
Abstract
Vinyl-terminated sulfonated poly (arylene ether sulfone) (VSPAES) and sulfonated polyvinyl alcohol containing vinyl group (VSPVA) have been prepared. The VSPVA and VSPAES were crosslinked by adding AIBN as initiator. The results of FT-IR and 1H-NMR indicated the existence of sulfonic acid groups and crosslinked structure in these crosslinked membranes. The microphase structures of the membranes are analyzed by SEM. The proton conductivities of membranes were above 10 mS cm−1, which indicated that it basically meet the needs of fuel cells. The methanol permeability coefficients of CSPAES membrane is 2.8 × 10–7 cm2 s−1, remarkably lower than neat SPAES (8.5 × 10–7 cm2 s−1) and Nafion117 (14.1 × 10–7 cm2 s−1). Moreover, the proton selecticity of CSPAES membrane is close to 2 times higher than that of Nafion 117 membrane. The crosslinked membrane showed better stability than the linear SPAES membrane.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
With the development of human civilization, the demand for energy was increasing day by day, but in the process of using these non-renewable energy sources, the environment was inevitably polluted. So, the renewable energy sources were receiving more and more attention [1]. Nowadays, wind energy, geothermal energy and solar energy had been put into use, but they were affected by environmental and climatic factors, and it was difficult to maintain the continuity and stability during use [2]. Therefore, the development a kind of energy storage system, which had higher energy conversion efficiency, larger storage capacity and lower price had become the key for the development of renewable energy [3]. Fuel cells were receiving more and more attention because it could convert chemical energy into electrical energy to supply load equipment through the redox reaction of positive and negative electrodes. As fourth-generation power generation device, the fuel cell can perform efficient energy storage and conversion. It had a wide range of fuel sources and could use hydrogen, methanol, ethanol, etc. as fuel. It was expected to achieve low pollution or zero emissions [4]. Proton exchange membrane fuel cells (PEMFC) was getting more and more attentions as a renewable energy source. Among them, the direct methanol fuel cell was widely used in automobiles, home appliances, aerospace and other fields [5]. PEM was one of the key materials of fuel cells used to conduct protons from anodic part to cathodic part as well as functioning as a barrier for the fuel cross-over, and its comprehensive performance will determine the performance and service life of fuel cells [6]. PEM was one of the key factors affecting the cost of fuel cells, which determined battery efficiency, output power and application performance [7]. The ideal PEM should had high proton conductivity, low fuel penetration and excellent chemical stability under various working conditions [8,9,10]. Nowdays, the most commonly used PEM was Dupont's Nafion perfluorosulfonic acid membrane series, which had higher proton conductivity, better mechanical strength and chemical stability than others [11, 12]. But Nafion membrane had the disadvantages of complicated preparation process, high cost, serious fuel penetration and reduced proton conductivity at higher temperatures (more than 80 ℃) and low humidity (less than 30% relative humidity), which seriously destoryed the energy density and energy output of fuel cells [13]. Therefore, the development of PEMs with excellent comprehensive performance to replace the Nafion membrane had become a hot spot in current research.
To solve the problems of perfluorosulfonic acid type PEM, such as high cost, non-fluorosulfonic acid type PEMs had been developed [14]. Among them, sulfonated aromatic polymer, which had excellent mechanical, chemical and thermal properties, was low in cost and relatively easy to prepare. Therefore the sulfonated aromatic polymer was often used as a substitute for perfluorosulfonic acid type PEM. They include: sulfonated polyphosphazenes [15], sulfonated polyimides [16, 17], sulfonated polyarylene ethers [18], phosphorylated or sulfonated polybenzimidazoles [19, 20], and the like. Shanmugam et al. [21] directly copolymerize sulfonated fluoroketone monomer with 4, 4′-biphenol to synthesize sulfonated polyether ketone. Xu et al. [22]. first synthesized a monomer with a sulfonic acid group, and then copolymerized with 4-carboxyphenylhydroquinone and bisphenol A to prepare polyarylether ketone sulfone with different degrees of sulfonation. However, these polymers exhibited lower proton conductivity than commercial membranes because their hydrophilic phases are generally small in size. Although a larger hydrophilic phase size could be formed by increasing the sulfonation degree (DS) of the polymer, thereby increasing the proton conductivity of the membrane, a high DS value will inevitably cause the polymer to easily absorb more water, leading The membrane ruptured due to the impact of stress during the operation of the fuel cell. [23]. Recently, our groups reported a method of grafting a sulfonated polyvinyl alcohol (SPVA) to a polyimide via an ester bond to prepare a grafting membrane [24]. Compared to Nafion117, the grafting membrane exhibits higher proton selectivity. Athough SPVA grafting polyimide membrane had some advantages, the ester bond was inevitably hydrolyzed during battery operation, resulting in a decrease in PEM stability. It was well known that crosslinking of polymers was one of the methods to improve the physical and chemical stability of PEM. Hui Na et al. [25] reported that a sulfonated poly(aryl ether ketone) containing double bonds was synthesized, followed by the use of dithiol as a crosslinking agent to achieve polymer crosslinking. As a result, the prepared crosslinked membrane greatly reduced water absorption and swelling. Jusung Han et al. [26] reported the preparation of a type of end-linked cross-linked SPAES. First, a thiol-terminated SPAES and a vinyl-containing polysulfone were synthesized, and then a cross-linking membrane prepared by a click chemical reaction showed improved oxidation stability compared to pure SPAES. However, it was well known that the carbon–sulfur bonds are extremely susceptible to oxidation. As a result, the oxidation stability of the membrane would be deteriorated if the membrane was used for a long time. Therefore, it was necessary to find a more resistant hydrolysis and oxidation method.
