
Abstract
Protactinium-233 (233Pa) was generated by neutron activation of thorium and isolated by column chromatography using an octanol-impregnated resin. Absolute activity standardization was performed on 233Pa using three independent methods, the results of which agreed within their associated uncertainties. The standardized 233Pa was used to calibrate a secondary standard ionization chamber and high purity germanium detectors to enable a rapid and traceable method for the production and quantification of this radiotracer.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Uranium radiochronometry is of great interest to the nuclear forensics community [1,2,3]. The 235U/231Pa isotope pair can be used to calculate the separation age of samples containing enriched 235U; a comparison with another radiochronometric pair, 234U/230Th, can provide additional information about the processing history of the material. Accurate dating using the 235U/231Pa nuclear chronometer typically requires access to the short-lived radiotracer 233Pa [t½ = 26.98(2) d] [4], which is used to quantify the long-lived 231Pa (t½ = 32.8 ky) by methods such as isotope dilution mass spectrometry [5, 6]. The short lived 233Pa is ideal for decay counting primary standardization using coincidence counting and liquid scintillation counting (LSC) methods.
At the present time, access to 233Pa is limited to the handful of laboratories around the world that possess significant quantities of 237Np (t½ = 2.14 My). As a decay product of 237Np, 233Pa forms continuously in situ, and can be isolated by complex chemical processing of these neptunium sources [5]. However, due to the long half-life of 237Np, significant quantities of 233Pa can only be generated after an interval of months from the previous source reprocessing, limiting its access upon immediate requisition. If the 235U/231Pa dating method is to be applied as a forensic tool in emergency response scenarios, access to 233Pa must be improved significantly.
The deliberate generation of 233Pa by bombarding natural thorium oxide with thermal neutrons was first reported more than 60 years ago [7]; but to our knowledge, this production route is not in use today. Although the reported post-irradiation processing is not appropriate for routine production due to the complex, multi-step chemical treatments and use of hazardous reagents including fluorine and hydrofluoric acid, the production physics of this process is near-ideal. Natural thorium is monoisotopic (100% 232Th), and thus the sole product of its thermal neutron activation is the short-lived radioisotope 233Th (t½ = 22 min), which undergoes beta decay to the desired 233Pa. The moderate cross-section for the 232Th(n, γ)233Th nuclear transformation (σ = 7.4 b) indicates that arbitrary (MBq) quantities of 233Pa can be generated using this production route even in low- and medium-flux nuclear research reactors. A re-investigation of the separation of 233Pa from neutron irradiated thorium to identify a methodology that is simple, rapid, and reproducible is therefore in order. The ideal approach would also avoid the use of reagents that require special handling, such as HF-assisted separations described elsewhere [8,9,10].
As one of the world’s leading producers and processors of uranium, Canada is interested in expanding its nuclear forensics capabilities for characterizing samples that are found outside of regulatory control. Towards this goal, the objectives of this work were two-fold: first, to investigate the feasibility of “on-demand” production of 233Pa from thorium at the McMaster Nuclear Reactor, the nation’s most powerful research reactor; and second, to conduct standardization measurements at the National Research Council of Canada. The resulting certified reference materials could be used to calibrate secondary radioisotope quantification instrumentation.
Experimental
Neutron irradiation of thorium
Note: As the decay product of 233Pa is the fissile species 233U, neutron irradiation times should be limited to no more than several hours in well-thermalized sites (in the absence of fast neutrons; where E > 1.2 MeV) to ensure that the 233U content of the sample is at a minimum while it is in the reactor core.
In a typical experiment, thorium (IV) nitrate pentahydrate (152.6 mg, 99.9%, Strem Chemicals) was heated at 120 °C for 3 h to generate thorium (IV) nitrate dihydrate as discrete white pellets (138.9 mg). The dihydrate was weighed into a polyethylene vial and heat-sealed; the vial was then placed within a larger polyethylene vial that was subsequently heat-sealed. The double-encapsulated target was irradiated for 6 h in site 9C (ϕth = 9.0–10 × 1012 n/cm2/s) of the McMaster Nuclear Reactor (MNR) at a nominal operating power of 3 MW. The activated thorium was stored underwater for a minimum of 4 h after end of irradiation to allow decay of 233Th and grow-in of 233Pa. The 233Pa activity (~ 86.2 MBq) was estimated using an AtomLab 400 Dose Calibrator.
