
Abstract
A new rapid method for the determination of 210Po in water samples has been developed at the Savannah River National Laboratory (SRNL) that can be used for emergency response or routine water analyses. If a radiological dispersive device event or a radiological attack associated with drinking water supplies occurs, there will be an urgent need for rapid analyses of water samples, including drinking water, ground water and other water effluents. Current analytical methods for the assay of 210Po in water samples have typically involved spontaneous auto-deposition of 210Po onto silver or other metal disks followed by counting by alpha spectrometry. The auto-deposition times range from 90 min to 24 h or more, at times with yields that may be less than desirable. If sample interferences are present, decreased yields and degraded alpha spectrums can occur due to unpredictable thickening in the deposited layer. Separation methods have focused on the use of Sr Resin™, often in combination with 210Pb analysis. A new rapid method for 210Po in water samples has been developed at the SRNL that utilizes a rapid calcium phosphate co-precipitation method, separation using DGA Resin® (N,N,N′,N′ tetraoctyldiglycolamide extractant-coated resin, Eichrom Technologies or Triskem-International), followed by rapid microprecipitation of 210Po using bismuth phosphate for counting by alpha spectrometry. This new method can be performed quickly with excellent removal of interferences, high chemical yields and very good alpha peak resolution, eliminating any potential problems with the alpha source preparation for emergency or routine samples. A rapid sequential separation method to separate 210Po and actinide isotopes was also developed. This new approach, rapid separation with DGA resin plus microprecipitation for alpha source preparation, is a significant advance in radiochemistry for the rapid determination of 210Po.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
210Po is a naturally-occurring alpha emitting radionuclide with a half-life of 138.38 days. It is used in industrial applications, specifically for static elimination and in neutron activation. 210Po is present in the environment as a result of decay within the 238U decay chain, and is considered to be one of the most toxic naturally-occurring radionuclides [1]. According to Scott [2], “an ingestion intake as small as 1 μg of 210Po may be lethal for the most radiosensitive members of the population; ingesting (or inhaling) a few tenths of a milligram would be expected to be lethal for all. Lethal intakes of 210Po would be expected to involve severe damage to the bone marrow, spleen, liver, kidney, skin, lymph nodes and possibly other sites in the body. The lung would also be an additional site of concern in the event of inhalation intake. Children would be expected to be at higher risk of harm than for adults for the same level of intake of 210Po because of their smaller body masses and relatively higher radiation doses”. It is therefore one of the most important environmental radionuclides due to its widespread distribution and potential for human radiation exposure through ingestion or inhalation.
Former Russian intelligence officer Alexander Litvinenko died in a London hospital on November 23, 2006. It was determined he was deliberately poisoned with 210Po. Police discovered that those involved in this crime had spread 210Po over many locations in London [3]. The incident caused widespread psychological concern and a heightened political and public health response.
Naturally-occurring radionuclides such as 226Ra and 210Po can also be used in a radiological dispersive device (RDD) as part of a terrorist attack. Analysis of international incident data shows that several acts of murder and terrorism with natural radionuclides have already been carried out in Europe and Russia [4–6]. The importance of detecting and quantifying the radionuclides present in environmental samples after a terrorist attack is well-known. Rapid analytical methods for 226Ra, 228Ra, actinide isotopes and 89,90Sr in various sample matrices have recently been reported by this laboratory [7–10].
Matthews et al. [11] have provided a good review of analytical techniques available for the assay of 210Po in a variety of environmental matrices. Most methods are based on spontaneous auto-deposition of polonium on silver disks. The auto-deposition times range from 90 min to 24 h, at times with yields that may be less than desirable. Many of the water methods involve evaporation of the sample, where much care must be exercised due the volatility of polonium ions. In addition, large water samples may require time-consuming destruction of silica accumulated during the evaporation step. Co-precipitation of 210Po has been achieved with MnO2 and Fe(OH)3 in combination with auto-deposition and separation methods. Direct preparation via auto-deposition separation steps has an advantage of simplicity, but may take a significant amount of time to complete, up to 24 h, depending on deposition time [12]. Without optimized heating during auto-deposition, polonium yields <60 % may occur. In addition, spontaneous auto-deposition, may suffer from interferences for certain samples. This is problematic, especially during a radiological emergency, where rapid detection of 210Po is critical and the safety of the public is at stake.