In this work, cross-linking membrane with excellent hydrolysis and oxidation resistance was attempted to prepare by free radical reaction. First, vinyl capped sulfonated poly(arylene ether sulfone) (VSPAES, the structure shown in Scheme 1) was prepared and a vinyl group was introduced into sulfonated polyvinyl alcohol (VSPVA, the structure shown in Scheme 2). Finally, a crosslinked sulfonated poly(arylene ether sulfone) membrane (CSPAES) was prepared through using the VSPAES and VSPVA through free radical reaction. The advantages of the method could be summarized as follows: 1) the crosslinked structures formed by carbon–carbon bond was much more stable than that by carbon–sulfur and ester bond, 2) forming a crosslinked network structure through free radical reaction which was simple and easy to operate, 3) SPVA synergizes with SPAES to enhance proton conduction, 4) the introduction of PVA was expected to greatly improve the alcohol resistance, because the PVA main chain contains a large amount of hydroxyl groups and was easy to combine with water molecules in the solution to effectively prevent methanol penetration [27]. The methanol penetration in direct methanol fuel cells was a bottleneck that hinders its development. In this paper, some properties of the prepared membranes, such as proton conductivity, methanol crossover, water absorption, swelling, mechanical strength, oxidation resistance, and micro-morphology, etc. were fully studied.

Synthesis of VSPAES

Synthesis of SPVA and VSPVA
To our best knowledge, VSPVA containing vinyl group and vinyl terminated VSPAES had been prepared for the first time. It could be predicted that the combination of the two structures may lead to a series of interesting results.
Experimental
Materials
Nafon117 membrane was obtained from Dupont Co (America). Dimethyl sulfoxide (DMSO), 4, 4′-difluorodiphenylsulfone (DFDPS) and 4, 4′-dihydroxybiphenyl (BP), bisphenol A (BPA), K2CO3, 4-hydroxybenzaldehyde, 3-bromopropene and deuterium reagent (DMSO‑d6 or CDCl3) were obtained from Aladin Reagents Co. Ltd (Shanghai, China). Concentrated sulfuric acid, fuming sulfuric acid with 50 wt% of sulfur trioxide (SO3), ethyl acetate, NaCl, hexanes, 1-methyl-2-pyrrolidone (NMP), azodiisobutyronitrile (AIBN), p-toluensulfonic acid and PVA 1799 (degree of hydrolysis of 99%) were purchased from Chengdu Kelong Reagents Co. Ltd (Chengdu, China). All the solvents and chemicals were used without further purification. 3, 3′-Disulfonate-4, 4′-difluorodiphenylsulfone (SDFDPS) was prepared from DFDPS [28].
Synthesis of vinyl sulfonated poly (arylene ether sulfone) (VSPAES)
First, SPAES was synthesized by the typical polycondensation (Scheme 1) using the BPA, DFDPS and SDFDPS [29]. BPA (2.2829 g, 10 mmol), DFDPS (1.5255 g, 6 mmol), SDFDPS (1.8333 g, 4 mmol), K2CO3 (2.07 g, 15 mmol), NMP (17 mL) and toluene (8.5 mL) were added to 100 mL three-neck flask. After the addition, the mixture was stirred at room temperature for 20 min to dissolve completely. Then the mixture was kept at 140 ℃ for 3–4 h to remove water from the system. The toluene was then completely removed by vacuum distillation, and the temperature of the system gradually increased to 190 ℃ and was kept at that temperature until a highly viscous solution was obtained. After the solution was cooled, excess NMP was added to reduce viscosity. The obtained solution was filtered to remove inorganic salts. The polymer was obtained by pouring the solution into acetone and washed with acetone and deionized water, respectively. Finally, the polymer was dried in 80 ℃ vacuum oven for at least 24 h. 1H-NMR (400 MHz, DMSO-d6, ppm): δ 8.25 (s, 1H), 7.9 (s, 1H), 7.80 (s, 1H), 7.27 (s, 1H), 6.9–7.1 (m, Ar–H), 1.56 (s, 1H).
The end group of SPAES was modified into vinyl group by a two-step nucleophilic substitution reactionto using BP and bromopropene as end capping agents [30]. VSPAES was synthesized as followed (Scheme 1): First, SPAES (5 g, 0.08 mmol) and NMP (45 ml) were added to a 50 ml completely dried threenecked round-bottomed flask and stirred at 60 ℃. When SPAES was completely dissolved, BP (0.06 g, 0.32 mmol) and K2CO3 (0.09 g, 0.64 mmol) were added. The reactants were refluxed under 160 ℃ for at least 12 h. After the reaction was completed, the mixture was cooled and poured into a lot of acetone. The white precipitate was filtered and washed with acetone and deionized water, respectively. Finally, the OH-SPAES was obtained by drying precipitate in 80 °C vacuum oven for at least 24 h. The VSPAES was obtained by modifing the end group of OH-SPAES using bromopropene. A 5 wt % solution of OH-SPAES (4 g, 0.064 mmol) was obtained in a flask by using NMP as the solvent. The mixture was completely dissolved, K2CO3 (0.036 g) and bromopropene (0.03 g, 0.256 mmol) were added. The mixture reacted under 70 °C for 12 h. Finally, VSPAES was obtained by precipitation polymer solution and washing the polymer solid. The structure of VSPAES could be confirmed by 1H-NMR spectra as shown in Fig. 1. The number-average molecular weight (Mn) and weight-average molecular weight (Mw) of VSPAES measured with the GPC were 52,000 and 98,000 g/mol.

