
Abstract
A new rapid method for the determination of 226Ra in environmental samples has been developed at the Savannah River Site Environmental Lab (Aiken, SC, USA) that can be used for emergency response or routine sample analyses. The need for rapid analyses in the event of a Radiological Dispersive Device or Improvised Nuclear Device event is well-known. In addition, the recent accident at Fukushima Nuclear Power Plant in March, 2011 reinforces the need to have rapid analyses for radionuclides in environmental samples in the event of a nuclear accident. 226Ra (T1/2 = 1,620 years) is one of the most toxic of the long-lived alpha-emitters present in the environment due to its long life and its tendency to concentrate in bones, which increases the internal radiation dose of individuals. The new method to determine 226Ra in environmental samples utilizes a rapid sodium hydroxide fusion method for solid samples, calcium carbonate precipitation to preconcentrate Ra, and rapid column separation steps to remove interferences. The column separation process uses cation exchange resin to remove large amounts of calcium, Sr Resin to remove barium and Ln Resin as a final purification step to remove 225Ac and potential interferences. The purified 226Ra sample test sources are prepared using barium sulfate microprecipitation in the presence of isopropanol for counting by alpha spectrometry. The method showed good chemical recoveries and effective removal of interferences. The determination of 226Ra in environmental samples can be performed in less than 16 h for vegetation, concrete, brick, soil, and air filter samples with excellent quality for emergency or routine analyses. The sample preparation work takes less than 6 h. 225Ra (T1/2 = 14.9 day) tracer is used and the 225Ra progeny 217At is used to determine chemical yield via alpha spectrometry. The rapid fusion technique is a rugged sample digestion method that ensures that any refractory radium particles are effectively digested. The preconcentration and column separation steps can also be applied to aqueous samples with good results.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
There is an increasing need to develop faster analytical methods for emergency response, including emergency environmental and food samples [1–3]. 226Ra can be determined indirectly using gamma spectrometry or radon emanation techniques [4, 5], however, both approaches require delays to allow for progeny ingrowth and require careful handling of gaseous radon. These methods can show negative biases due to loss of radon or possibly due to homogeneity issues regarding progeny location sealed containers for gamma counting. 226Ra can also be measured by inductively-coupled plasma mass spectrometry (ICP-MS), but isobaric polyatomic interferences such as 88Sr and 138Ba must be removed [6].
There are a number of analytical methods reported that use ion exchange/extraction chromatography plus alpha spectrometry to determine 226Ra in environmental samples. Chabaux [7] used multiple, large cation exchange columns (25 ml, 10 ml resin) followed by a Sr Resin column to remove interferences prior to measurement of 226Ra in volcanic rocks. Lariviere [8] tested several different methods, including the large cation resin column method by Chabaux, as well as sulfate precipitation and manganese dioxide approaches. The overall results were good, but the column rinse volumes were very large, requiring long processing times. The chemical yields in some cases were low.
Crespo [9] applied a similar ion exchange separation approach to geological samples using 225Ra tracer, with large anion exchange and cation exchange columns followed by electrodeposition.
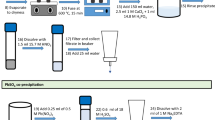
Manganese dioxide precipitation and the use of MnO2 Resin has been applied successfully to water samples [10–13]. One of the challenges, however, of applying this separation approach to solid samples is that when the pH of a solid sample digest containing iron is increased to the optimal pH 7 range for using MnO2 Resin, co-precipitation of Ra occurs. As Fig. 1 shows Ra, along with other alkaline earth metal ions, will co-precipitate along with iron hydroxide, and be separated prior to loading to MnO2 Resin. Manganese dioxide precipitation could be used, but calcium would also precipitate along with the iron hydroxide. For solid samples containing iron, a different approach seems advantageous.