Isolation of 233Pa
The 233Pa was separated from the thorium nitrate target material using methods loosely adapted from recent work by Jerome et al. with protactinium-231 [11]. Octanol-impregnated solid support (TK400 resin, 100–150 μm) was used as received from TrisKem. Acids utilized in the conditioning and elution tests such as concentrated hydrochloric acid (12 M, > 99.999% trace metals) and sulfuric acid (18 M, 95–98%) were obtained from Sigma-Aldrich and diluted with ultra-pure water (> 18 MΩ cm) as required. While several elution chemistries were tested, two main methods were selected for the final production.
Method A: elution in sulfuric acid
TK400 resin (2.00 g) was slurried in water and poured into a glass chromatography column (o.d. = 10 mm). An irradiated target (5–10 MBq, 40–60 mg) was dissolved in 6 M HCl (500 uL). The resulting clear pale yellow solution was loaded onto the resin column, and was washed with 6 M HCl (25 mL) to remove thorium and uranium. The 233Pa was eluted in 2.5 M H2SO4 (3 × 6 mL) with an efficiency of greater than 95%.
Method B: elution in hydrochloric acid
TK400 resin (2.00 g) was poured into a glass chromatography column (o.d. = 10 mm) and conditioned with 10 M HCl (10 mL). An irradiated target (30–85 MBq, 40–60 mg)) was dissolved in 10 M HCl (1 mL) and loaded onto the resin column. The resin was washed with 10 M HCl (20 mL) to remove the thorium and uranium; greater than 95% of the 233Pa was subsequently eluted in 1.0 M HCl (10 mL), collecting in 1 mL fractions to maximize the activity concentration. Typically, 70–95% of the 233Pa was recovered in only three fractions (3 × 1 mL).
The identity and radionuclidic purity of the resulting 233Pa solutions were verified using an Ortec GMX N-type High Purity Germanium (HPGe) coaxial radiation detector (30% efficiency, 70 mm endcap). Gamma spectra were acquired using GammaVision for Windows Version 5.31 and analyzed using the Aptec MCA Application Version 7.04 (Canberra Co., 2002) software package. The efficiency of the detector was determined over the energy range 0.10–1.4 MeV using a mixed 152/154/155Eu multi-gamma standard disc source (Canberra). Small quantities (< 5 MBq) of 233Pa were initially quantified on the HPGe detector based on its efficiency curve. For larger activities and routine quantification, an aliquot of 233Pa was quantified using the HPGe detector efficiency curve, then measured in an AtomLab 400 ion chamber Dose Calibrator on the 60Co setting to determine an approximate, uncalibrated, dial value (36.6). At the conclusion of this work, a certified reference calibration source was measured in the AtomLab 400 at MNR, yielding a calibrated dial value of 33.7 ± 0.3 at the 95% confidence level,Footnote 1 which will facilitate the rapid and accurate determination of future 233Pa production batches. The uncertainty on this value only reflects the uncertainty of the calibrant.
Standardization of 233Pa
The most concentrated fraction from Method B (approximately 25 MBq in 1 mL) was sent to the National Research Council (NRC) of Canada in Ottawa, Ontario for standardization. Upon receipt, the bulk 233Pa was diluted to 30 mL and pycnometers were used to dispense the solution to produce aliquots contained in silanized glass ampoules. Thin film (VYNSFootnote 2) sources and a set of ten quenched LSC vials were produced using a pycnometer. The thin film sources were made by depositing 10–50 mg drops of the solution onto a VYNS film with the addition of a single drop of a surfactant (Catanac SN) and left to dry in a 40 °C air stream. The quenched LSC set was prepared with 15 mL of cocktail (Ultima Gold). Roughly 10–50 mg drops of the 233Pa solution were deposited in each vial. Increasing amounts of nitromethane (0–100 μL) were added to act as the quenching agent. No additional reagents were added.
4πβ-γ coincidence counting
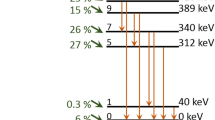
NRC implements a 4πβ-γ coincidence and anti-coincidence [12] counting method for the standardization of radionuclides consisting of a β-emission followed immediately by a γ. The NRC 4πβ-γ primary detection system consists of a pressurized proportional gas chamber (PPC), in which the VYNS sources are inserted. The PPC is filled with P10 counting gas to a pressure of 10 atm and registers the β-emissions. The PPC is sandwiched between two NaI crystals coupled to PMTs, that act together to form a γ-channel. The analog signals are counted with scalars and analyzed for coincidence/anti-coincidence as described in [12]. An electronic threshold was utilized to vary the β efficiency. The PPC β inefficiency was traced by centering the γ gate on the 311.904(5) keV transition of the 233Pa decay scheme.