1H-NMR spectrum of VSPAES in DMSO-d6
Synthesis of 4-(allyloxy)benzaldehyde
4-(Allyloxy)benzaldehyde was synthesized by a similar method as previously described [31]. A typical procedure could be described as follows: 4-hydroxybenzaldehyde (5 g, 41 mmol), K2CO3 (6.79 g, 49 mmol), bromopropene (4.25 mL, 49 mmol) and anhydrous acetone (80 mL) were added to 250 mL three-neck flask. The reactants were refluxed and the course of reaction was observed through thin layer chromatography (TLC) until complete reaction of 4-hydroxybenzaldehyde (about 6 h). The filtrate was filtered to remove K2CO3 and concentrated under reduced pressure. The crude product was extracted from ethyl acetate three times and the organic layer was dried with anhydrous MgSO4. Following removal of the solvent by rotary evaporation, the residue was pured by a silica gel column chromatography with ethyl acetate/hexanes (v/v = 1: 10) as elution. Finally, a corlerless oil was obtained in nearly 90% yield. The structure of 4-(allyloxy)benzaldehyde could be confirmed by 1H-NMR spectra (as shown in Fig. 2). 1H-NMR (400 MHz, CDCl3): d = 10 (s, 1 H), 7.81 (d, J = 8.8 Hz, 2 H), 7.02 (d, J = 8.8 Hz, 2 H), 6.03 (m, 1 H), 5.41 (dq, J = 17.2, 1.5 Hz, 1 H), 5.29 (dq, J = 12.0, 1.5 Hz, 1 H), 4.63 (dt, J = 5.2, 1.4 Hz, 2 H).

1H-NMR (above) and 13C-NMR (below) spectra of 4-(allyloxy)benzaldehyde in CDCl3
Synthesis of VSPVA
First, SPVA is synthesized by the esterification of PVA with H2SO4 as followed (Scheme 2) [32]: PVA (5 g) and deionized water (100 ml) were added into a 250 ml single round-bottomed with stirring under 90 ℃. When PVA was completely dissolved, the mixture was cooled to below 0 ℃, H2SO4 (60 g) was added dropwise into the flask. After, the mixture was heated to 50 ℃ and maintained for 5 h. The solution was cooled and slowly dripped into methanol. The white precipitate was filtered and washed with methanol thoroughly until the pH of solution become around 7. The precipitate was collected and dried under vacuum at 40 ℃ for all night.
VSPVA was synthesized by the acetal reaction of SPVA with 4-(allyloxy)benzaldehyde [24] as followed (Scheme 2): SPVA (1 g) and DMSO (20 ml) were added into a 50 ml threenecked flask with stirring under 80 ℃. When SPVA was dissolved, p-toluensulfonic acid (0.1 g) and 4-(allyloxy)benzaldehyde (0.36 ml) were successively added. The mixture was heated to 80 ℃ for 8 h. The solution was cooled and slowly dripped into methanol to isolate polymer. The pH of solution was adjusted to 7–8 with NaOH. The VSPVA was collected and washed with methanol thoroughly. Finally. VSPVA was obtained by drying under vacuum at 60 ℃ for 24 h. The Mn and Mw of VSPVA are about 71,000 and 106,000 g/mol.
Preparation of SPAES, SPAES/SPVA and CSPAES-30 membranes
The CSPAES-30 (where 30 indicated the weight percent of VSPVA compared with total weight) were prepared by solution casting and free radical reaction containing AIBN as an initiator [25]. The preparation of CSPAES-30 was described as follows: VSPAES (0.42 g), VSPVA (0.18 g), AIBN (0.018 g) and DMSO (3.4 g) were added into a penicillin bottle with stirring. The uniform solution was cast onto the clean glass plate and heated at 70–80 ℃ for 12 h. During the heating process, the crosslinked structure of CSPAES-30 was formed by the free radical reaction between the vinyl groups of VSPAES and VSPVA, respectively. SPVA/SPAES composite membrane was prepared as followed: SPAES (0.42 g), SPVA (0.18 g) and DMSO (3.4 g) were added into a penicillin bottle with stirring. The solution was cast onto the clean glass plate and dried to remove the solvent. The fabrication method of the pure SPAES membrane was the same as SPAES/SPVA membranes. All the membranes were took down from the substrate by immersing in deionized water. All the membranes were immersed in 1 M H2SO4 aqueous solution at 25 ℃ for 24 h to convert to an acid polymer and immersed in deionized water for at least 24 h before using.
Characterization and measurements
1H-NMR was tested by 400 MHz Unity INVOA 400 nuclear magnetic resonance instrument using deuterated (CDCl3 or DMSO-d6) as a solvent and tetramethylsilane (TMS) as an internal standard. FTIR was measured by Nicolet 560 FTIR spectrometer of Nicolet company in the United States. The molecular weight of SPAES was determined by Gel Permeation Chromatography (GPC), using an Agilent 1100 column, tetrahydrofuran (THF) as eluent and polystyrene standards were used for calibration. Thermogravimetry analysis (TGA) was carried out by DuPont TGA 2100 analyzer. Under nitrogen flow, the heating rate was 10 ℃/min, and the stable range was 100–800 ℃. The morphology of some membranes were determined with SEM (FEI quanta 250).
mechanical strength
The mechanical strength of the membranes were measured by the 4302 universal material testing machine of Instron company in the United Kingdom. The sample size was 4 × 1 cm2 of membrane and the tensile speed was 10 mm/min. The experimental data was the average of 3–5 samples.
