
Abstract
To improve the impact property of Nylon 6 at temperature below 0°C, Nylon 6 was toughened by the blending of Pebax® Rnew (PEBA) and compatibilized with poly(styrene-co-maleic anhydride) (SMA). The effect of the content of soft segment of PEBA, polytetramethylene oxide (PTMO), on the toughening properties was investigated. The impact properties of the resulting blends were measured at 23°C and − 20°C. With the increase of PTMO content, the impact strength increased, whereas the tensile and flexural properties decreased. The thermal stability of these blends was not affected significantly. Rheological properties revealed that these blends exhibited predominantly viscous behavior at 240°C. Furthermore, higher PTMO content led to higher storage modulus (G’), lower loss modulus (G”) and complex viscosity (η*). The higher PTMO content of PEBA also improved the toughness more. The DSC results showed that the glass transition temperature (Tg) of Nylon 6 decreased with the increase of PTMO content. The crystallization temperature and the rate of crystallization of the blends were not much affected by the PTMO content, yet the relative amounts of α and γ crystals formed were affected by the type of PEBA added. Based on the Avrami model, the non-isothermal crystallization of Nylon 6 in the blends was a diffusion-controlled crystallization.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
As an important engineering plastic, Nylon 6 is a semi-crystalline thermoplastic exhibiting high mechanical strength, resistance to heat and chemicals, low coefficient of friction and wearability. However, at temperature below 0°C, Nylon 6 becomes brittle and notch-sensitive, thus limiting its application at low temperature. To overcome this limitation, Nylon 6 is often toughened through melt-blending with other polymers, as reported in the literature [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28].
Melt blending is a common process for modifying the properties of polymers. Usually, two or more polymers are compounded with a twin screw extruder to achieve the good melt blending. This modification process is fast and thus is widely employed in the industry, and is frequently discussed in related textbooks [29, 30]. Toughened Nylon 6 can be used in applications such as office furniture, sport equipment, luggage, strollers and automobile parts, etc.
In the literature, toughening agents for Nylon 6 are usually chosen from soft thermoplastic elastomers including polyethylene-octene elastomer (POE) [1,2,3,4,5,6,7], ethylene-propylene diene copolymer (EPDM) [8,9,10,11,12,13], ethylene-propylene rubber (EPR) [6, 14,15,16,17], natural rubber (NR) [18, 19], fluoroelastomer [20], styrene ethylenebutylene styrene rubber (SEBS) [9, 14, 15, 21,22,23,24], ethylene vinyl acetate (EVA) [25,26,27]. Because Nylon 6 is a polar material while those elastomers are non-polar, maleic anhydride is often employed as the compatibilizer to improve the toughening performance. In addition, the toughness of Nylon 6 can also be enhanced by a core-shell structured polyacrylic modifier [28].
Pebax® Rnew resins are bio-based thermoplastic polyamide elastomers manufactured by Arkema, France. The Pebax® Rnew is the poly(ether-block-amide) (PEBA) copolymer consisted of flexible polytetramethylene oxide (PTMO) and rigid polyamide 11 (PA11) constituents, and derived from renewable castor beans [31, 32]. The flexibility of PEBA depends on the ratio of PTMO to PA11. Higher PTMO content makes PEBA more flexible and less rigid. On the other hand, higher PA11 content makes PEBA more rigid and less flexible, due to the intermolecular hydrogen bonding [33].
In our previous study [34], it was shown that by blending Pebax® Rnew 40R53 as the toughening agent and using poly(styrene-co-maleic anhydride) (SMA) as the compatibilizer, the toughness of Nylon 6 was effectively improved. In this work, the effect of flexibility of PEBA on the properties of Nylon 6 was studied and the mechanical, impact, thermal, rheological and crystalline behaviors of different blends were characterized.
Experimental
Materials
Table 1 summarizes the properties of the materials used in this study. Neat Nylon 6 (Zisamide TP4208, ZigSheng, Taiwan) has a relative viscosity (RV) of 2.4 (−Mw = 20.5kD with PDI 1.38) and the density of amino end-groups was 44 μeq g−1. Polyether block amides (PEBA) (Pebax® Rnew series) were obtained from Arkema, France. The compatibilizer, SMA, was purchased from Sigma-Aldrich, USA, with a mole ratio of styrene to maleic anhydride of 1.3 to 1.
Blending
Table 3 lists the formulation of the blends. Before processing, all materials were dried for 24 h at 85°C in a vacuum oven to ensure removal of water. Blends were prepared in a co-rotating twin screw extruder (L/D = 42, L = 1.05 m) (ZE 25A × 42D, Thermo-Haake, Germany) operating at a screw speed of 250 rpm and a temperature profile of 205°C, 220°C, 225°C, 230°C, 235°C, 240°C, 245°C, 250°C, and 255°C (die). Table 4 lists the details of the processing conditions.
The molecular weight of Nylon 6 in Table 3 was estimated from the relative viscosity (RV), which was measured by dissolving the sample in 96% H2SO4 to a concentration of 0.01 g/mL at 25°C according to ISO 307. The resultant RV was then converted to Mw based on the datasheet from BASF.