Liquid scintillation counting
Primary standard LSC techniques require a model of the physical and chemical processes involved in light emission and of the statistics of photon emission and detection [13]. The CIEMAT/NIST [14] efficiency tracing method was used to determine the absolute activity of the 233Pa standard. This method requires the input of an experimental counting efficiency of a tracer nuclide which in this analysis was determined using a NIST traceable 3H standard. The 233Pa efficiencies were calculated for the CIEMAT/NIST method using the MICELLE2 code [15]. Two commercial counters, a Wallac 1410 and a Hidex-300SL-METRO, were utilized for the LSC measurements.
Results and discussion
Increasing regulation of thorium—a substance that is subject to international safeguards restrictions—has resulted in decreased availability of thorium-containing compounds from standard chemical suppliers. Consequently, thorium (IV) nitrate hydrate Th(NO3)4·5H2O was procured for this project despite its low thermal stability and resultant unsuitability as an irradiation target. Fortunately, the thermal behavior of this compound has been well characterized: loss of water of hydration occurs in two discrete steps, and is followed upon further heating by step-wise denitration, ultimately yielding thorium (IV) oxide [16, 17]. Unfortunately, as the thermal stability of this thorium series increases, the complexes’ solubility in aqueous media decreases. The desire for a highly water soluble target material to facilitate post-irradiation processing must therefore be balanced against the need for a thermally stable thorium complex that meets regulatory requirements for neutron irradiation materials.
Two irradiation targets were investigated in detail: thorium (IV) dinitro oxide Th(O)(NO3)2, and thorium (IV) nitrate dihydrate Th(NO3)4·2H2O, both of which were prepared by heating thorium (IV) nitrate pentahydrate as described in [16, 17]. As gamma heating and neutron recoil can cause localized temperature increases within a target, all neutron irradiations were conducted using a large-volume sample irradiation tube with sufficient headspace to avoid pressurization in the event of target outgassing.
Initial irradiation experiments employed short activation times (< 2 h) and focused on the more thermally stable mixed oxide/nitrate complex. While separation of 233Pa was achieved for this novel irradiation target using both solvent extraction [18] and ion exchange chromatography [19] strategies, process optimization focused exclusively on the TK400 resin approach [11] due to its simplicity and high recovery yields. Both hydrochloric and sulfuric acid were found to be effective in eluting 233Pa from this resin; however, the radioisotope is recovered in smaller volumes using dilute hydrochloric acid. The resulting 233Pa solutions were subjected to neutron activation analysis to quantify any residual thorium. The thorium content of the post-processing 233Pa was below the limit of detectionFootnote 3 which is consistent with the observation in reference 11 that thorium washes through TK400 during sample loading, and protactinium is selectively retained until dilute acid is introduced. Furthermore, no other gamma emitting radionuclidic impurities were detected (e.g. 233U, fission products) as well-thermalized irradiation sites were used for the thorium activation. Having successfully completed several neutron activations with no indication of heating within the oxide/nitrate irradiation target, a brief trial irradiation and processing of the more thermally sensitive thorium (IV) nitrate dihydrate was undertaken. No pressurization of the target vial or thermal degradation of the target material was detected. No difference in quality of the post-processing 233Pa was observed compared to the mixed oxide/nitrate target. However, the fraction of the pre-processing activity recovered was 10–30% higher, at approximately 95% of the total activity. The higher yields associated with the hydrated target material are likely due to its greater solubility in aqueous media. Complete dissolution of the mixed oxide/nitrate irradiation target prior to column loading was difficult to achieve. An additional advantage of thorium (IV) nitrate dihydrate is its rapid (3 h) preparation time from a commercially available complex. As a result of these experiments, thorium (IV) nitrate dihydrate was identified as the preferred target material for the production of 233Pa.
Having identified a preferred target material, irradiation site, and post-irradiation separation process, the scale of the 233Pa production was gradually increased to approximately 85 MBq by increasing the irradiation time to 6 h. No pressurization of the target vial was observed, indicating that this process can be scaled up further, if necessary, by increasing the mass of target material.
A batch of the 233Pa prepared by the optimized procedure described here (Method B) was used for the standardization measurements using three complementary approaches. The anti-coincidence result defined the absolute activity determination with the other primary methods acting as validation measurements. The thin film sources had a very high measured efficiency between 60 and 90% indicating the high quality of the produced counting artifact. The uncertainty budgets for the coincidence and anti-coincidence primary methods are given in Table 1. The uncertainty is dominated by the uncertainty of the extrapolation. The massic activity for the 233Pa was 0.784 ± 0.008 MBq/g (k = 2) on November 21, 2017 at 12:00 EST.