Ra precipitation vs pH with iron hydroxide data provided courtesy of Dr. Dan McAlister, P&G Research Foundation, Lisle, IL, USA
Based on a survey of the literature, there still seems to be a need for improvements in a rapid 226Ra method, particularly for solid environmental samples. The use of 225Ra tracer (measuring the alpha emitting 217At progeny) is very promising because it does not exist naturally in the environment (unlike 223Ra) and it eliminates problems associated with using 133Ba to determine Ra chemical yield. Ba and Ra may not always behave in an identical fashion, thus a Ra isotopic tracer offers advantages over a 133Ba. In addition, when 133Ba is used, native barium, which can adversely affect alpha spectrometry resolution, cannot be removed. This effectively limits the sample aliquot size when native barium is present. Native barium is typically present in solid samples such as soil or concrete, and therefore small sample aliquots, 0.25 g or less of soil or concrete, must be used if 133Ba tracer is employed. Since a larger sample aliquot is often useful, removal of native barium is important to allow that approach.
A new rapid method to determine 226Ra in environmental samples has been developed in the Savannah River Site Environmental Lab (Aiken, SC, USA). This new approach has reduced the sample preparation time for soil, concrete, and brick matrices to <6 h for a batch of ten samples, including fusion, preconcentration and column separation steps. This method can be used in emergency response situations and offers advantages for routine sample analysis as well. For samples such as vegetation, a pre-treatment furnace step was used to destroy the organic content. This furnace ashing step adds about 2 h to the sample preparation. For concrete, brick, soil, and glass fiber air filter samples, the rapid sodium hydroxide fusion was applied directly, followed by precipitation steps including a calcium carbonate matrix removal step, followed by a cation exchange column to remove most of the calcium. Sr Resin was used to remove stable barium, when native barium removal was required. Ln Resin was used as a final polishing step to remove 225Ac and sample matrix interferences. Vacuum box technology was used to allow rapid flow rates and a stacked resin cartridge approach was employed to reduce separation times. Alpha spectrometry sources were prepared using a barium sulfate microprecipitation technique in the presence of isopropanol which provides high resolution alpha spectra, much improved over the barium sulfate seeding suspension method [10] for counting by alpha spectrometry. This new method showed good chemical recoveries and effective removal of interferences. It avoids the problem associated with MnO2 Resin separation applied to solid samples, which requires the raising of the pH of a digested sample and the potential loss of Ra along with Fe(OH)3 precipitation when the pH is decreased to pH 7. The fusion, unlike fusions that are performed one at a time over a burner, can be performed simultaneously in a furnace or multiple furnaces using relatively inexpensive zirconium crucibles. An adaptation of this method without the fusion may be applied to aqueous samples as well. Another possible advantage of this approach is that this method does not result in large amounts of manganese ions that typically occur when using MnO2 precipitations or MnO2 Resin, usually undesirable for ICP-MS measurement of 226Ra.
Experimental
Reagents
The resins employed in this work are cation resin (50 WX8, hydrogen form, 200–400 mesh), Ln Resin® (bis (2-ethylhexyl) phosphoric acid) and Sr Resin (4, 4′, 5′) di-t-butylcyclohexane-18-crown-6), available from Eichrom Technologies, Inc. (Lyle, Illinois, USA). Nitric and hydrofluoric acids were prepared from reagent-grade acids (Fisher Scientific, Inc.). All water was obtained from a Milli-Q2™ water purification system. All other materials were ACS reagent grade. Radiochemical isotope tracer 229Th (225Ra) and 226Ra were obtained from Eckert Ziegler/Analytics, Inc. (Atlanta, GA, USA) and diluted to approximately 3.66 and 0.37 Bq ml−1, respectively.
Procedures
Column preparation
Cation exchange resin (Eichrom 50WX8, 200–400 mesh) was obtained as bulk resin and columns were prepared by weighing out the resin amounts in large ion exchange column reservoirs. (Environmental Express, Mount Pleasant, SC, USA). Sr Resin and Ln Resin cartridges containing 2 ml of each resin were obtained from Eichrom Technologies, Inc. (Lisle, IL). Small particle size (50–100 micron) resin was employed, along with a vacuum extraction system (Eichrom Technologies) that will handle 24 samples at a time. Flow rates of 1–2 ml min−1 were typically used.