The International Atomic Energy Agency (IAEA) ALMERA (Analytical Laboratories for the Measurement of Environmental Radioactivity) network administered proficiency testing for the rapid analysis of 210Po in water after the Litvinenko event. It is interesting to note that only 70 % of the results reported by the 33 labs that participated in the proficiency test exercise met the requirements of the IAEA performance test guidelines. The ALMERA network, established by the IAEA in 1995, is a technical collaboration of existing institutions and makes available to Member States a worldwide network of analytical laboratories capable of providing reliable and timely analysis of environmental samples in the event of an accidental or intentional release of radioactivity. It is also somewhat of a concern that only 19 of the 33 labs participating reported the 210Po results within the 1 week target. The IAEA report stated “The PT results demonstrated that 22 of 33 participants were able to report results which fit the purpose of rapid detection of 210Po in water. However, although the matrix was a relatively straightforward one and the activity concentrations were relatively high, 15 % of the reported results failed to pass the proficiency test criteria. In a few cases positive results were reported for the blank sample which suggests a possibility of false positive reporting”. It was also noted that “19 laboratories should improve their repeatability and reproducibility of their determinations” [13]. Reporting times were usually about 10 days for most labs, but some labs took over 20 days and still did not report acceptable results.
Many of the labs provided very good results on these relatively straightforward water samples containing high levels of 210Po activity. But in light of the difficulty many of the labs had with this emergency exercise, it would seem that a new rapid method would have value, especially if that method could provide rigor, reliability and rapid turnaround times. If a radiological emergency involving 210Po occurs, it is important to have rapid sample preparation methods that can preconcentrate polonium ions without long evaporation times. Low temperature heating, which can be time-consuming, is often used to minimize the risk of 210Po volatilization. It is also very important to eliminate the risk of sample matrix interferences and poor sample count sources.
If sample interferences are present, decreased yields and degraded spectrums can occur, due to an unpredictable increase in thickness in the deposited layer. Separation methods to address interferences and chemical yield issues include using solvents such as isopropyl ether, methyl isobutyl ketone and tributyl phosphate, as well as anion exchange. The most popular solid phase extraction resin for 210Po extraction has been Sr Resin™ (4,4′,(5′)di-t-butylcyclohexane-18-crown-6 coated resin from Eichrom Technologies or Triskem-International), often used in combination with 210Pb analysis. Vajda was the first to report this novel approach, with reported yields of 50–70 % for polonium [14]. This method is effective, but does not lend itself easily to a rapid microprecipitation option to prepare the counting source. Polonium is typically eluted from Sr resin with 6M HNO3. Evaporation of this solution would be time-consuming, since higher temperatures risk the loss of polonium due to its volatility. The lack of a rapid, effective microprecipitation option for 210Po in the last 60 or more years of radiochemistry is apparent, so in this laboratory an effort was made to develop a new method that included this option.
A new method has been developed at the Savannah River National Laboratory (SRNL) to allow the rapid measurement of 210Po in environmental water samples. The method utilizes a rapid calcium phosphate precipitation, separation on DGA resin [15], and microprecipitation using bismuth phosphate for final alpha spectrometry source preparation. The method can be applied to small sample aliquots or larger water aliquots to achieve a much lower minimum detectable activity (MDA). The sample preparation for this method is as follows: ~15 min to 1 h depending on sample aliquot size (coprecipitation), ~1 h to perform the DGA resin separation, and ~15 min for final source preparation. For 200 mL samples, for example, samples may be completed in about 1.5–1.75 h, with assurance of the removal of sample interferences and good chemical yields. DGA resin has an extremely high affinity for polonium ions, and can be used to effectively remove a wide range of interferences. For example, Pb, Bi, Ra, Th, U, Pu, Am and Cm isotopes can be removed rapidly using DGA resin.
By using a DGA cartridge which contains resin with 50–100 μm particle size, rapid flow rates may be used. Although the combination of 210Pb analysis was not investigated for this work, it would seem that Sr resin or Pb resin cartridges (either 1 or 2 mL cartridges) could be attached below the DGA resin cartridge to allow the simultaneous purification and assay of 210Pb. The microprecipitation option for 210Po is rapid and convenient, with very good alpha peak resolution. This new technology seems to be a significant step for emergency sample analysis and can offer productivity improvements for routine analysis of water samples for 210Po as well.
In addition, a new sequential method is presented that uses a stacked column consisting of TRU Resin™ plus DGA resin cartridges that allows rapid collection and separation of not only 210Po, but plutonium, uranium, americium, and curium isotopes as well. This sequential method could also be modified to a uranium/polonium sequential method if only naturally-occurring radionuclides are desired.
Experimental
Reagents
The extraction chromatography resin employed in this work is DGA Resin® (N,N,N′,N′ tetraoctyldiglycolamide) and TRU-Resin® [tri-n-butylphosphate (TBP) and octyl (phenyl) N,N-diisobutylcarbamoylmethylphosphine oxide (CMPO)], available from Eichrom Technologies, Inc., (Lisle, IL, USA) or Triskem International (Bruz, France). Nitric and hydrochloric acids were prepared from reagent-grade acids (Fisher Scientific, Inc., Pittsburgh, PA, USA). All water was obtained from a Milli-Q2™ water purification system. All other materials were ACS reagent grade and were used as received. The radiochemical isotopes 209Po, 210Po, were obtained from Eckert–Ziegler Analytics, Inc. (Atlanta, GA, USA) and diluted to the appropriate levels. Radiochemical isotope tracers 242Pu, 243Am, and 232U, obtained from Eckert–Ziegler Analytics, Inc. (Atlanta, GA, USA) and diluted to approximately 0.37 Bq/mL, were employed to enable yield corrections. 232U tracer was prepared to be self-cleaning, removing its 228Th daughter using barium sulfate precipitation [16].