The calculated efficiency range for the LSC quenched set was very high, above 95%. An ionization quenching parameter kB value of 0.0075 cm MeV−1 was used in the modelling. Variation of the kB value between 0.005 and 0.012 cm MeV−1 had a small effect (0.24%) on the calculated activities but nevertheless knowledge of kB was one of the main contributers to the total uncertainty of the massic activity determined by this method (see Table 1 for a complete uncertainty budget). Furthermore, LSC counting indicated no other alpha or beta-emitting radionuclidic impurities were present in the sample post-processing.
The agreement between the three different primary methods was excellent and is summarized graphically in Fig. 1. Further validation can also be derived from an international comparison between NRC (publication in preparation), NIST [20] and NPL on the massic activity of 233Pa material prepared by LLNL; which was derived from a legacy 237Np source. In this case the solution solvent was 2 M HNO3 and 0.1 M HF. Nevertheless, the agreement between these groups was reported to be excellent providing further confidence in the results contained in this study.Footnote 4

Activity concentration for the 233Pa tracer as a function of primary method used. All uncertainty bars are expanded uncertainties at 95% confidence level (k = 2)
Single isotopic radioactive certified reference material is typically disseminated through the shipment of a liquid artifact contained in glass ampoules. Due to the adhesive nature of protactinium on glass, the ampoules were silanized before filling. Multiple ampoules were prepared at the time of standardization and then subsequently opened at later dates and re-measured. When opened, gravimetric dilutions were performed prior to measurement. The dilution factors were also determined radiometrically through measurement in calibrated ion chambers [21]. The massic activity could be recovered if the radiometric dilution factors were used but significant losses were being observed otherwise. This was an indication of the adsorption of the 233Pa onto the glass despite its pretreatment prior to filling. The addition of HF lessened the loss but it was still significant. (See Fig. 2).

Radioactive losses of 233Pa due to storage in 1 M HCl in salinized glass as a function of time of containment. The * corresponds to a solution of 1 M HCl and 0.1 M HF. All uncertainty bars are expanded uncertainties at 95% confidence level (k = 2)
As a result of the evidence of sticking of the 233Pa to the silanized glass, an unopened the ampoule retains its traceability and function as a calibrant but is not a suitable tracer for the calibration of an atom counting 231Pa standard or for any other radiochemistry analysis. Monte Carlo calculations indicate that in the highly unlikely event that 233Pa leaches out of the solution entirely and deposits on the inner layer of the glass ampoule, the calibration factor would change by 0.7%. This is within the 95% confidence limit that is set for the standardized activity and the major contributor to the ionization chamber calibration factor. However, all experimental evidence to date indicates that less than 1% of the 233Pa plates out. Thus, these MC calculations represent a worst-case scenario that is unlikely to occur.
Measurements over the period of several months indicated that the total activity could be recovered, accounting for isotope decay. Therefore for this exercise we are confident that the calibrant returned to MNR for calibration of their ionization chamber could be used and act suitably as a traceable calibrant. Nevertheless, work will continue to stabilize the isotope in solution resulting in a proper and indefinite standard. Several approaches to stabilizing the 233Pa in solution to eliminate deposition are currently being evaluated, and will be presented in a future publication.
Conclusions
Neutron irradiation of natural thorium represents a convenient and facile route to MBq quantities of the important forensic radiotracer 233Pa. Standardization of this material enabled the production of certified 233Pa sources for instrument calibration, so that this radioisotope can be quantified rapidly and traceably in the future. Canada’s nuclear forensics capabilities have been enhanced significantly by this work, as our laboratories can now provide certified 233Pa calibrants within days of a request. Further experimental work is ongoing to fully eliminate the deposition behavior of 233Pa on glass surfaces, and will ultimately enable the production and distribution of certified 233Pa radiotracer solutions.
Notes
The uncertainty associated with this instrument in this energy range is approximately 10%, which is likely greater than any minor geometry-induced errors accounting for < 1% deposition of 233Pa on the walls of the glass ampoule.
Poly-vinyl chloride poly-vinyl acetate.
A precise value for the limit of detection is strongly dependent on the sample matrix (approximately 1–10 μg). For example, the presence of hydrochloric acid significantly increases the limit of detection due to the formation of 38Cl, whereas it is much lower in nitric or sulfuric matrices.
Richard Essex, NIST (2018) Private Communication.