Sample preparation
Replicate sample aliquots of various environmental samples were added to 250 ml zirconium crucibles. Concrete and brick samples were ground to a fine powder with a mortar and pestle in this work but a ball mill apparatus with sieving or drill sampling could have been used. Soil samples were dried, ground in a ball mill, sieved, and blended. For concrete, brick and soil samples, 1 g of sample was added to each crucible. To test the method on vegetation samples, 5 g of a blank vegetation matrix (hay) was added for each crucible. A 47-mm glass fiber filter was added to each crucible to test the method on air filter samples. To each replicate sample, 99.4 pCi of 229Th tracer (in equilibrium with daughter 225Ra) was added to and samples were dried on a hotplate. The amount of tracer added can be varied depending on the number of 217At counts desired in the tracer region and how long an ingrowth time is desired prior to counting. A blank replicate was also analyzed with each batch so that the 226Ra results could be corrected for the native content of 226Ra. To each spiked vegetation, soil, brick, concrete, and air filter sample, a known amount of 226Ra was added. To demonstrate this approach may also be adapted to aqueous samples, tap water samples were also prepared for analysis.
Figure 2 shows the rapid fusion and precipitation steps used to digest the solid environmental samples and preconcentrate the 226Ra from the alkaline fusion matrix. For the 5 g vegetation aliquots, the crucibles were placed in a furnace at 300 °C and ramped immediately to 700 °C and ashed for ~2 h. After crucibles were removed and cooled, 5 ml of concentrated nitric acid and 30 wt% hydrogen peroxide was added to each crucible and the ashed samples were dried on a hot plate. The crucibles were placed back in a furnace at 600 °C and ashed for ~10 min to ensure the samples were ashed completely. The crucibles containing ashed vegetation were removed from the furnace and were ready for the rapid fusion. From this point on, the vegetation, brick, concrete, soil and air filter samples fusion, and preconcentration steps were the same, except for the amounts of calcium that were added for the calcium carbonate precipitation. To fuse the samples, 10 g NaOH were added to each crucible. The crucibles were covered with a zirconium lid and placed into a furnace already heated to 600 °C for ~15 min.

Rapid 226Ra sample preparation for solid environmental samples
After removing the crucibles from the furnace, the crucibles were cooled for about 10 min, water was added to each and the crucibles were heated on a hot plate to dissolve and transfer the solids to 225 ml centrifuge tubes. The residual solids were removed from the crucibles by adding water and heating the crucibles further on the hot plate as needed. The samples were diluted to 150 ml with water and cooled in an ice bath to room temperature.
Figure 3 shows the rapid precipitation steps that can be used for aqueous environmental samples to preconcentrate the 226Ra from a water matrix. Water sample aliquots of 150 ml were acidified to ~pH 2 in 225 ml centrifuge tubes using nitric acid. To each replicate, 99.4 pCi of 229Th (225Ra) tracer and 73.8 mBq 226Ra were added to each replicate A blank replicate was also analyzed with each batch so that the 226Ra results could be corrected for the native content of 226Ra. 10 ml of concentrated ammonium hydroxide was added to each tube and 150 mg Ca was added to each sample. Larger aliquots could have been processed in large beakers, allowed to settle and then transferred to centrifuge tubes, but for this test 150 ml water replicates were added directly to centrifuge tubes.