Water uptake and swelling ratio
The water uptake and swelling ratio of the samples were evaluated by comparing the changes in weight and surface area [33]. The dry membranes were cut into 2 × 2 cm2 and placed into deionized water for 24 h (at 25 ℃). After quickly wiped off the excess water of samples, the changes in their weight and surface area were recorded. The water uptake and swelling ratio of samples were evaluated with the following equation:
where Wwet and Wdry are the weights of the sample in wet and dry state, respectively.
where Awet and Adry are the surface area of the wet and dry samples, respectively.
Ion Exchange Capacity (IEC)
The IEC value of samples was tested by the method of acid–base titrationtitration [22]. The dried sample was immersed in a saturated NaCl solution for 48 h to release protons from the polymer. The phenolphthalein was then used as an indicator and the solution was titrated with 0.05 M NaOH solution. NaOH solution is calibrated with potassium hydrogen phthalate before use to obtain the actual concentration. The IEC (mmol/g) was defined as the milliequivalents of sulfonic groups per gram of dry membrane. The formula for calculating IEC was shown as follows:
where: Md was the dry quality of the membrane and CNaOH and VNaOH were the molar concentration (mol/L) and volume (ml) of the NaOH solution, respectively.
Chemical stability and hydrilysis stability
The chemical stability of the samples were evaluated by the Fenton's reagent (3 wt % H2O2 aqueous solution and 4 ppm FeSO4) test under 80 ℃. The chemical stability of sample was confirmed by recording T1 and T2, which were the times when the sample began to be degradated into pieces and completely dissolved, respectively. The hydrolytic stability was tested by soaking the membrane sample in hot water at 60 ℃ for 200 h. The hydrolytic stability of the membrane was evaluated by recording the weight loss percentage of the membrane.
Proton conductivity
Proton conductivity of the sample was confirmed by alternating current (AC) impedance method [34]. The sample was placed between two silver electrodes to form a loop state. The ionic conductivity of the sample was tested with a CHI660E electrochemical workstation (Shanghai Chenhua Instruments Co., China). The AC perturbation was 5.0 mV and frequency range was 1.0 Hz to 100 kHz. All the membranes were soaked in 1 M H2SO4 for at least 24 h and rinsed thoroughly with distilled water. The proton conductivity (σ) of samples was determined by using the following equation:
where: d and S were the thickness and surface area of the samples, respectively. The R was the tested resistance of the samples.
Permeability of methanol
The permeability of methanol of the samples was tested through a glass diffusion cell. The device consisted of two reservoirs with a capacity of 100 ml. They were separated by a ion exchange membrane. At the same time, 100 mL of 2 M methanol solution and 100 mL of deionized water were added to Room A and Room B, respectively. The Room A and Room B were stirred simultaneously. Methanol concentration within Room B was monitored at different time using a gas chromatograph (Agilent 7820A). The methanol permeability of samples was confirmed by following method:
where: P was the methanol permeability cofficiency, CA and CB were the methanol concentrations of Room A and Room B, respectively. A, L and VB were the effective surface area, thickness of membrane and volume of diffusion cell, respectively. The proton selectivity of samples was confirmed by the ratio of σ and P.
Results and discussion
Synthesis of VSPAES and VSPVA
The VSPAES with DS of 40 mol % was prepared according to Scheme 1. At first, SPAES was synthesized through a polycondensation reaction. Because the molar ratio of sulfonated monomer to nonsulfonated monomer was designed to be 4:6, the DS of SPAES was deemed to be 40 mol %. The Mn and Mw of SPAES measured from the GPC were 62,000 and 116,000 g mol−1, respectively. The terminal group of the SPAES was transfered to the vinyl group through a two-step termination process using BP and bromopropene as end-capped reagents. At first, the end group of the SPAES was transfered to the hydroxyl group by using BP as end-capped reagent. After that, the end group of the OH-SPAES was transfered to the vinyl group by using bromopropene as end-capped reagent. 1H-NMR spectra of VSPAES was shown in Fig. 1. On the 1H-NMR spectrum of VSPAES, four small peaks at 7.60, 7.40, 7.05 and 6.80 ppm were attributed to the hydrogen atoms on the BP units which were located at the end of the polymer chain [35]. The three new peaks at 4.5, 5.2 and 6.0 ppm were attributed to the hydrogen atoms of p, r and q, respectively, which indicates that a vinyl group was introduced into SPAES [36].
The synthesis of VSPVA was prepared by a simple three-step process. Frist, PVA is sulfonated by H2SO4 as a sulfonating reagent to obtain SPVA. Second, p-hydroxybenzaldehyde and bromopropene reactions with acetone as solvent and the course of reaction was observed through TLC until complete reaction of 4-hydroxybenzaldehyde. The raw product was puried through a silica gel column. The chemical structures of 4-(allyloxy)benzaldehyde was confirmed by 1H NMR and 13C-NMR spectra (Fig. 2). All the characteristic peaks of hydrogen and carbon atoms are perfectly assigned, indicating that 4-(allyloxy)benzaldehyde is successfully synthesized. Finally, 4-(allyloxy)benzaldehyde and SPVA were subjected to acetalization to prepare VSPVA. 1H-NMR spectrum confirmed that the acetal rate of 4-(allyloxy)benzaldehyde was controlled at 6% through comparing with the areas of peak at 6.2 ppm assigned to the proton of 3 and 1.5 ppm assigned to the proton of 11, respectively (Fig. 3). Above results indicated the successful preparation of VSPAES and VSPVA.