Mechanical properties
The extruded pellets were injection molded into standard 0.318 cm thick Izod (ASTM D256) and tensile (ASTM D638 with type I) bars using an injection molding machine (VS-100 M, Victor Taichung Machinery Works Co., Taiwan). In addition, the flexural specimen bars were prepared as specified by ASTM D790. The barrel temperature was set up at 255°C (nozzle) and the molding temperature at 80°C. An injection pressure of 8 MPa and a holding pressure of 3.5 MPa were applied. The details of the processing conditions were described in Table 4.
Notched Izod impact strength was determined using an impact tester (Cs137–25, Atlas Co., Germany). The tensile testing was performed using an Instron tester (type 5567) at a crosshead speed of 50 mm/min for strength measurements and 5 mm/min for modulus measurements. In addition, flexural testing was performed also using an Instron tester (type 5567) at a crosshead speed of 14 mm/min for strength measurements and 1.4 mm/min for modulus measurements. The fractured surfaces of Nylon 6/SMA/PEBA blends were examined using a scanning electron microscope (JSM-6150, JEOL, Japan) at an accelerating voltage of 10 kV.
Thermogravimetric analyses
The thermal stabilities of Nylon 6/SMA/PEBA blends were analyzed using a thermogravimetric analyzer (TGA) (Q-500, TA Instrument, USA). The measurements were performed in a temperature range of 30 to 600°C at a heating rate of 10°C/min under a nitrogen atmosphere(40 mL/min). The mass of the samples was about 10 mg.
Rheological characterization
Rheological measurements of the Nylon 6/SMA/PEBA blends were conducted with a rheometer (HR-2, TA Instrument, USA). Frequency sweep for the Nylon 6/SMA/PEBA samples was carried out using 25 mm plate-plate geometry. The gap distance between the parallel plates was 0.8 mm for all tests. A strain sweep test was initially conducted to determine the linear viscoelastic region of the materials. The amplitude of the oscillation was 1%. The angular frequency range used during testing was 10–200 rad s−1. The temperature was operated at 240°C.
DSC analyses
The thermograms were measured using Perkin-Elmer DSC-7 in the nitrogen environment with a nitrogen flow rate of 40 ml/min. The specimens were heated at a heating rate of 10°C/min from −40°C to 230°C (1st heating scan), held at 230°C for 5 min to erase the thermal history, subsequently cooled to −40°C at a cooling rate of either 5 or 10°C/min, and then again heated to 230°C at a heating rate of 10°C/min (2nd heating scan). The DSC thermal characteristics of the each sample were averaged from at least three specimens with hermetic aluminum (Al) pans. The indium standard and the baseline obtained from the empty Al pan were used to calibrate the thermograms acquired.
The corrected crystallinity of the samples was calculated as follows,
where Xc is the crystallinity, ΔHcn is the nominal heat of crystallization determined by DSC, ΔHref is the heat of fusion of theoretical 100% crystalline Nylon 6 (241 J/g) and w is the weight fraction of Nylon 6 in the samples. [34]
The kinetics of crystallization can be described by the Avrami equation as follows [35].
or
where k is the apparent rate constant, n is the Avrami exponent, Xt is the relative crystallinity at time t, defined as follows,
where q is the heat flow in the cooling thermogram at temperature T, and t is the elapse time of the crystallization,
where T0 is the onset temperature of crystlization, and Rc is the cooling rate (°C/min).
Results and discussion
Mechanical properties
Table 5 summarizes the mechanical properties of Nylon 6/SMA/PEBAs blends, including impact properties, tensile properties and flexural properties. Comparing the properties of TN-0 and TN-1, the addition of SMA improved both the tensile properties and flexural properties, while reduced slightly the impact strength.
Impact properties
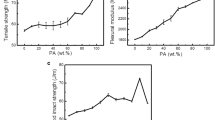
Figure 1 shows the notched Izod impact strength of the blends at 23°C and − 20°C. According to the well-known toughening mechanism [9, 36,37,38,39,40], the elastomers are dispersed in the plastic matrix as fine particles; they activate the plastic layer surrounding them, and the motion of plastic segments consumes a major part of impact energy, resulting in toughening. The compatibilizer (SMA) would react with the end groups of Nylon 6, thus extending the molecular chains, resulting higher tensile strength and lower impact strength [41]. Because the SMA content was only 1%, the effect on the impact strength was insignificant. However, the impact strength was greatly improved by the addition of 15 wt% of PEBA. In the case of TN-2, although the soft segment (PTMO) in PEBA was only about 5%, the impact strength was 100% and 86% of that of TN-1, respectively at 23°C and − 20°C. In addition, the impact strength increased with increasing soft segment in the PEBA. In particular, sample TN-5 exhibited an impact strength 3.8 folds (238.5 ± 9.1 J/m) of that without PEBA (TN-1) at 23°C, and 3.5 folds (168.3 ± 8.3 J/m) at −20°C. This indicates that the presence of higher soft-segment (PTMO) content in PEBA can greatly improve the impact strength of Nylon 6.