Rapid 226Ra sample preparation for aqueous samples
The amounts of calcium were varied for each sample matrix type, with the goal of total calcium being 100–150 mg Ca. The following amounts of Ca were added: for air filters, 2.5 ml 1.25 M Ca(NO3)2 (125 mg Ca) was added; for concrete/brick samples, 0.5 ml 1.25 M Ca(NO3)2 (25 mg Ca) was added; for soil and vegetation samples, 1 ml 1.25 M Ca(NO3)2 was added (50 mg Ca), and for water samples, 3 ml 1.25 M Ca(NO3)2 (150 mg Ca) was added. In addition, to lower the alkalinity each tube slightly, 10 ml 12 M HCl was added to each tube (fused sample matrix only) and each was capped and mixed well. To form the calcium carbonate precipitate, 25 ml 2 M sodium carbonate was added to each tube and the tubes were mixed well. The samples were cooled in an ice bath for ~10 min. The tubes were centrifuged at 3,500 rpm for 5 min and the supernatant was discarded. The remaining solids were dissolved in 10 ml of 1.5 M HCl, and transferred to a 50 ml centrifuge tube. The 225 ml tube was rinsed well with 10 ml of 1.5 M HCl and this rinse solution was added to each dissolved sample. Additional HCl (1 ml 12 M HCl) was added to ensure the brick samples were dissolved in an acidic matrix. The sample solutions were heated briefly in a hot water bath and then centrifuged at 3,500 rpm for 5 min. If any residual solids remained, they were rinsed well by mixing with 5 ml 1.5 M HCl, and centrifuging to remove any residual solids. This rinse was added to the original sample solution.
To minimize the impact of any Fe3+ present on the cation resin step, 3 ml 1.5 M ascorbic acid was added to each tube to reduce Fe3+ to Fe2+. Gravity flow was typically sufficient to achieve a flow rate of ~1 drop/s, however, vacuum was applied if needed.
Column separation
Figure 4 shows the column separation sequence used. The Ra was retained on cation resin (5 g), calcium was removed by rinsing with 3 M HCl, and Ra was stripped from the cation resin with 8 M HNO3 and evaporated to dryness on medium/low heat. The sample was redissolved in 3 M HNO3 and passed through Sr Resin. For samples where barium removal may not required (example, water, air filter, and vegetation samples), the Sr Resin separation would likely not be necessary for alpha spectrometry assay. The 3 M HNO3 solution with Ba/Sr removed was evaporated to dryness on medium/low heat, then ashed once more to dryness with 2 ml 1 M HCl and 2 ml 30 wt% H2O2. Gentle heating of these solutions just as the samples were going to dryness at each of this evaporation steps was found to be very important. The heating steps were such that the solutions were allowed to go to dryness after the beakers were taken off the hotplate, to minimize oxide formation and maximize chemical yields.

Rapid 226Ra column separation for environmental samples
The samples were redissolved in 2 ml 0.1 M HCl, warmed on a hot plate, diluted with 8 ml water, and reheated briefly. After cooling, the samples were passed through Ln Resin to remove 225Ac and any other possible interferences, rinsing Ln Resin with 10 ml 0.02 M HCL. The 0.02 M acidity was set to ensure Ra was eluted and any residual Ca was retained. Two separation times are important. The first time is related to adding the tracer 229Th/225Ra in equilibrium. It was found that 229Th was removed from 225Ra at the cation elution step. At this point the 225Ra is unsupported and starts to decay, however, if the time between the cation resin elution and the Ln Resin separation that removes 225Ac the correction is minimal (~1% for a 4 h time difference).
For soil samples it was found that improved yields were obtained if 6 g cation resin (35 ml 3 M HCl cation resin/30 ml 8 M HNO3 cation resin elution), presumably due to additional cation capacity required for the soil matrix.
Microprecipitation
To each final purified solution containing 20 ml 0.02 M HCl, 3 ml 12 M HCl was added to increase the acidity to ~ 1.5 M HCL. 3 g of ammonium sulfate were added to each tube and mixed well to dissolve completely. 50 μg of barium and the solution was mixed well. 5 ml isopropanol were added to each tube and mixed again. The tubes were iced for 15 min, periodically vortexed during that time (beginning, middle and on removal from ice). The solutions were filtered onto 0.1 micron 25-mm polypropylene filters (Resolve-Filter-Eichrom Technologies), rinsing the filters with 20% isopropanol. The filters were dried under a heat lamp and counted by alpha spectrometry. Alpha spectrometry was used for this testing, but the purified 0.02 M HCl solution could have been measured using ICP-MS, perhaps using 228Ra as a tracer to monitor yield.