References
Stanley EE (2012) A beginner’s guide to uranium chronometry in nuclear forensics and safeguards. J Anal At Spectrom 27:1821–1830
Mayer K, Wallenius M, Varga Z (2013) Nuclear forensic science: correlating measurable material parameters to the history of nuclear material. Chem Rev 113:884–900
Keegan E, Kristo MJ, Toole K, Kips R, Young E (2016) Nuclear forensics: scientific analysis supporting law enforcement and nuclear security investigations. Anal Chem 88:1496–1505
Bé MM, Chisté V, Dulieu C, Mougeot X, Browne E, Chechev V, Kuzmenko N, Kondev F, Luca A, Galan M, Nichols AL, Arinc A, Huang X (2010) Table of radionuclides vol. 5. Monographie BIPM-5. Bureau International des Poids et Mesures, Sèvres
Eppich GR, Williams RW, Gaffney AM, Schorzman KC (2013) 235U–231Pa age dating of uranium materials for nuclear forensic investigations. J Anal At Spectrom 28:666–674
Rolison JM, Treinen KC, McHugh KC, Gaffney AM (2017) Application of the 226Ra–230Th–234U and 227Ac–231Pa–235U radiochronometers to uranium certified reference materials. J Radioanal Nucl Chem 314:2459–2467
Fudge AJ, Woodhead JL (1956) The isolation and determination of protactinium-233. Analyst 81:417–426
Knight AW, Nelson AW, Eitrheim ES, Forbes TZ, Schultz MK (2016) A chromatographic separation of neptunium and protactinium using 1-octanol impregnated onto a solid phase support. J Radioanal Nucl Chem 307:59–67
van der Meulen NP, Steyn GF, van der Walt TN, Shishkin SV, Vermeulen C, Tretyakova SP, Guglielmetti A, Bonetti R, Ogloblin AA, McGee D (2006) The separation of Pa from a Th target by means of ion exchange chromatography. Czechoslov J Phys 56:D357–D362
Radchenko V, Engle JW, Wilson JJ, Maassen JR, Nortier MF, Birnbaum ER, John KD, Fassbender ME (2016) Formation cross-sections and chromatographic separation of protactinium isotopes formed in proton-irradiated thorium metal. Radiochim Acta 104:291–304
Jerome SM, Collins SM, Happel S, Ivanov P, Russell BC (2018) Isolation and purification of protactinium-231. Appl Radiat Isot 134:18–22
Baerg AP, Munzenmayer K, Bowes GC (1976) Live-timed anti-coincidence counting with extending dead-time circuitry. Metrologia 12:77–80
Broda R, Cassette P, Kossert K (2005) Radionuclide metrology using liquid scintillation counting. Metrologia 44:36–52
Grau Malonda A, Garcia-Toraño E (1982) Evaluation of counting efficiency in liquid scintillation counting of pure β -ray emitters. Int J Appl Radiat Isot 33:249–253
Kossert K, Grau Carles A (2010) Improved method for the calculation of the counting efficiency of electron-capture nuclides in liquid scintillation samples. Int J Appl Radiat Isot 68:1482–1488
Hussein GAM, Ismail HM (1995) Texture assessment of thoria as a final decomposition product of hydrated thorium nitrate and oxycarbonate. Colloids Surf A Physicochem Eng Asp 99:129–139
Dash S, Kamruddin M, Ajikumar PK, Tyagi AK, Raj B, Bera S, Narasimhan SV (2000) Temperature programmed decomposition of thorium nitrate pentahydrate. J Nucl Mat 278:173–185
Kumari N, Pathak PN, Prabhu DR, Manchanda VK (2012) Solvent extraction studies of protactinium for its recovery from short-cooled spent fuel and high-level waste solutions in thorium fuel cycle using diisobutyl carbinol (DIBC) as extractant. Desalin Water Treat 38:46–51
Kuroda R, Ishida K (1965) Cation-exchange separation of protactinium-233 from irradiated thorium. J Chromatogr 18:438–440
Fitzgerald R, Pibida L (2018) Primary standardization of the massic activity of a protactinium-233 solution. J Radioanal Nucl Chem. (SI: MARC XI. LOG 585)
Galea R, Gameil K (2016) Renewing the radiopharmaceutical accuracy check service for Canadian radionuclide calibrators. App Radiat Isot 109:254–256
Acknowledgements
Funding for this research was provided by the Canadian Safety and Security Program, Project No. CSSP-2016-TI-2223.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Naperstkow, Z., Moore, K., Szames, D. et al. Production and standardization of an on-demand protactinium-233 tracer. J Radioanal Nucl Chem 318, 703–709 (2018). https://doi.org/10.1007/s10967-018-6068-x
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-018-6068-x