Procedures
Column preparation
DGA resin and TRU resin columns were obtained as cartridges containing 2 mL of each resin from Eichrom Technologies, Inc. (Lisle, IL, USA). Small particle size (50–100 μ) resin was employed, along with a vacuum extraction system (Eichrom Technologies). Flow rates of ~1–2 mL/min were typically used for this work, slower on sample loading and final elution steps, faster for the rinses used to remove sample matrix interferences.
Sample preparation for 210Po in water
Figure 1 shows the rapid sample preparation method used to rapidly collect 210Po from water samples in this work. This method was performed on ground water samples obtained from wells on the U.S. Department of Energy Savannah River Site (Aiken, SC, USA) using 200 mL and 1 L sample aliquots. Tests were also performed on 2 L sample aliquots of laboratory tap water. To each sample aliquot, 4.66 pCi of 209Po tracer and varying levels of 210Po were added. To ensure Po ions were in the Po4+ valence state, 30 wt% H2O2 was added to each sample aliquot as follows: 0.1 mL 30 wt% H2O2 to the 200 mL aliquots, 1 mL 30 wt% H2O2 to the 1 L sample aliquots, and 2 mL 30 wt% H2O2 to the 2 L sample aliquots. Addition of hydrogen peroxide is important to ensure 209Po tracer and 210Po are in valence state equilibrium in the sample.

Rapid sample preparation method for 210Po in water
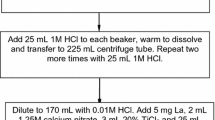
To perform the rapid calcium phosphate co-precipitation, 1 mL 1.25M calcium nitrate (50 mg Ca) and 3 mL 3.2M ammonium hydrogen phosphate were added to each sample. For the 200 mL samples, the sample aliquots were placed directly in 225 mL centrifuge tubes. The pH was adjusted to ~pH 10 with concentrated ammonium hydroxide using a pink phenolphthalein endpoint. The samples were centrifuged at 3,500 rpm for ~6 min and the supernatant was discarded.
For the 1 L sample aliquots tested, the sample aliquots were heated on a hot plate for about 30 min with the addition of calcium, phosphate, hydrogen peroxide and ammonium hydroxide. For the 2 L tap water samples it was found that chemical yields were enhanced if 1.5 mL 1.25M calcium nitrate (75 mg Ca) were added instead of 50 mg Ca. The beakers were removed from the hot plate and the precipitate was allowed to settle. The supernatant was poured off down to just less than 200 mL volume so that the remaining sample and solids could be transferred to 225 mL centrifuge tubes. The samples were centrifuged at 3,500 rpm for ~6 min and the supernatant was discarded.
The calcium phosphate precipitate was dissolved in 10 mL 9M HCl and the dissolved samples were transferred to 50 mL centrifuge tubes. The 225 mL tubes were rinsed with 10 mL 2M HCl so that an approximate HCl concentration of ~4.5M HCl was achieved. If any residual solids remained (from unfiltered well water samples), they were rinsed well by mixing with 3 mL 2M HCl, and centrifuged to remove any residual solids. This rinse was added to the original sample load solution.
Figure 2 shows why DGA resin was selected. The k′ for Po4+ with DGA resin is between 10,000 and 100,000 for Po4+ ions in HCl solutions, much higher than the affinity of Po4+ ions for Sr resin in 2M HCl (k′ of ~100) (Fig. 3). The k′ values were measured using the batch extraction procedure and calculations described in [15]. Po4+ separation methods on Sr resin typically use 2M HCl as column load solutions [13].

Retention of Po4+ on DGA resin in HCl and HNO3

Retention of Po4+ on Sr resin in HCl
Calcium and phosphate ions are present in the column load solution in this method as a result of the co-precipitation step. Figure 4 shows that the Ca2+ and PO4 3− ions have little impact on Po4+ retention on DGA resin. It should be noted that while Fe3+ is not typically present at significant levels in filtered water samples, Fe3+ would have minimal impact, as the k′ still exceeds 1,000 even with significant amounts of Fe3+ present. In addition, other common sample matrix ions such as Na+, K+, Mg2+ and Al3+ have no significant impact on Po4+ retention on DGA resin.

Impact of Ca2+, PO4 3− and Fe3+on Po4+ retention on DGA resin
Column separation for 210Po
DGA cartridges were placed on the vacuum box and 50 mL centrifuge tubes were used to collect rinse or final purified fractions. Figure 5 shows the rapid column separation method used.