1H-NMR spectrum of VSPVA in DMSO-d6
Preparation of CSPAES-30 and FTIR analysis
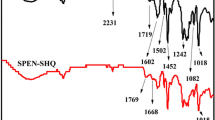
The CSPAES-30 membrane were prepared through solution casting and heating process by using the DMSO as solvent of the VSPAES and VSPVA and a bit of thermal initiator (AIBN). The FTIR spectra of VSPAES, PVA, VSPVA and CSPAES-30 are presented in Fig. 4. For VSPVA, the peaks centered at 3330 cm−1 and 2940 cm−1 were assigned to the -OH stretching vibration and the CH2 symmetrical stretching vibration, respectively. The new peaks at 1515 and 1586 cm−1 were assigned to the vibration of benzene ring of VSPVA. Compared to pristine PVA, new two absorption bands 1185 cm−1 and 1020 cm−1 attributed to the stretching vibration of O = S = O of sulfonic acid groups [37]. This shown that PVA was successfully sulfonated. The new peak at 917 cm−1 was attributed to the vibration of the allyl groups showed that C = C bond was induced into VSPVA and VSPAES [38]. After free radical reaction, this peak at 917 cm−1 was almost disappeared as in the FT-IR spectrum of CSPAES-30. The above results indicated that CSPAES-30 was successfully prepared.

FTIR spectra of PVA, VPVA, VSPAES and CSPAES-30
Oxidative and hydrolytic stability
It was important for PEMs to possess excellent chemial stability [39]. So, Fenton reagent (contained some free radicals such as ·OH, ·OOH) test was conducted to evaluate the oxidative stability of the samples. Figure 5a showed that the chemical stability of the SPAES, SPAES/SPVA and CSPAES-30 membranes determined by recording the T1 and T2. The T1 of CSPAES-30, SPAES/SPVA and SPAES membrane was 170 min, 110 min and 80 min, respectively. The T2 of CSPAES-30, SPAES/SPVA and SPAES membrane was 230 min, 150 min and 80 min, respectively. Figure 5a clearly shown that the SPAES/SPVA and CSPES-30 membranes were much more stable than pure SPAES membrane under the same condition. Compared to SPAES membrane, oxidative stability of SPAES/SPVA membrane increases because the introduction of SPVA results in more hydrogen bonding interactions within the molecules. Compared to SPAES/SPVA membrane, oxidative stability of CSPAES-30 membrane increases because the form of crosslinked structure [40]. The results indicated that the crosslinked membrane prepared by the radical initiated reaction greatly improves the oxidation stability.

(a) Oxidative stability and (b) mechanical properties of SPAES, SPAES/SPVA and CSPAES-30 membranes
The hydrolytic stability of the membranes was tested by immersing the membrane in hot water at 60 ℃ for 200 h to record the weight loss percentage as shown in Table 1. The weight loss percentage of SPAES, SPAES/SPVA and CSPAES-30 is 0.85%, 1.5% and 1.2%, respectively. The introduction of SPVA led to a slight decrease in the hydrolytic stability of SPAES and crosslinking can improve the hydrolysis stability of SPVA.
Mechanical property and thermal stabilities
The mechanical properties of the samples were shown in Fig. 5b and Table 1. The tensile strength and elongation at break values of Nafion 117 membrane were 41 MPa and 200%, respectively. It was clear that all membranes had higher tensile strength than Nafion117 membrane. The pure SPAES membrane exhibited tensile strength and elongation at break values of 49 MPa and 20%. Compared with SPAES, SPAES/SVA exhibited higher tensile strength and elongation at break, reaching 75 MPa and 91%, respectively. This was due to the introduction of SPVA resulting in more hydrogen bonding interactions between the polymer chains. Compared with SPAES and SPAES/SPVA, CSPAES-30 exhibited highest tensile strength and lower elongation at break, 75 MPa and 91% because CSPAES-30 membranes had the structure of cross-linking, which can inhibit the movement of the polymer chain.
Thermal stability of the samples in the acid form was measured by TGA under N2 atmosphere. The TGA curves of SPAES, SPAES/SPVA and CSPAES-30 membranes were shown in Fig. 6. It could be seen that all the samples displayed three weight loss stages. Clearly, all the samples exhibit mass loss about 100 ℃, which could be assigned to the loss of moisture and solvent of membrane. For the SPAES membrane, the second stage (280–400 ℃) shown the weight loss of the sulfonic acid groups. The third stage (400–500 ℃) shown the decomposition of the backbone of SPAES. The SPAES/SPVA and CSPAES-30 membranes shown a similar behavior, the evaporation of moisture and solvent at about 100 ℃. The second stage (220–380 ℃) shown the decomposition of the hydroxyl groups of SPVA and sulfonic acid groups of SPVA and SPAES. The third step (400–500 ℃) shown the decomposition of the backbone of SPVA and SPAES. It is worth noting that the mass residual rate of CSPAES-30 at 800 ℃ is significantly higher than that of SPAES and SPAES/SPVA, which is caused by the introduction of benzene ring in VSPVA. The result of TGA indicated better thermal stability of the crosslinked membrane than SPAES/SPVA membrane.

TGA curves of SPAES, SPAES/SPVA and CSPAES-30 membranes
IEC, water uptake and swelling ratio
IEC value was closely related to the number of protons that could be exchanged in the proton exchange membrane. For SPAES membrane, the total IEC of the membrane resulted from the sulfonic groups of main chain of SPAES. But for SPAES/SPVA and CSPAES-30, the IEC of the samples resulted from the sulfonic groups of main chain of SPAES and SPVA. The IEC value of the SPAES, SPAES/SPVA, CSPAES-30 and Nafion117 membranes was shown in Table 2. The IEC values of CSPAES-30 and SPAES/SPVA membranes showed decreasing trend compare with SPAES membrane. The highest IEC was assigned to SPAES membrane (1.4 mmol/g). The introduction of SPVA resulted in a decrease in IEC because the main chain of SPVA had a much lower sulfonic acid group content than SPAES, resulting in less IEC. Compared with SPAES/SPVA, the IEC value of CSPAES-30 was slightly reduced because the presence of crosslinked structure makes the sulfonic acid group difficult to move.