The Izod impact strength of Nylon 6/SMA/PEBAs blends at 23 °C and − 20 °C
Figure 2 shows the SEM images of the fractured surfaces of the samples after impact test. The morphologies for TN-0 ~ TN-3 appeared homogeneous. However, for TN-4 and TN-5, submicron particles evenly dispersed in the matrix were observable. Even so, these particles (PEBA) were well embedded in the major phase (Nylon 6), indicating that SMA did compatibilize Nylon 6 and PEBA, hence improving the impact strength.

SEM images of the fractured surfaces of the samples. TN-0, TN-1, TN-2, TN-3, TN-4, TN-5
Tensile properties
Figure 3 shows the effect of the addition of 15 wt% PEBAs on the tensile strength and modulus of the blends with 1 wt% SMA. The tensile strength decreased from 64.2 (TN-1) to 54.8 MPa (TN-5). This can be attributed to the presence of lower-strength PEBAs. On the other hand, the tensile modulus decreased from 2.81 (TN-1) to 1.79 GPa (TN-5), a reduction of ~36%, indicating that higher soft elastomeric phase (PTMO) in PEBA making the blend more flexible. As indicated in the literature, flexible molecular chains can make the blend softer [9, 34, 42, 43].

The tensile strength and modulus of Nylon 6/SMA/PEBAs blends
Flexural properties
Figure 4 shows the flexural strengths and modulus of the blends. Similar to the tensile properties, the flexural properties exhibited the same trend. The flexural strength decreased from 104.0 (TN-1) to 62.6 MPa (TN-5). In addition, the flexural modulus decreased from 2.05 (TN-1) to 1.21 GPa (TN-5), a reduction of ~40%. With the increase in PTMO content in PEBA, the flexural properties of blends decreased. In general, toughening elastomer has a low modulus; inferior to that of Nylon6 and therefore decrease the modulus of the blends [34, 44].

The flexural strength and modulus of Nylon 6/SMA/PEBAs blends
Thermal stability
The thermal degradation behavior of Nylon 6/SMA/PEBA blends was examined via TGA analysis. Figures 5 and 6 show the thermogravimetric curves and the derivative weight for blends separately. The thermal decomposition temperatures (Td) at a 50 wt.% weight loss and (Tp) from the derivative weight were listed in Table 6. These decomposition temperatures are the temperature where the sample decomposes chemically [45].