Rapid column separation method for 210Po in water
To ensure polonium ions were present as Po4+, 100 μL 30 wt% H2O2 was added to each column load solution and mixed well. After the valence adjustment, the sample solution was loaded onto the DGA resin cartridge at approximately ~1 drop/s. After the sample was loaded, a tube rinse of ~5 mL 2M HCl was transferred to the DGA column and allowed to pass through the resin at ~1–2 drops/s. A rinse of 5 mL 2M HCl was added directly to the DGA column at ~2 drops/s. To remove any americium, curium, uranium or thorium present, 15 mL 0.25M HCl was added to the DGA cartridge at ~1–2 drops/s. To ensure removal of chloride ions, which form a strong complex with Po4+ ions and could interfere with Po elution, 5 mL 6M HNO3 was added to each column at ~1–2 drops/s. Po was then eluted from the DGA cartridge with 15 mL 0.05M HNO3 at ~1 drop/s.
Microprecipitation of 210Po
To each purified solution of 15 mL HNO3 containing eluted Po4+ ions, 50 μL 30 wt% H2O2 was added to ensure Po4+ was maintained. To co-precipitate Po, 100–125 μg Bi (100–125 μL of 1,000 μg Bi/mL standard in 2 % nitric acid) was added to each solution. To optimize the pH slightly, improving both chemical yield and peak resolution, 200 μL 2.9M NH4OH was added to each tube. To form the bismuth phosphate micro-precipitate, 750 μL 3.2M ammonium hydrogen phosphate was added to each tube. Each tube was mixed briefly using a vortex mixer, and after ~15 min, the solution was filtered using a 25 mm polypropylene filter (Resolve™ filter, Eichrom Technologies). Each tube was rinsed with ~5 mL deionized water and added to the filter funnel while the original solution was still present in the filter reservoir, followed by ethanol to facilitate drying. The filters were air-dried instead of heating to avoid any problems associated with polonium volatility.
Polonium alpha source filters were counted by alpha spectrometry for approximately 8–16 h, but shorter count times (<2 h) can also be performed for emergency response samples using higher level tracers, depending on the detection limit needed.
Sample preparation for 210Po and actinides in water
Figure 6 shows the sequential method to separate and purify actinide isotopes and 210Po using a single sample aliquot from a water sample. Tracers (242Pu, 243Am, 232U and 209Po) were added to each 200 mL groundwater sample aliquot already in 225 mL centrifuge tubes. To test for analyte recovery, spikes were added to each 200 mL aliquot to prepare analyte levels of 370 mBq/L 238Pu, 370 mBq/L 241Am, 327.5 mBq/L 244Cm, 654.9 mBq/L 238U and 1583.6 mBq/L 210Po in each sample aliquot. In addition, 237Np and 230Th were spiked into each sample aliquot at the 370 and 740 mBq/L levels respectively to test for interference removal.

Rapid sequential separation method for actinides and 210Po in water
To each tube, 200 μL 30 wt% H2O2 was added to ensure Po4+. To perform the rapid calcium phosphate co-precipitation, 1 mL 1.25M calcium nitrate (50 mg Ca) and 3 mL 3.2M ammonium hydrogen phosphate were added to each sample. The pH was adjusted to ~pH 10 with concentrated ammonium hydroxide using a pink phenolphthalein endpoint. The samples were centrifuged at 3,500 rpm for ~6 min and the supernatant was discarded.
To dissolve each precipitate, 10 mL 8M HNO3 and 3 mL 2M Al(NO3)3 were added to each tube and 100 μL 30 wt% H2O2 was added. The sample load solutions using this approach contained ~ 7.5 M total nitrate, which would result in a k′ of > 300 on DGA resin, as shown in Fig. 2. The high nitrate approach as chosen to also reduce calcium affinity and adverse impact of calcium ions on polonium retention on DGA resin [15].
Sequential column separation for 210Po and actinides
Figure 6 shows the sequential separation approach that was developed. TRU resin cartridges and DGA resin cartridges were stacked and placed on the vacuum box (TRU resin on top) and 50 mL centrifuge tubes were used to collect rinse or final purified fractions.
After the valence adjustment using hydrogen peroxide, the sample solution was loaded onto the TRU resin + DGA resin stacked column at approximately ~1 drop/s After the sample was loaded, a tube rinse of ~5 mL 8M HNO3 was transferred to the TRU resin + DGA resin column and allowed to pass through the resin at ~1–2 drops/s. A rinse of 10 mL 10 M HNO3 was added to each column to ensure Po was eluted from TRU resin onto DGA resin at ~1 drop/s. Figure 7 shows the Po4+ retention on TRU resin as HNO3 is increased and illustrates why 10 M HNO3 was selected to elute polonium onto DGA resin (although 8M HNO3 may also be effective). Then 15 mL 4 M HCl was added directly to the TRU + DGA column at ~1–2 drops/s to elute Am from TRU resin onto DGA resin. The TRU resin and DGA resin cartridges were split and processed separately from that point on two separate vacuum boxes for efficiency.