Water uptake and swelling ratio will affect the dimensional stability of the membrane in the working state. Water molecules were used for facilitating acid dissociation, thus transporting proton. However, excessive water absorption and swelling could cause the dimensional stability and mechanical property of the membrane to decrease, which was detrimental to the operation of the PEM. Therefore, proper water absorption was very important for the stability of the membrane. As shown in Table 2, all membranes exhibited moderate water uptake with the largest value being less than 35%, indicating their good dimensional stability. With an introduction of SPVA, the water uptake dramatically increased for all the membranes. This was because the PVA backbone contains a large amount of hydroxyl groups which were hydrophilic groups. After soaking in deionized water for 24 h at room temperature, the water absorption and swelling ratio of SPAES/SPVA membrane reached the maximum, 33% and 12.8% respectively. This was mainly because hydrophilic sulfonic acid groups and hydroxyl groups of SPVA. Compared to Nafion117 membrane, all membranes significantly increased water absorption, which would facilitate the conduction of protons within the membrane. It was worth noting that CSPAES-30 membrane had the lowest water absorption and swelling ratio, 25.8% and 10.2%, respectively, even lower than SPAES. Crosslink of SPVA and SPAES greatly inhibited the movement and swelling of the polymer chain. Obviously, Crosslinking SPVA and SPAES did not obviously reduced proton conduction while increasing the dimensional stability of the membrane.
Morphological study
The properties of membrane were related to their microstructures. The microstructure and the dispersion of microphase within membrane were evaluated by SEM. The SEM photomicrographs of SPAES, SPAES/SPVA and CSPAES-30 membranes were shown in Fig. 7. Clearly, all of the samples shown a homogenous structure, which indicated that no phase separation occurs in both SPAES and SPVA. Predictably, the cross section of neat SPAES was smooth, as shown in Fig. 7a. However, the cross section of SPAES/SPVA and CSPAES-30 membranes appear rougher than pure SPAES membrane because of the introduction of SPVA, as shown in Fig. 7b, c. This results from the decreased crystallinity of SPVA lead to the toughness fracture in the membrane. It was noted that the cross section of CSPAES-30 appears to be more rough than SPAES/SPVA membrane, which was caused by the formation of a crosslinked structure between SPAES and SPVA [26]. The results of SEM could fully demonstrate that obvious phase separation did not occur in both SPAES/SPVA and CSPAES-30 membranes.

SEM photomicrographs of the cross section of (a) SPAES, (b) SPAES/SPVA and (c) CSPAES-30 membranes
Proton conductivity, methanol permeability and protn selecitivity
The proton conductivities (σ), permeability of methanol (P) and proton selectivity (S) of the SPAES, SPAES/SPVA, CSPAES-30 and Nafion117 membranes were shown in Table 3 at 25 °C and 100% RH conditions, respectively. In the study of methanol fuel cells, the alcohol resistance of PEMs was one of the most important parametere. Methanol infiltration could cause a certain energy loss caused by self-discharge of the battery and reduce the energy efficiency of the battery. It was well known that methanol penetration of Nafion117 membrane was severe (14.1 × 10–7 cm2 s−1). Obviously, all membranes showed lower methanol permeability than Nafion 117 membrane. For example, SPAES membrane with the highest methanol permeability coefficients was 8.5 × 10–7 cm2 s−1, which was lower than Nafion membrane. Compared with SPAES membrane, the SPAES/SPVA and CSPAES-30 membranes had lower methanol permeability, which were 5.1 × 10–7 cm2 s−1 and 2.8 × 10–7 cm2 s−1, respectively. Clearly, the introduction of SPVA hindered the methanol crossover prominently. This was because the PVA main chain contains a lot of hydroxyl groups which was easy to combine with water molecules in the solution to effectively prevent methanol penetration. It was noted that CSPAES-30 membrane had lowest methanol permeability coefficients (2.8 × 10–7 cm2 s−1), which might be attributed to the lower water uptake and the existence of crosslinking network.
The proton conductivities of membranes were above 10 mS cm−1, which indicated that it basically meet the needs of fuel cells [22]. Compared with SPAES membrane, the introduction of SPVA led to a decrease in the proton conduction of the membrane, which was due to the number of sulfonic acid groups in the SPVA backbone was much less than SPAES. Compared with SPAES/SPVA membrane, the proton conduction of CSPAES-30 membrane decreased due to the presence of the crosslinked structure, which limited the movement of the sulfonic acid group.
PEMs needed to strike a balance between proton conductivity and methanol permeability to effectively improve battery performance. The relative selectivity of the SPAES, SPAES/SPVA, CSPAES-30 and Nafion117 membranes were shown in Fig. 8. The proton conductivity of SPAES, SPAES/SPVA and CSPAES-30 membranes was slightly lower than Nafion117 membrane, but they showed much lower methanol permeability than Nafion117 membrane. Therefore, their proton selectivity was higher than Nafion117 membrane, especially, proton selectivity of CSPAES-30 membrane (4.6 × 104 S s cm−3) was almost twice higher than that of Nafion117 membrane (2.4 × 104 S s cm−3). It was confirmed that end group cross-linking SPAES and SPVA was a efficient method to increase the proton selectivity.