TGA curves of weight percent (wt%) for Nylon 6/SMA/PEBA blends

DTG curves of Nylon 6/SMA/PEBA blends
In TGA, the sample losses weight on heating due to evaporation or oxidation into gases. Nylon 6 degrades through fast and slow decomposition. Pure Nylon 6 decomposes into cyclic caprolactam. Initially, fast decomposition due to backbiting occurs at the end of molecular chain, resulting in decomposed product and active end group, allowing the decomposition to repeat continuously. On the other hand, slower decomposition is due to the cyclic transformation through the inchworm movement in the molecular chain. This reaction occurs all over the molecular chain, thus its occurrence is more frequent than the backbiting reaction [46].
Figures 5 and 6 show that the onset of the decomposition became more conspicuous with increase in the soft-segment (PTMO) in PEBA, as TN-4 and TN-5 shown in Fig. 6, attributing to the decomposition of PTMO [33]. When the hard-segment (PA11) increased, as TN-2 and TN-3 in Fig. 5, their TGA curves became similar to that of TN-0 (Neat Nylon 6). Based on the TGA results, those samples containing PEBA with higher PA11 content exhibited higher thermal stability. This can be attributed to the intermolecular hydrogen boding between PA11 chains [33].
Table 6 shows the peak decomposition temperature (Tp) ranges between 447~453°C. Thus the effect of PEBA on the thermal stability of Nylon 6 was small especially when the processing was carried out below 300°C.
Rheological properties
The rheological behavior of polymer melt affects greatly the processing of polymer blends. Polymer melt exhibits viscoelasticity, that is, the deformation recovers gradually with the time after removing the applied stress [47]. The viscosity is expressed with G’ (storage modulus) and G” (loss modulus) in dynamic rheological experiments. Figures 7 and 8 show that both G’ and G” for the Nylon 6/SMA/PEBAs depend on the flexibility of PEBA.

Plots of storage modulus versus angular frequency for Nylon 6/SMA/PEBA blends

Plots of loss modulus versus angular frequency for Nylon 6/SMA/PEBA blends
Both G’ and G” of Nylon 6/SMA/PEBAs blends increased with increasing angular frequency. The storage modulus of Nylon 6/SMA/PEBAs increased with the flexibility of PEBA within the frequency range of 10–200 rad s−1, whereas the loss modulus showed the reverse trend. The dynamic rheological results revealed that PEBAs with a higher PTMO content are more elastic because the presence of flexible PTMO confers a higher elasticity to the blends. On the other hand, a higher PA11 content confers a higher rigidity to the blends due to an extensive intermolecular hydrogen bonding network that exists between the PA chains in the hard blocks, leading to higher loss modulus (G”).
The complex viscosity η* can be obtained from G’ and G” as follows:
where ω is the angular frequency in rad·s−1.
Figure 9 illustrates the relationship between complex viscosity η* and angular frequency of Nylon 6/SMA/PEBAs blends at 240°C. It can be seen that existence of 15 wt% PEBAs had a dramatic effect on the complex viscosity of the blends. The complex viscosity decreased with the increase of angular frequency, indicating the shear-thinning behavior. This is because at higher angular frequency, the molecular chains are more oriented in the flow, thus reducing the degree of entanglement, and leading to lower viscosity.

Plots of complex viscosity versus angular frequency for Nylon 6/SMA/PEBA blends
At low frequency, the viscosity of the melt is dominated by the entanglement, which is related to the chain length of the polymer. Thus at 10 rad/s, η* decreased with the increase of molecular weight of Nylon 6.
At higher frequency, the viscosity gradually levelled off, indicating disentanglement occurred, and the interaction between molecular chains became important. In Pebax®, the interchain hydrogen bond occurs between –NH and C=O groups in the PA11 segments [33]. The PTMO segments would reduce the interaction strength in the melt. At 200 rad/s, the ratio of η* for TN-5 to that of TN-1 was 0.83, while the ratio of molecular weight of Nylon 6 for TN-5 to that of TN-1 was 0.82. Thus η* decreased slightly more than the molecular weight ratio, indicating the reduction in the intermolecular interaction due to the PTMO content was not as important as the molecular weight of Nylon 6.
Figure 10 shows the loss factor (tanδ) of Nylon 6/SMA/PEBA varies with the frequency. For a viscoelastic melt, tanδ, the ratio of dynamic loss modulus (G”) to dynamic storage modulus (G’) in an oscillatory flow has been employed to evaluate the viscoelasticity. G’ represents the elastic aspect of the material and G” is the viscous aspect. When tanδ equals 1 (G“/G’=1), indicating the transition from more viscous (G”/G’ > 1) to more elastic (G”/G’ < 1) [48, 49]. Because all the values of tanδ were greater than 1, these samples exhibited a predominantly viscous behavior rather than an elastic behavior, especially during all the frequencies (10–200 rad s−1). Figure 10 shows that the values of tanδ decreased with the increase of PTMO content. This indicates that flexible PTMO can increase the elastic aspect of the blends.