Retention of Po4+ on TRU resin in HNO3
After placing a new column reservoir on the TRU resin cartridge, Pu was eluted from TRU resin using 12 mL 3M HCl–0.02M TiCl3 slowly at ~1 drop/s. TRU resin was then rinsed with 5 mL 8M HNO3 at ~1–2 drops/s to ensure all the uranium is in the U+6 valence state. To remove thorium and neptunium, 15–20 mL 1.5M HCl–0.15M HF was added to rinse TRU resin at 1–2 drops/s. By ensuring uranium was in the U+6 valence state, losses during the thorium/neptunium removal step are minimized. Uranium was eluted using 15 mL 0.1M ammonium bioxalate at ~1 drop/s. Column connector tips were changed to eliminate any possibility of cross-contamination from the Th rinse solution that passes through prior to U elution.
To prepare the purified Pu samples for alpha spectrometry counting, 10 mL of water was added to reduce the acid strength and 0.5 mL 30 wt% H2O2 was added to oxidize any uranium ions that might be present to prevent uranium co-precipitation. After adding 50 μg Ce and 2 mL 28 M HF to the Pu eluent solution and waiting 15 min, the solution was filtered using a 25 mm polypropylene filter (Resolve™ filter). Each tube was rinsed with ~5 mL deionized water, followed by ethanol to facilitate drying. The filters were heated briefly under a heat lamp to ensure dryness. For the uranium eluent solutions, 50 μg Ce, 0.5 mL 10 % TiCl3 (to reduce U+6–U4+) and 1 mL 28 M HF were added and the tubes mixed well. After 15 min, the solutions were filtered as described above.
The DGA resin separation for Am and Po is described below. Five milliliters of 3M HCl were added to each DGA resin cartridge at ~2 drops/s to ensure complete Ca removal. To remove any thorium present, 12 mL 3M HNO3–0.25M HF was added at ~1–2 drops/s. Next, 5 mL 4M HCl was added to remove the any residual fluoride ions. Am and Cm isotopes were eluted with 12 mL 0.25M HCl at ~1 drop/s. Polonium ions remain on DGA resin during the elution of Am and Cm. To prepare Am/Cm eluent solutions for microprecipitation, 50 μg Ce and 200 μL of 30 wt% H2O2 were added to each tube, followed by 1 mL 28 M HF. The 30 wt% H2O2 is added to ensure any residual uranium ions, if present, are oxidized to U+6 and do not precipitate. After 15 min, the solutions were filtered as described above.
To ensure removal of chloride ions, 5 mL 6M HNO3 was added to each column at ~1–2 drops/s. Polonium was then eluted from the DGA cartridge with 15 mL 0.05M HNO3 at ~1 drop/s. Polonium microprecipitation was performed as described earlier. If any Pu ions bled to DGA resin, is retained on DGA resin in 0.05M nitric acid. Bi isotopes are also retained during this final elution step. While a weaker strength nitric acid could have been used, 0.05M nitric acid was selected so that both Pu and Bi isotopes would still be retained [15].
Apparatus
Polonium isotope measurements were performed by alpha-particle pulse-height measurements using Passivated Implanted Planar Silicon (PIPS) detectors. The PIPS detectors have an active surface area of 450 mm2. The nominal counting efficiency for these detectors is ~25 %. The distance between the sample and detector surface is ~3 mm.
Polycarbonate vacuum boxes with 24 positions and a rack to hold 50 mL plastic tubes were used. Two boxes were connected to a single vacuum source by using a T-connector and individual valves on the tubing to each box.
Results and discussion
Table 1 shows the individual results for the determination of 210Po in groundwater samples (spiked at 315.6 mBq/L) in six 200 mL groundwater samples using this rapid separation method and bismuth phosphate microprecipitation as source preparation for counting with alpha spectrometry. The 210Po results were corrected for 209Po tracer yield. The average 210Po result for the 200 mL groundwater samples was 308.1 mBq/L, with a −2.4 % bias and 1SD (standard deviation) of 4.8 mBq/L. The average tracer recovery for 209Po was 87.4 % ± 5.8 % at 1SD. The sample preparation and column separation steps, including the final microprecipitation step using bismuth phosphate took <1.5 h. Since the microprecipitation efficiency is at ~92–95 %, the chemical yield across the sample preparation and DGA separation steps was ~90 %.
Table 2 shows the individual results for the determination of 210Po in groundwater samples (spiked at 1261.7 mBq/L) in seven 200 mL groundwater samples using this rapid method. The 210Po results were corrected for 209Po tracer yield. The average 210Po result for the 200 mL groundwater samples was 1288.8 mBq/L, with a 2.2 % bias and SD of 5.5 mBq/L. The average tracer recovery for 209Po was 82.3 % ± 3.9 % at 1SD.