The relative selectivity of the SPAES, SPAES/SPVA, CSPAES-30 and Nafion117 membranes
Conclusions
For the first time, VSPVA containing vinyl group and vinyl terminated VSPAES had been prepared. The VSPVA and VSPAES were crosslinked by adding AIBN as initiator. The crosslinked membrane CSPAES significantly improved physicochemical stability. The proton conductivities of membranes were above 10 mS cm−1, which indicated that it basically meet the needs of fuel cells. All membranes displayed a lower methanol permeability than Nafion 117 membrane. Moreover, the SPAES/SPVA and CSPAES membranes exhibited better mechanical strength and higher proton selectivity than neat SPAES and Nafion 117 membranes. Especially, the proton selecticity of CSPAES-30 was close to 2 times higher than that of Nafion 117 membrane. We strongly believed that crosslinked membrane CSPAES was expected to be promising candidate PEM applied in direct methanol fuel cell.
References
Rao V, Hariyanto CC, Stimming U (2007) Investigation of the Ethanol Electro-Oxidation in Alkaline Membrane Electrode Assembly by Differential Electrochemical Mass Spectrometry. Fuel Cells 7(5):417–423
Chiari L, Zecca A (2011) Constraints of fossil fuels depletion on global warming projections. Energy Policy 39(9):5026–5034
Joerissen L, Garche J, Fabjan C, Tomazic G (2004) Possible use of vanadium redox-flow batteries for energy storage in small grids and stand-alone photovoltaic systems. J Power Sources 127(1–2):98–104
Meng H (2008) Numerical studies of cold-start phenomenon in PEM fuel cells. Electrochim Acta 53(22):6521–6529
Beydaghi H, Javanbakht M, Salarizadeh P, Bagheri A, Amoozadeh A (2017) Novel proton exchange membrane nanocomposites based on sulfonated tungsten trioxide for application in direct methanol fuel cells. Polymer 119:253–262
Neelakandan S, Kanagaraj P, Sabarathinam RM, Nagendran A (2015) Polypyrrole layered SPEES/TPA proton exchange membrane for direct methanol fuel cells. Appl Surf Sci 359:272–279
Shin DW, Lee SY, Kang NR, Lee KH, Guiver MD, Lee YM (2013) Durable Sulfonated Poly(arylene sulfide sulfone nitrile)s Containing Naphthalene Units for Direct Methanol Fuel Cells (DMFCs). Macromolecules 46(9):3452–3460
Mondal AN, He Y, Ge L, Wu L, Emmanuel K, Hossain MM, Xu T (2017) Preparation and characterization of click-driven N-vinylcarbazole-based anion exchange membranes with improved water uptake for fuel cells. RSC Adv 7(47):29794–29805
Sutradhar SC, Jang H, Banik N, Yoo J, Ryu T, Yang H, Yoon S, Kim W (2017) Synthesis and characterization of proton exchange poly (phenylenebenzophenone)s membranes grafted with propane sulfonic acid on pendant phenyl groups. Int J Hydrog Energy 42(17):12749–12758
Wang G, Guiver MD (2017) Proton exchange membranes derived from sulfonated polybenzothiazoles containing naphthalene units. J Membr Sci 542:159–167
Pang J, Feng S, Yu Y, Zhang H, Jiang Z (2014) Poly(aryl ether ketone) containing flexible tetra-sulfonated side chains as proton exchange membranes. Polym Chem 5(4):1477–1486
Lai AN, Zhuo YZ, Lin CX, Zhang QG, Zhu AM, Ye ML, Liu QL (2016) Side-chain-type phenolphthalein-based poly(arylene ether sulfone nitrile)s anion exchange membrane for fuel cells. J Membr Sci 502:94–105
Rozière J, Jones DJ (2003) Non-Fluorinated Polymer Materials for Proton Exchange Membrane Fuel Cells. Ann Rev Mater Res 33(1):503–555
Kludský M, Vopička O, Matějka P, Hovorka Š, Friess K (2018) Nafion® modified with primary amines: chemical structure, sorption properties and pervaporative separation of methanol-dimethyl carbonate mixtures. Eur Polym J 99:268–276
Dong Y, Xu H, Fu F, Zhu C (2016) Preparation and evaluation of crosslinked sulfonated polyphosphazene with poly(aryloxy cyclotriphosphazene) for proton exchange membrane. J Energy Chem 25(3):472–480
Kowsari E, Ansari V, Moradi A, Zare A, Mortezaei M (2015) Poly(amide-imide) bearing imidazole groups/sulfonated polyimide blends for low humidity and medium temperature proton exchange membranes. J Polym Res 22:77
Rehman W, Liaqat K, Fazil S, Saeed S, Waseem M, Shakeel M, Mir S, Bibi I, Guo C-Y (2019) Chemically tethered functionalized graphene oxide based novel sulfonated polyimide composite for polymer electrolyte membrane. J Polym Res 26(3):82
Chen L, Pu Z, Yang J, Yang X, Liu X (2013) Synthesis and properties of sulfonated polyarylene ether nitrile copolymers for PEM with high thermal stability. J Polym Res 20(1):45
Wang K, Yuan X, Zhan M (2017) Comparison between microwave and thermal curing of a polyimide adhesive end-caped with phenylethynyl groups. Int J Adhes Adhes 74:28–34
Singha S, Jana T, Modestra JA, Naresh Kumar A, Mohan SV (2016) Highly efficient sulfonated polybenzimidazole as a proton exchange membrane for microbial fuel cells. J Power Sources 317:143–152
Oh K, Ketpang K, Kim H, Shanmugam S (2016) Synthesis of sulfonated poly(arylene ether ketone) block copolymers for proton exchange membrane fuel cells. J Membr Sci 507:135–142