Plots of tanδ of Nylon 6/SMA/PEBA blends versus angular frequency
Crystallization characteristics
The crystallization behaviors of blends with different PEBA were obtained from the DSC cooling thermograms shown in Fig. 11 and the resultant crystallization characteristics were listed in Table 7. For all blends, a large exothermal peak starting from about 185~188°C (Tonset) and with the peak (Tc) near 181~184°C was observed due to the crystallization of Nylon 6 constituent. An additional small peak near 165°C attributing to the crystallization of PA11 [50,51,52,53] from the added PEBA was also observed in the thermograms of TN-2 and TN-3 because of the higher PA11 content (13%–14%). The blends with PEBA (TN-2 ~ TN-5) all exhibited lower Tonset and Tc than the unblended sample TN-1 (with Tonset = 188.2°C and Tc = 184.7°C), indicating that the addition of PEBA would impede the crystallization rate of Nylon 6 [34].

The 1st cooling thermograms at a cooling rate of 10 °C/min
In Table 7, the time to 50% relative crystallization (t1/2) for the Nylon 6/SMA/PEBA blends (TN-2 ~ TN-5) was significantly longer than that of Nylon 6/SMA (TN-1), indicating that the addition of PEBA did impede the crystallization. This can be attributed to the flexible PTMO and rigid PA11 chains in PEBA that could impede the crystallization of Nylon 6. In Table 8, the effect of flexible PEBA was proved by the lower glass transition temperatures (Tg) obtained from TN-2 ~ TN-5 than that of TN-1. It was also shown previously that PEBA has a much lower Tonset and Tc, and a much smaller ΔHcc [34]. Consequently, Nylon 6 diluted by PEBA exhibited slower crystallization rate (larger t1/2).
Interestingly, the corrected crystallinities of Nylon 6 in the blends were little affected by the PEBA added. As observed in Table 7, the nominal crystallization enthalpy (ΔHcn) of TN-2~TN-5 (52.9 ~ 57.0 J/g) were all smaller than that of TN-1 (63.7 J/g) because the amounts of Nylon 6 constituent in the blends were diluted by PEBA. After corrected by the actual content of Nylon 6, the corrected crystallization enthalpy (ΔHcc) of the blends (63.0 ~ 67.8 J/g) were not too different than that of TN-1 (64.3 J/g) when the standard deviations were considered. Accordingly, the crystallinities (Xc) calculated from ΔHcc by using 241 J/g as the theoretical heat of fusion of perfectly crystallized Nylon 6 [54, 55] were between 26% and 28% which also implied that the crystallinity of the Nylon 6 constituent was little affected by the presence of PEBA.
Figure 12 shows the 2nd heating thermograms of Nylon 6/SMA/PEBA and the corresponding melting characteristics are summarized in Table 8. As shown in Fig. 12, all thermograms exhibited two large melting peaks (Tm2 and Tm3) near 212 and 220°C corresponding to the melting of γ and α crystals of Nylon 6, respectively [34]. The thermogram of TN-1 exhibited a small melting shoulder (Tm1) near 186°C due to the melting of the formed small crystallites. For TN-2 and TN-3, the Tm1 peak shifted to 181°C attributing to the melting of PA11 that overwhelmed the original Tm1 of Nylon 6. On the other hand, for TN-4 and TN-5, the melting peak of PA11 was unobservable in Fig. 12 because of the abundance of amorphous PTMO in the PEBA added. Consequently, the small Tm1 shoulder of Nylon 6 was exposed in the thermograms of TN-4 and TN-5. In addition, regardless of the type of PEBA, Tm2 (210 ~ 212°C) and Tm3 (219 ~ 220°C) of these five blends differed little, indicating that the formation of γ and α crystals of Nylon 6 constituent was not much affected by the type of PEBA.