Table 3 shows the individual results for the determination of 210Po in groundwater samples (spiked at 63.3 mBq/L) in six 1 L groundwater samples using this rapid method. The 210Po results were corrected for 209Po tracer yield, which had an average tracer recovery of 85.0 % ± 8.2 % at 1SD. The average 210Po result for the 1 L groundwater samples was 61.5 mBq/L, with a −2.8 % bias and SD of 5.1 mBq/L. The 1 L samples took about 30–45 min longer to process due to the heating and settling steps used for larger aliquot samples.
Table 4 shows the individual results for the determination of 210Po in tap water samples (spiked at 63.3 mBq/L) in five 1 L samples using this rapid method. The 210Po results were corrected for 209Po tracer yield, which had an average tracer recovery of 80 % ± 9.6 % at 1SD. The average 210Po result for the 2 L groundwater samples was 61.1 mBq/L, with a −3.4 % bias and SD of 6.2 mBq/L. The tracer yields were slightly lower for sample aliquot 1, but the average yield was still 80 % and the 210Po results were very good. This demonstrates that sample aliquots >1 L can be taken so that much lower detection limits can be achieved, if needed.
Table 5 shows the individual results for the determination of 210Po in groundwater samples (spiked at 1583.6 mBq/L) in six 200 mL groundwater samples using the rapid sequential method for actinides and 210Po. The 210Po results were corrected for 209Po tracer yield, which had an average tracer recovery of 81.5 ± 2.6 % at 1 SD. The average 210Po result for the 200 mL groundwater sample aliquots was 1,660.4 mBq/L, with a 4.9 % bias and SD of 2.9 mBq/L. The separation procedure using TRU resin (Pu, U) and DGA resin (Am, Cm, Po), took about 2 h longer than the Po only method.
Table 6 shows the results for 238Pu in the six 200 mL groundwater samples using the rapid sequential method for actinides and 210Po spiked with 238Pu at the 370 mBq/L level. The average 242Pu yield of 93.4 % was very good. The average 238Pu result for the 200 mL groundwater sample aliquots was 381.1 mBq/L, with a 3.0 % bias and SD of 4.0 mBq/L. The plutonium alpha spectra did not show any presence of 230Th or 237Np.
Table 7 shows the results for 241Am in the six 200 mL groundwater samples using the rapid sequential method for actinides and 210Po spiked with 241Am at the 370 mBq/L level. The average 243Am yield of 100.2 % was excellent. The average 241Am result for the 200 mL groundwater sample aliquots was 380.8 mBq/L, with a 2.9 % bias and SD of 2.9 mBq/L. The resolution of the 243Am and 241Am peaks was very good and no indication of 230Th was observed in the alpha spectra. Table 8 shows the 244Cm results, for the samples spiked at the 327.5 mBq/L level, also determined from the Am/Cm fraction using the 243Am yields. The average 244Cm result for the 200 mL groundwater sample aliquots was 327.9 mBq/L, with a 0.1 % bias and SD of 3.7 mBq/L.
Table 9 shows the results for 238U in the six 200 mL groundwater samples using the rapid sequential method for actinides and 210Po spiked with 238U at the 654.9 mBq/L level. The average 232U yield of 96.6 % was excellent. The average 238U result for the 200 mL groundwater sample aliquots was 626.8 mBq/L, with a −4.3 % bias and SD of 3.7 mBq/L. No indication of 230Th or 237Np alpha emissions was observed in the uranium alpha spectra.
Optimization of the bismuth phosphate microprecipitation of Po from 15 mL 0.05M nitric acid was achieved by varying the conditions described earlier: 100–125 μg Bi, 200 μL 2.9M NH4OH, 50 μL 30 wt% H2O2 and 750 μL 3.2M ammonium hydrogen phosphate. Bismuth content in excess of 125 μg does not significantly improve the Po recovery, while having a negative impact on the full width half maximum (FWHM). ammonium hydrogen phosphate in excess of 750 μL decreases Po recovery, likely by increasing the solubility of Bi. The ammonium hydrogen phosphate buffers the pH between 6.4 and 6.6. However, addition of a small amount of 2.9M NH4OH (200 μL) improves the Po yields while decreasing the FWHM from 40–50 to 30–40 keV. The FWHM values for the microprecipitation are not quite as good as autodeposition (25–30 keV) [11]. However, the 32–45 keV FWHM values are more than sufficient to resolve the 209Po (4,883 keV) and 210Po (5,304 keV) alpha peaks. In contrast to autodeposition, sample matrix interferences that can affect the alpha spectrometry source preparation are removed and the final source preparation step is very rapid.