Xu J, Ni H, Wang S, Wang Z, Zhang H (2015) Direct polymerization of a novel sulfonated poly(arylene ether ketone sulfone)/sulfonated poly(vinylalcohol) crosslinked membrane for direct methanol fuel cell applications. J Membr Sci 492:505–517
Guan R, Gong C, Lu D, Zou H, Lu W (2005) Development and characterization of homogeneous membranes prepared from sulfonated poly(phenylene oxide). J Appl Polym Sci 98(3):1244–1250
Zhao H, Yang C, Li N, Yin J, Feng Y, Liu Y, Li J, Li Y, Yue D, Zhu C, Liu X (2019) Electrical and mechanical properties of polyimide composite films reinforced by ultralong titanate nanotubes. Surf Coat Technol 360:13–19
Jiang H, Guo X, Zhang G, Ni J, Zhao C, Liu Z, Zhang L, Li M, Xu S, Na H (2013) Cross-linked high conductive membranes based on water soluble ionomer for high performance proton exchange membrane fuel cells. J Power Sources 241:529–535
Han J, Kim K, Kim J, Kim S, Choi S-W, Lee H, Kim J-j, Kim T-H, Sung Y-E, Lee J-C (2019) Cross-linked highly sulfonated poly(arylene ether sulfone) membranes prepared by in-situ casting and thiol-ene click reaction for fuel cell application. J Membr Sci 579:70–78
Maiti J, Kakati N, Lee SH, Jee SH, Viswanathan B, Yoon YS (2012) Where do poly(vinyl alcohol) based membranes stand in relation to Nafion® for direct methanol fuel cell applications? J Power Sources 216:48–66
Park J-y, Kim T-H, Kim HJ, Choi J-H, Hong YT (2012) Crosslinked sulfonated poly(arylene ether sulfone) membranes for fuel cell application. Int J Hydrog Energy 37(3):2603–2613
Chandra Sutradhar S, Ahmed F, Ryu T, Yoon S, Lee S, Rahman MM, Kim J, Lee Y, Kim W, Jin Y (2019) A novel synthesis approach to partially fluorinated sulfonimide based poly (arylene ether sulfone) s for proton exchange membrane. Int J Hydrog Energy 44(22):11321–11331
Zhang X, Tian J, Ren Z, Shi W, Zhang Z, Xu Y, Gao S, Cui F (2016) High performance thin-film composite (TFC) forward osmosis (FO) membrane fabricated on novel hydrophilic disulfonated poly(arylene ether sulfone) multiblock copolymer/polysulfone substrate. J Membr Sci 520:529–539
Liu J, Qiu J, Wang M, Wang L, Su L, Gao J, Gu Q, Xu J, Huang SL, Gu LQ, Huang ZS (1840) Li D (2014) Synthesis and characterization of 1H-phenanthro[9,10-d]imidazole derivatives as multifunctional agents for treatment of Alzheimer’s disease. Biochim Biophys Acta 9:2886–2903
Li HQ, Liu XJ, Wang H, Yang H, Wang Z, He J (2020) Proton exchange membranes with cross-linked interpenetrating network of sulfonated polyvinyl alcohol and poly(2-acrylamido-2-methyl-1-propanesulfonic acid): Excellent relative selectivity. J Membr Sci 595:117511
Ko T, Kim K, Lim M-Y, Nam SY, Kim T-H, Kim S-K, Lee J-C (2015) Sulfonated poly(arylene ether sulfone) composite membranes having poly(2,5-benzimidazole)-grafted graphene oxide for fuel cell applications. J Mater Chem A 3(41):20595–20606
Zhiwei W, Hao Z, Qiang C, Sumei Z, Feng Y, Jian K, Jinyao C, Ya C, Ming X (2019) Preparation and characterization of PVA proton exchange membranes containing phosphonic acid groups for direct methanol fuel cell applications. J Polym Res 26:200
Lee H-S, Roy A, Lane O, Dunn S, McGrath JE (2008) Hydrophilic–hydrophobic multiblock copolymers based on poly(arylene ether sulfone) via low-temperature coupling reactions for proton exchange membrane fuel cells. Polymer 49(3):715–723
Alami M, L’Hermite N, Peyrat J-F, Hildgen P (2008) Brion J-D (2008) Synthesis of Poly(aryl propargyl ether) (PAPE) Stars and Evaluation of Their Cytotoxic Properties. Synthesis 07:1049–1060
Zhang Y, Wan Y, Pan G, Shi H, Yan H, Xu J, Guo M, Wang Z, Liu Y (2017) Surface modification of polyamide reverse osmosis membrane with sulfonated polyvinyl alcohol for antifouling. Appl Surf Sci 419:177–187
Chen X, Lü H, Lin Q, Zhang X, Chen D, Zheng Y (2018) Partially fluorinated poly(arylene ether)s bearing long alkyl sulfonate side chains for stable and highly conductive proton exchange membranes. J Membr Sci 549:12–22
Xu J, Ma L, Han H, Ni H, Wang Z, Zhang H (2014) Synthesis and properties of a novel sulfonated poly(arylene ether ketone sulfone) membrane with a high β-value for direct methanol fuel cell applications. Electrochim Acta 146:688–696
Cheng H, Xu J, Ma L, Xu L, Liu B, Wang Z, Zhang H (2014) Preparation and characterization of sulfonated poly(arylene ether ketone) copolymers with pendant sulfoalkyl groups as proton exchange membranes. J Power Sources 260:307–316
Acknowledgements
This work was supported by the National Natural Science Foundation of China [grant number 51173115].
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Yuan, C., Wang, Y. Synthesis and characterization of a crosslinked membrane based on sulfonated poly(aryl ether sulfone) and sulfonated polyvinyl alcohol applied in direct methanol fuel cells. J Polym Res 27, 329 (2020). https://doi.org/10.1007/s10965-020-02305-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-020-02305-z