Apparatus
226Ra measurements were performed by alpha-particle pulse-height measurements using Passivated Implanted Planar Silicon (PIPS) detectors. The PIPS detectors have an active surface of 450-mm2. The nominal counting efficiency for these detectors is 0.30. The distance between the sample and detector surface is ~3-mm.
Polycarbonate vacuum boxes with 24 positions and a rack to hold 50 ml plastic tubes were used.
Results and discussion
Table 1 shows the individual results for the determination of 226Ra in five 5 g vegetation samples using this rapid separation method and alpha spectrometry. The results were corrected for 225Ra (via the 217At progeny) tracer yield. The average 226Ra result for the 5 g vegetation samples was 72.8 mBq smp−1, with a −1.2% bias and 1 SD (standard deviation) of 5.1 mBq smp−1. The measured values were corrected for 9.17 mBq 226Ra found in the unspiked vegetation sample. The high 225Ra (217At) tracer recoveries and excellent results for the analytes versus known values indicate the sample preparation and measurement steps for the vegetation samples were effective. The average tracer recovery for 225Ra was 87.1% ± 5.7 at 1 SD. The samples were counted for 8 h and the 217At ingrowth time to midpoint of the count was 9.03 h. The amount of time for ingrowth can be varied and is dependent on the amount of 225Ra added and the number of counts desired in the 217At tracer region. For this work a relatively large amount of 225Ra was added so that a minimal ingrowth time was required. It should be noted that when short ingrowth times are used that very careful time measurements must be applied to minimize error in 217At ingrowth calculations.
Table 2 shows the individual results for the determination of 226Ra in five 1 g concrete samples using this new method with alpha spectrometry. The results were corrected for 225Ra (217At) tracer yield. The average 226Ra result for the 1 g concrete samples was 180.6 mBq smp−1, with a −2.1% bias and 1 SD of 8.0 mBq smp−1. The measured values were corrected for 26.8 mBq 226Ra found in the unspiked concrete sample. The average tracer recovery for 225Ra (217At) was 84.6% ± 6.8 at 1 SD. The samples were counted for only 4 h and the 217At ingrowth time to midpoint of the count was 11.86 h. The method offers some flexibility in terms of count times and ingrowth time periods and much less tracer may be added, but longer ingrowth times are required under those conditions.
Table 3 shows the individual results for the determination of 226Ra in six 1 g brick samples using this new method. The average 226Ra result for the 1 g brick samples was 77.8 mBq smp−1, with a 5.5% bias and 1 SD of 4.6 mBq smp−1. The measured values were corrected for 29.5 mBq 226Ra found in the unspiked brick sample. The average tracer recovery for 225Ra (217At) was 86.5% ± 6.6 at 1 SD. The samples were counted for 4 h and the 217At ingrowth time to midpoint of the count was 12.15 h.
Table 4 shows the individual results for the determination of 226Ra in five 47-mm glass fiber filter samples using this new method. The average 226Ra result for the air filter samples was 77.1 mBq smp−1, with a 4.5% bias and 1 SD of 4.0 mBq smp−1. The measured values were corrected for 3.7 mBq 226Ra found in the unspiked air filter sample. The average tracer recovery for 225Ra (217At) was 76.7% ± 4.2 at 1 SD. The samples were counted for 4 h and the 217At ingrowth time to midpoint of the count was 12.47 h. It should be noted that these air filters were glass fiber and were fused directly. Cellulose filters can be analyzed using this method, but would require a short furnace ashing step prior to fusion.