Figure 8 shows an example of the spectra of 210Po using bismuth phosphate microprecipitation with 100 μg Bi carrier. The 209Po tracer recovery was 95.4 % and the FWHM was 32.4 keV, showing acceptable alpha peak resolution and good tracer recovery. Figure 9 shows an example of the spectra of 210Po using bismuth phosphate microprecipitation with 125 μg Bi carrier. The 209Po tracer recovery was 97.6 % and the FWHM was 38.6 keV, showing acceptable alpha peak resolution and good tracer recovery. The alpha peaks for Po isotopes showed very good peak resolution using either 100 μg or 125 μg Bi, typically with a FWHM of ~35–45 keV. The peak resolution was slightly better using 100 μg Bi carrier, but still very good with 125 μg Bi. Figure 10 shows an example of the final alpha spectrometry spectra from a 1 L groundwater sample spiked with 210Po, using 125 μg Bi. The 209Po tracer recovery was 87.0 % and the FWHM was 46 keV.

Alpha spectra for Po isotopes using bismuth phosphate (100 μg Bi)

Alpha spectra for Po isotopes using bismuth phosphate (125 μg Bi)

Po isotopes—bismuth phosphate (125 μg Bi)—1 L groundwater sample
In this new rapid method, 100–125 μg Bi, 200 μL 2.9M NH4OH, and 0.75 mL 3.2M (NH4)2HPO4 were added to the 15 mL 0.05M HNO3 eluent solution, with a 15 min wait time, to optimize chemical yield and FWHM. To ensure Po was present as Po4+, 50 μL 30 wt% H2O2 was also added. These conditions maximize the precipitation yield for BiPO4 and result in excellent alpha peak resolution.
The MDA for the actinide isotopes by alpha spectrometry were calculated according to equations prescribed by Currie [17]:
where B = total background counts, = BKG (rate) * BKG count time; CT is the sample count time (min); R is the chemical recovery; V is the sample volume (liters); EFF is the detector efficiency; 0.060 is the conversion from dpm to mBq.
In low-level counting, where a zero background count is quite common, the constant 3 is used to prevent an excessively high false positive rate.
The MDA for the alpha spectrometry results can be adjusted as needed, depending on the sample aliquot and count time. For a 200 mL sample aliquot, the MDA for an 8 h count time is ~6.3 mBq/L. For a 1 L sample aliquot, the MDA for an 8 h count time is ~0.4 mBq/L. The alpha spectrometry samples were counted for 8–16 h in this work, but emergency level samples can be counted for less than 1 h if activity levels are high enough, which may be the case for emergency incident samples.
The 210Po results test results show very good quality when compared to the reference values. The rapid separation method can be performed in less time than many of the reported methods for 210Po using autodeposition on silver or other disks that use 4–24 h deposition times. For 200 mL samples, for example, sample preparation and alpha spectrometry source preparation may be completed in about 1.5–1.75 h, with the assurance of removal of sample interferences and interferences. Removal of interferences is extremely important, especially during a radiological emergency, where rapid detection of 210Po is critical and the safety of the public is at stake.
This DGA resin separation method was designed to effectively remove radiological interferences. Ra and Pb isotopes are not retained on DGA resin in HCl solutions and U, Th, Am and Cm isotopes are removed during the 0.25M HCl rinse step. Pu4+ ions and Bi isotopes remain on DGA resin during column loading and rinsing with HCl solutions, and during the Po elution step using 0.05M HNO3 [15]. To ensure effective removal of interferences, column reservoirs and connector tips in the vacuum box lid are changed prior to the final elution step.
This new method is not only rapid and effective in removing interferences, but it can be used to assay actinides and 210Po in a sequential manner, which saves time and reduces labor costs for these analyses. The actinides and polonium sequential method can likely be streamlined to simplify rinsing if only uranium and polonium are desired. There is also the potential of adding a sequential determination of 210Pb by adding Sr resin or Pb resin in tandem with DGA resin. Due to the very high affinity of polonium for DGA resin, it may be possible to use 1 mL DGA resin cartridges (instead of 2 mL) to reduce costs, scaling back column volumes proportionally. Having a reliable method that removes interferences for emergency or routine analyses is extremely important, and this method provides 210Po results on water samples very rapidly with rigorous removal of interferences.
In addition to providing a rapid method for use in a radiological emergency, the assay of 210Po in well water has become more important. In 2007, very high levels of 210Po were discovered in drinking water from a number of private wells in Lahontan Valley, Churchill County, in northern Nevada. Of the 60 private wells and three public supply wells sampled in the valley, more than one-third of the private wells had 210Po levels exceeding 0.55 Bq/L, and 10 % had levels that exceeded 2 Bq/L [18].
The only applicable drinking water standard for 210Po in the United States is the adjusted gross alpha radioactivity (GAR) standard of 15 pCi/L. Seiler notes that “additional information on the volatility of 210Po is needed because GAR is an inappropriate method to screen for volatile radionuclides. About 25 % of the [Nevada] samples had 210Po activities that exceed the level associated with a lifetime total cancer risk of 1 × 10(−4) (1.1 pCi/L) without exceeding the GAR standard. In cases where the 72-h GAR exceeds the uranium activity by more than 5–10 pCi/L, an analysis to rule out the presence of 210Po may be justified to protect human health even though the maximum contaminant level for adjusted GAR is not exceeded” [19].