The 2nd heating thermograms at 10 °C/min after cooling at 10 °C/min for samples TN-0 ~ TN-5
In Table 8, the nominal heat of fusion (ΔHmn) and corrected ΔHmc of TN-1 ~ TN-5 were generally following the similar trend of those of ΔHcn and ΔHcc shown in Table 7. All the values of ΔHmc for TN-1~TN-5 were around 70 J/g, corresponding to a crystallinity of 30% for Nylon 6 [54]. Thus, the type of PEBA caused little effect on the crystallinity of Nylon 6. It is also interesting to note that the values of ΔHmc in Table 8 were all greater than the values of ΔHcc in Table 7. This is attributed to the well-known cold crystallization and secondary crystallization occurred during the 2nd heating process that increased Xc by about 3%.
Although Tm and Xc of blends were affected little, the ratios of α and γ crystals were affected by the type of PEBA. As shown in Fig. 12, the relative intensities of Tm2 and Tm3 peaks changed with the type of PEBA. The presence of PEBA favored the formation of α over γ crystals. It was indicated that the processing conditions of Nylon 6 greatly affected the type of crystals formed. Faster crystallization rate would benefit the formation of γ crystals, while slower rate would favor the formation of α crystals [56,57,58,59,60]. From t1/2 obtained previously, TN-2 ~ TN-5 had slower crystallization rates than TN-1, henceforth, they had more α crystals formed during the cooling cycle. Consequently, the small differences of Tc and t1/2 among TN-2 ~ TN-5 also resulted in the variations of the relative intensities of Tm2 and Tm3 peaks observed in these four blends.
Figure 13 shows the cooling thermograms at a slower cooling rate of 5°C/min and the related characteristics are listed in Table 9. In addition to the major exothermic peaks of Nylon 6 similar to those in Fig. 11, a small exothermic peak at about 167°C corresponding to the crystallization of PA11 were also observed from TN-2 and TN-3. Comparing with those in Table 7, all Tonset and Tc cooling at 5°C/min were higher while the differences among the samples were much smaller. This can be attributed to the kinetics of crystallization, i. e., a slower cooling rate led to a longer crystallization time (confirmed by the larger t1/2 obtained in Table 10). In addition, a longer crystallization time starting at a higher temperature obtained from the slower cooling rate resulted in a higher crystallinity, confirmed by the higher ΔHcc and Xc in Table 9. It is also interesting to note that the melting peaks at Tm2 from the melting of γ crystals in Fig. 14 were much larger than those in Fig. 12 due to the combining effects of crystallization rates, crystallization temperatures, and time for crystallization on the phases of crystals formed.

The 1st cooling thermograms at a cooling rate of 5 °C/min for samples TN-0 ~ TN-5

The 2nd heating thermograms at 10 °C/min after cooling at 5 °C/min for samples TN-0 ~ TN-5
The crystallization behaviors of different blends were further analyzed by using the Avrami equation on non-isothermal DSC cooling thermograms. Figure 15 shows the Avrami plot of ln(−ln(1-Xt)) vs ln(t) at three cooling rates calculated from DSC thermograms of TN-1 where Xt is the relative crystallinity of the sample crystallized at time t. Obviously the data from these three cooling rates fitted well to Avrami equation. Figure 16 shows similar result for TN-3. The linear regions in the Fig. 16 still covered a large range of Xt (10%~90%). Thus, the Avrami equation is applicable to compare the crystallization kinetics of TN-1~TN-5.