Table 5 shows the individual results for the determination of 226Ra in four 1 g soil samples. The average 226Ra result for the water samples was 184.9 mBq smp−1, with a 0.2% bias and 1 SD of 6.2 mBq smp−1. The measured values were corrected for 49.8 mBq 226Ra found in the unspiked soil sample. The average tracer recovery for 225Ra (217At) was 75.3% ± 1.9% at 1 SD. The samples were counted for 8 h and the 217At ingrowth time to midpoint of the count was 11.61 h.
Table 6 shows the individual results for the determination of 226Ra in four 150 ml water samples using an adaptation of this new method. The results were corrected for 225Ra (217At) tracer yield. The average 226Ra result for the water samples was 70.9 mBq smp−1, with a −3.9% bias and 1 SD of 3.7 mBq smp−1. The measured values were corrected for 9.6 mBq 226Ra found in the unspiked water sample. The average tracer recovery for 225Ra (217At) was 91.8% ± 6.7 at 1 SD. The samples were counted for 6 h and the 217At ingrowth time to midpoint of the count was 33.9 h. The samples could have been counted much sooner, but a longer ingrowth period was used simply to demonstrate flexibility in that area.
The minimum detectable activity (MDA) for the actinide isotopes by alpha spectrometry were calculated according to equations prescribed by Currie [14]:
MDA = [3 + 4.65√B]/(CT × R × W × Eff × 0.060).
where B is the total background counts, = BKG (rate) × BKG count time; CT is sample count time (min); R is chemical recovery; W is sample aliquot (g or l); Eff is detector efficiency; 0.060 is conversion from dpm to mBq
In low-level counting, where a zero background count is common, the constant 3 is used to prevent an excessively high false positive rate.
The MDA for the alpha spectrometry results can be adjusted as needed, depending on the sample aliquot and count time. This method provides a typical MDA of ~1.5 mBq g−1 for an 8 h count time for 1 g sample, 0.15 mBq g−1 for a 10 g sample. Longer count times can be used to lower MDA levels as needed. For water samples, MDA levels are also dependent on the aliquot taken and count time. For a 150 ml sample aliquot and 16 h count time, for example, the MDA is ~5 mBq l−1.
Figure 5 shows an example of the spectra of a concrete sample. The key thing to notice is that the peak resolution is very good (typically 30–50 keV), much better than the peak resolution reported when using the barium seeding suspension approach. Because the isopropanol reduces solubility, only 50 μg of barium was added, resulting in very good peak resolution.

Alpha spectra for 226Ra sample
New resin cartridges were used for each analysis to minimize any chance of cross-contamination of samples or unexpected degradation of performance, which can occur over time and may be different than the anticipated reuse rate depending on real world sample matrix variation. Some laboratories, however, have had success reusing resins. It is anticipated that Sr Resin, used when barium removal is needed, can be reused after rinsing the resin cartridges with water.
The initial sample ashing step for 5 g vegetation aliquots takes about 2 h for a batch of ten samples. The rapid fusion method plus precipitation steps take about 1.5 h, followed by column separation steps that take about 4–5 h to complete (depending on flow rates used). Samples may be counted by alpha spectrometry for 4–16 h in an emergency. It is also possible to apply ICP-MS measurement technology if desired, since isobaric interferences and isobaric polyatomic interferences such as 88Sr138Ba have been removed using Sr Resin.
The method uses much less cation resin and rinse volumes than other published methods, and the combination of column separations using small particle size resin and vacuum flow is rapid and effective. The use of 225Ra (217At) to determine chemical yield worked very well and the removal of native barium eliminated the need to minimize sample aliquots and/or characterize the samples for native barium. It also eliminated the need for an additional count using a different analysis, such as 133Ba by gamma spectrometry.
The sodium hydroxide fusion was found to be rapid and rugged, and the calcium carbonate precipitation steps were effective in the preconcentration of 226Ra from the alkaline fusion matrix. The amounts of Ca were adjusted to obtain a 100–150 mg total for optimal yields, taking into account the ~100 mg per gram native Ca content in concrete and brick samples, for example. It should be noted that when reagent blanks (empty crucibles) are analyzed along with solid samples where a blank sample matrix is not available that calcium should be added simulate the approximate the Ca in the sample matrix, such as 100–150 mg Ca for concrete, brick or soil.