The determination of 210Po using a rapid, reliable method that eliminates interferences and is not adversely affected by polonium volatility would seem to be useful, not only for water samples after a radiological emergency, but to determine 210Po in well water samples with potentially high 210Po activity. This is particularly important since GAR screening may not accurately reflect 210Po levels present.
Conclusion
A new rapid separation method that allows separation and preconcentration of 210Po in emergency or routine water samples was developed. The method is rapid and provides very good chemical recoveries with excellent removal of interferences. Testing of the method at various levels of 210Po activity showed very good results versus reference values. In an emergency, method rigor and minimizing rework are extremely important. This is also very important for processing routine samples efficiently. By using this rapid method, the potential for interferences causing decreased chemical yields and degraded alpha spectrometry spectrums is eliminated. This method provides rapid source preparation using bismuth phosphate microprecipitation, a rapid option to spontaneous autodeposition on metal disks.
References
Radiation Studies Branch, Division of Environmental Hazards and Health Effects, National Center for Environmental Health, Centers for Disease Control and Prevention, Atlanta. http://emergency.cdc.gov/radiation/isotopes/polonium/clinicians.asp
Scott B (2007) Health risk evaluations for ingestion exposure of humans to polonium-210. Dose Response 5(2):94–122
Miller C, Whitcomb R, Ansari A, McCurley C, Nemhauser N, Jones R (2012) Murder by radiation poisoning. implications for public health. J Environ Health 74(10):8
Steinhäusler F, Rydell S, Zaitseva L (2007) Risk due to radiological terror attacks with natural radionuclides. In: The natural radiation environment: 8th international symposium (NRE VIII). AIP conference proceedings 1034, Buzios, Rio de Janeiro, 07–12 Oct 2007, pp 3–15. doi:http://dx.doi.org/10.1063/1.2991254
Gleick P (2006) Water and terrorism. Water Policy 8:481–503. http://www.pacinst.org/reports/water_terrorism.pdf
Nemhauser JB (2010) The polonium-210 public health assessment: the need for medical toxicology expertise in radiation terrorism events. J Med Toxicol 6:355–359
Maxwell S, Culligan B (2012) Rapid determination of Ra226 in environmental samples. J Radioanal Nucl Chem 293(1):149–156
Maxwell S, Culligan B (2013) Rapid method for determination of 228Ra in water samples. J Radioanal Nucl Chem 295:2188–2191
Maxwell S, Culligan B, Noyes G (2010) Rapid separation method for actinides in emergency air filter samples. Appl Radiat Isot 68:2125
Maxwell S, Culligan B, Kelsey-Wall A (2012) Rapid determination of actinides in emergency food samples. J Radioanal Nucl Chem 292(1):339
Matthews K, Chang-Kyu K, Martin P (2007) Determination of 210Po in environmental materials: a review of analytical methodology. Appl Radiat Isot 65:267–279
Clayton R, Bradley E (1995) A cost-effective method for the determination of 120Po and 210Pb in environmental samples. Sci Total Environ 173/174:23–28
International Atomic Energy Agency (2006) Report on the second ALMERA coordination meeting and the ALMERA soil sampling intercomparison exercise—IAEA/SIE/01, IAEA/AL/164, p 36
Vajda N, La Rosa J, Zeisler R, Danesi P, Kis-Benedek GY (1997) A novel technique for the simultaneous determination of 210Pb and 210Po using a crown ether. J Environ Radioact 37:355–372
Horwitz P, McAlister D, Bond A, Barrans AB Jr (2005) Novel extraction chromatographic resins based on tetraalkyldiglycolamides: characterization and potential applications. Solvent Extr Ion Exch 23(3):319
Sill C (1974) Purification of radioactive tracers for use in high sensitivity alpha spectrometry. Anal Chem 46(11):1426
Currie LA (1968) Limits for qualitative and quantitative determination. Anal Chem 40:586
Frequently asked questions on USGS data on polonium-210 in wells in Lahontan Valley, Churchill County, Nevada. http://nevada.usgs.gov/polonium/FAQ.pdf. Accessed 25 July 2011
Seiler RL (2011) 210Po in Nevada groundwater and its relation to gross alpha radioactivity. Ground Water 49(2):160–171
Acknowledgments
This work was performed under the auspices of the Department of Energy, DOE Contract No. DE-AC09-96SR18500. The authors wish to acknowledge Rebecca Chavous, Jack Herrington and Staci Britt for their assistance with this work, as well as Dr. Dave Diprete of SRNL for providing the 210Po standard used in this work. The work by Dr. Dan McAlister at PG Research Foundation is very much appreciated.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Maxwell, S.L., Culligan, B.K., Hutchison, J.B. et al. Rapid determination of 210Po in water samples. J Radioanal Nucl Chem 298, 1977–1989 (2013). https://doi.org/10.1007/s10967-013-2644-2
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-013-2644-2