The plot of ln(−ln(1-Xt)) vs ln(t) from DSC the thermograms of TN-1

The plot of ln(−ln(1-Xt)) vs ln(t) from DSC the thermograms of TN-3
Figures 15 and 16 show the Avrami plots for TN-1 and TN-3, respectively. The resultant values of k and n as well as the values of Tc and t1/2 were summarized in Table 10. TN-1 exhibited greater k than those four blended samples, suggesting the addition of PEBA would retard the crystallization of Nylon 6. Furthermore, the values of k increased with the cooling rate, because higher cooling rate could increase the nucleating speed, henceforth, increased the crystallization rate [35]. These k values can be used to estimate the crystallization half-time (t1/2) as follows
Those estimated values of t1/2 were comparable to those experimental values, as shown in Table 10, suggesting that Avrami Equation can fit the non-isothermal crystallization kinetics of Nylon 6 although it was originally for isothermal crystallization.
All values of n were between 2 and 3, indicating that the non-isothermal crystallization of TN-1~TN-5 was a diffusion-controlled crystallization in the fitted regions. In addition, the values of n of all five samples decreased with increasing cooling rate. This suggests that at a higher cooling rate, the crystallization follows a platelet growth more than a spherulitic growth [61].
Conclusions
The toughness of Nylon 6 were effectively improved by melt blending Pebax® Rnew resins (PEBA) as the toughening agent and compatibilized with poly(styrene-co-maleic anhydride) (SMA). The soft-segment (PTMO) content in PEBA was the key factor for toughness enhancement. As revealed that the presence of higher soft-segment (PTMO) content in PEBA can greatly improve the impact strength of Nylon 6. In particular, the blend of Nylon 6 containing 1 wt% SMA with 15 wt% of PEBA 25R53 exhibited an impact strength 3.8 folds of that without PEBA at 23°C, and 3.5 folds at −20°C. Unlike impact strength, the tensile and flexural properties of the blends decreased with the increase of the PTMO content in PEBA. However, the extent of reduction was around 20 ~ 40%, which was acceptable considering the improvement in the impact strength.
The results of TGA indicated the effect of PEBA on the thermal stability of the blend of Nylon 6 with 1 wt% SMA was small especially when the processing was carried out below 300°C. In addition, rheological properties revealed that these blends exhibited predominantly viscous behavior. Furthermore, higher PTMO content led to higher storage modulus (G’), lower loss modulus (G”) and complex viscosity (η*), indicating that flexible PTMO can increase the elastic aspect of blends.
The DSC results showed that the glass transition temperature (Tg) of Nylon 6 decreased with the increase of PTMO content. The addition of PEBA caused the decrease of the rate of crystallization. However, the crystallization temperature and the rate of crystallization of the blends were not much affected by the type of PEBA, while the relative amounts of α and γ crystals formed were affected. The crystallization behaviors of different blends were analyzed by using the Avrami equation on non-isothermal cooling thermograms. All values of Avrami exponent (n) were between 2 and 3, indicating that the non-isothermal crystallization of Nylon 6/SMA/PEBA blends was a diffusion-controlled crystallization in the fitted regions.
References
Yu Z, Ou Y, Qi Z, Hu G (1998). J Polym Sci B Polym Phys 36:1987–1994
Li QF, Kim DF, Wu DZ, Lu K, Jin RG (2001). Polym Eng Sci 41:2155–2161
Wang X, Feng W, Li H, Jin R (2003). J Appl Polym Sci 88:3110–3116
Huang JJ, Paul DR (2006). Polymer 47:3505–3519
Kim JG, Lee J, Son Y (2014). Mater Lett 126:43–47
Yu ZZ, Ke YC, Ou YC, Hu GH (2000). J Appl Polym Sci 76:1285–1295
Premphet-Sirisinha K, Chalearmthitipa S (2003). Polym Eng Sci 43:317–328
Borggreve RJM, Gaymans RJ, Schuijer J, Housz JFI (1987). Polymer 28:1489–1496
Wu D, Wang X, Jin R (2004). Eur Polym J 40:1223–1232