Conclusions
A new rapid method to determine 226Ra in environmental samples has been developed and tested for vegetation, soil, concrete, brick, air filter, and water samples that allows the separation of 226Ra with high chemical yields and effective removal of interferences. 225Ra (via alpha counting of 217At progeny) provides an excellent measurement of chemical yield that offers some advantages over 133Ba. The barium microprecipitation method in the presence of isopropanol was found to provide excellent alpha peak resolution. Since Sr/Ba are removed in this method, ICP-MS may also be used if desired.
References
Larivière D, Cumming T, Kiser S, Li C, Cornett R (2008) Automated flow injection system using extraction chromatography for the determination of plutonium in urine by inductively coupled plasma mass spectrometry. J Anal At Spectrom 23:352
Stricklin DL, Tjarnhage A, Nygren U (2002) Application of low energy gamma-spectrometry in rapid actinide analysis for emergency preparedness. J Radioanal Nucl Chem 251(1):69
Maxwell S, Culligan B, Noyes G (2010) Rapid separation method for actinides and radiostrontium in vegetation samples. J Radioanal Nucl Chem 286(1):273–282
Wardaszko T, Grzybowska D, Nidecka J (1986) 222Rn and 226Ra in fresh waters: measurement method and results. Nucl Instrum Methods Phys Res B 17(5–6):530–534
Escobar VG, Tome F, Lozano J, Sanchez A (1996) Determination of 222Ra and 226Ra in aqueous samples using a low level liquid scintillation counter. Appl Radiat Isot 47(9/10):861–867
Benkhedda K, Larivière D, Scott S, Evans D (2005) Hyphenation of flow injection on-line preconcentration and ICP-MS for the rapid determination of 226Ra in natural waters. J Anal At Spectrom 20:523–528
Chabaux F, Othman B, Birck JL (1994) A new Ra–Ba chromatographic separation and its application to Ra mass-spectrometric measurement in volcanic rocks. Chem Geol 114(3–4):191–197
Larivière D, Brownell DK, Epov VN, Cornett RJ, Evans RD (2007) Determination of 226Ra in sediments by ICP-MS: a comparative study of three sample preparation approaches. J Radioanal Nucl Chem 273(2):337–344
Crespo MT (2000) On the determination of 226Ra in environmental and geological samples by alpha-spectrometry using 225Ra as yield tracer. Appl Radiat Isot 53(1–2):109–114
Maxwell SL (2006) Rapid method for 226Ra and 228Ra analysis in water samples. J Radioanal Nucl Chem 270(3):651–655
Burnett WC (2004) Radium-228 determination of natural waters via concentration on manganese dioxide and separation using Diphonix ion exchange resin. Appl Radiat Isot 61:1173
Burnett WC, Moon DS, Nour S, Horwitz P, Bond A (2003) Preconcentration of radium isotopes from natural waters using MnO2 resin. Appl Radiat Isot 59:255–262
Sharabia G, Lazarb B, Kolodny Y, Teplyakov N, Halicz L (2010) High precision determination of 228Ra and 228Ra/226Ra isotope ratio in natural waters by MC-ICPMS. Int J Mass Spectrom 294(2–3):112–115
Currie LA (1968) Limits for qualitative and quantitative determination. Anal Chem 40:586
Acknowledgment
This work was performed under the auspices of the Department of Energy, DOE Contract No. DE-AC09-96SR18500. The authors wish to acknowledge Staci Britt, Jack Herrington and Becky Chavous for their assistance with this work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Maxwell, S.L., Culligan, B.K. Rapid determination of 226Ra in environmental samples. J Radioanal Nucl Chem 293, 149–156 (2012). https://doi.org/10.1007/s10967-012-1627-z
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-012-1627-z