determination of lanthanides and yttrium in various uranium hydrometallurgical products. J Radioanal Nucl Chem 251(2):233–239
Dwivedi VN, Mahanta PL, Premadas A (2003) An integrated approach to the complete chemical analysis of magnesium or sodium diuranate (yellow cake) sample. J Radioanal Nucl Chem 258(3):575–581
Kyser K, Chipley D, Bukata A, Polito P, Fitzpatrick A, Alexandre P (2003) Application of laser ablation and high resolution ICPMS to the analysis of metal contents of three rings, ages of uranium-rich minerals, and Se content in sulphide ores. Can J Anal Sci Spectrosc 48(5):258–268
Oliveira OP, Sarkis JES (2002) Determination of impurities in uranium oxide by ICPMS by the matrix matching method. J Radioanal Nucl Chem 254(3):519–526
ASTM C1287-10 (2010) Standard test method for determination of impurities in nuclear grade uranium compounds by inductively coupled plasma mass spectrometry
Quemet A, Brennetot R, Chevalier E, Prian E, Laridon AL, Mariet C, Fichet P, Laszak I, Goutelard F (2012) Analysis of twenty five impurities in uranium matrix by ICP-MS with iron measurement optimized by using reaction collision cell, cold plasma or medium resolution. Talanta 99:207–212
Bürger S, Riciputi LR, Bostick DA (2007) Determination of impurities in uranium matrices by time-of-flight ICP-MS using matrix-matched method. J Radioanal Nucl Chem 274(3):491–505
Švedkauskaitė-LeGore J, Rasmussen G, Abousahl S, Van Belle P (2008) Investigation of the sample characteristics needed for the determination of the origin of uranium-bearing materials. J Radioanal Nucl Chem 278(1):201–209
Varga Z, Katona R, Stefánka Z, Wallenius M, Mayer K, Nicholl A (2010) Determination of rare-earth elements in uranium-bearing materials by inductively coupled plasma mass spectrometry. Talanta 80:1744–1749
Wolf SF, Bowers DL, Cunnane JC (2005) Analysis of high burnup spent nuclear fuel by ICP-MS. J Radioanal Nucl Chem 263(3):581–586
Doubek N, Bagliano G, Deron S (1984) Report on intercomparison exercise SR-54. Determination of impurities in U3O8. IAEA, Vienna. IAEA/RL/110
Doubek N, Bagliano G, Deron S (1985) Report on intercomparison exercise SR-64. Determination of impurities in U3O8. IAEA, Vienna. IAEA/RL/116
IAEA (1985) Reference sheet SR-54: impurities in uranium oxide (U3O8)
Granier G, Balsley SD, Bulyha S, Aregbe Y, Roudila D (2012) Round robin “impurities in uranium matrix”: a success for CETAMA and IAEA. Procedia Chem 7:666–672
Currie LA (1968) Limits for qualitative detection and quantitative determination: application to radiochemistry. Anal Chem 40:586–593
Currie LA (1995) Nomenclature in evaluation of analytical methods including detection and quantification capabilities (IUPAC recommendations). Pure Appl Chem 87(10):1699–1723
IRMM (1997) Certificate of isotopic composition. Reference number SMS 7267, sample identification # 9064-01-B
ISO 17294-2 (2003) Water quality: application of inductively coupled plasma mass spectrometry. Part 2: determination of 62 elements
DIN 38406-29 (1999) Determination of 61 elements by inductively coupled plasma mass spectrometry (E29)
ASTM C1347-08 (2008) Standard practice for preparation and dissolution of uranium materials for analysis
Link DD, Walter PJ, Kingston HM (1998) Development and validation of the new EPA microwave-assisted leach method 3051A. Environ Sci Technol 32:3623–3628
JCGM 100:2008 (2008) Evaluation of measurement data: guide to the expression of uncertainty in measurement
ISO Guide 35 (1989) Certification of reference materials: general and statistical principles
Bürger S, Mathew KJ, Mason P, Narayanan U (2009) Reference materials characterized for impurities in uranium matrices: an overview and re-evaluation of the NBL CRM 124 series. J Radioanal Nucl Chem 279(2):659–673
Zawisza B, Pytlakowska K, Feist B, Polowniak M, Kita A, Sitko R (2011) Determination of rare earth elements by spectroscopic techniques: a review. J Anal At Spectrom 26:2373–2390
Varga Z, Wallenius M, Mayer K (2010) Origin assessment of uranium ore concentrates based on their REE impurity pattern. Radiochim Acta 98:771–778
Riciputi L (2012) Utilization of REE and trace elements in UOC. In: IAEA technical meeting on analysis of elemental impurities in uranium samples, Vienna
Peńkin M (2012) Use of REE patterns in safeguards data evaluations. In: IAEA technical meeting on analysis of elemental impurities in uranium samples, Vienna
CETAMA (2011) Reference materials catalogue
NBL (1991) Certificate of analysis. CRM 123(1–7): uranium (normal) oxide
NBL (1983) Provisional certificate of analysis. CRM 124(1–7): uranium (normal) oxide
CANMET (1988) Certificate of analysis. CUP-2: uranium ore concentrate
Acknowledgments
Authors are grateful to Mikhail Ryzhinskiy for the in-depth discussions of the quality of analysis results on impurities in uranium samples, which led to the initiation of this interlaboratory comparison, and for his advice regarding the selection of test materials. Also acknowledged is the contribution of Zsolt Varga (ITU) who thoroughly reviewed the manuscript and provided valuable comments.
Author information
Authors and Affiliations
Corresponding author
Additional information
S. Bürger: deceased.
Rights and permissions
About this article

Cite this article
Bürger, S., Boulyga, S.F., Peńkin, M.V. et al. Quantifying multiple trace elements in uranium ore concentrates: an interlaboratory comparison. J Radioanal Nucl Chem 301, 711–729 (2014). https://doi.org/10.1007/s10967-014-3224-9
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-014-3224-9