Okada O, Keskkula H, Paul DR (2004). J Polym Sci B Polym Phys 42:1739–1758
Wang BB, Wei LX, Hu GS (2008). J Appl Polym Sci 110:1344–1350
Wang Y, Wang W, Peng F, Liu M, Zhao Q, Fu PF (2009). Polym Int 58:190–197
Kumar S, Ramanaiah BV, Maiti SN (2007). Soft Mater 4:85–100
Gonzhlez-Montiel A, Keskkula H, Paul DR (1995). Polymer 36:4621–4637
Lu M, Keskkula H, Paul DR (1995). J Appl Polym Sci 58:1175–1188
Corté L, Beaume F, Leibler L (2005). Polymer 46:2748–2757
Ahn Y, Paul DR (2006). Polymer 47:2830–2838
Tanrattanakul V, Sungthong N, Raksa P (2008). Polym Test 27:794–800
Carone Jr E, Kopacak U, Goncalves MC, Nunes SP (2000). Polymer 41:5929–5935
Banerjee SS, Bhowmick AK (2013). Polymer 54:6561–6571
Kayano Y, Keskkula H, Paul DR (1997). Polymer 38:1885–1902
Wu D, Wang X, Jin R (2006). J Appl Polym Sci 99:3336–3343
Xie T, Yang G (2004). J Appl Polym Sci 93:1446–1453
Huang JJ, Keskkula H, Paul DR (2006). Polymer 47:639–651
Yu H, Zhang Y, Ren W (2009). J Polym Sci B Polym Phys 47:877–887
Liu H, Xie T, Hou L, Ou Y, Yang G (2006). J Appl Polym Sci 99:3300–3307
Yu H, Zhang Y, Ren W, Hoch M, Guo S (2011). J Appl Polym Sci 121:3340–3346
Xu C, Ma J, Li G, Wang N, Zhang Q, Grami ME, Qu X (2017). J Polym Res 24:147
Utracki LA (2003) Polymer Blends Handbook. Kluwer Academic Publishers, Dordrecht
Newman S, Paul DR (1978) Polymer blends: volumes I and II. Academic Press, New York
Ponnamma D, Sadasivuni KK, Wan C, Thomas S, Alma’adeed MA (2015) Flexible and stretchable electronic composites: electronic applications of polyamide elastomers and its composites. Springer Verlag, Berlin
Hoffendahl C, Fontaine G, Bourbigot S (2013). Polym Degrad Stabil 98:1247–1255
Todros S, Natali AN, Piga M, Giffin GA, Pace G, Noto VD (2013). Polym Degrad Stabil 98:1126–1137
Chen WM, Yang MC, Hong SG, Hsieh YS (2017). J Polym Res 24:40
Rinawa K, Maiti SN, Sonnier R, Lopez Cuesta JM (2015). Bull Mater Sci 38:1315–1327
Wu S (1985). Polymer 26:1885
Angola JC, Fujita Y, Sakai T, Inoue T (1988). J Polym Sci B Polym Phys 26:807–816
Margolin A, Wu S (1988). Polymer 29:2170
Wu S, Margolin A (1990) Reply to comments. Polymer 31:972–974
Sjooerdsma SD (1989). Polym Commun 30:106
Kim BK, Park SJ (1991). J Appl Polym Sci 43:357–363
Nouparvar H, Hassan A, Mohamad Z, Wahit MU (2014). J Elastom Plast 46:269–283
Navid N, Mat UW, Azman H, Shaya M (2011). Key Eng Mater 471-472:518–523
Ogunsona EO, Misra M, Mohanty AK (2017). RSC Adv 7:8727–8739
Thanakkasaranee S, Kim D, Seo J (2018). Polymers 10:225
Lehrle RS, Parsons IW, Rollinson M (2000). Polym Degrad Stabil 67:21–33
Macosko CW (1994) Rheology-principles, measurements, and applications. Wiley-VCH
Pang H, Liao B, Huang Y, Cong G (2002). J Appl Polym Sci 86:2140–2144
Li X, Ai X, Pan H, Yang J, Gao G, Zhang H, Yang H, Dong L (2018). Polym Advan Technol 29:1706–1717
Latko P, Kolbuk D, Kozera R, Boczkowska A (2016). J Mater Eng Perform 25:68–75
Bourmaud A, Duigou AL, Gourier C, Baley C (2016). Ind Crop Prod 84:151–165
Qi F, Chen N, Wang Q (2017). Mater Des 131:135–143
Ma YL, Hu GS, Ren XL, Wang BB (2007). Mat Sci Eng A-Struct 460-461:611–618
Fornes TD, Paul DR (2003). Polymer 44:3945–3961
Illers KH (1978). Makromol Chem 179(2):497–507
Ren X, Wu G, Zhang X (2011) 2011 2nd international conference on chemistry and chemical engineering. IPCBEE 14:125–129
Liu T, Tjiu WC, He C, Na SS, Chung TS (2004). Polym Int 53:392–399
Dasgupta S, Hammond WB, Goddard III WA (1996). J Am Chem Soc 118:12291–12301
Liu Y, Cui L, Guan F, Gao Y, Hedin NE, Zhu L, Fong H (2007). Macromolecules 40(17):6283–6290
Katoh Y, Okamoto M (2009). Polymer 50:4718–4726
Lam RSH, Rogers MA (2011). Cryst Eng Comm 13:866–875
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Chen, WM., Yang, MC., Hong, SG. et al. Effect of soft segment content of Pebax® Rnew on the properties of Nylon-6/SMA/PEBA blends. J Polym Res 26, 25 (2019). https://doi.org/10.1007/s10965-018-1684-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-018-1684-2