
Abstract
The atomic-level AI topological indices and the modified Xu (mXu) index were utilized for quantitative structure–property relationship (QSPR) modeling of the infinite dilution activity coefficients of 108 oxo compounds in water at 298.15 K. Stepwise multiple linear regression (SMLR) analysis using the topological descriptors resulted in a model with R2 = \({\mathrm{R}}_{\mathrm{a}\mathrm{d}\mathrm{j}}^{2}\)= 0.9904, SE = 0.3769, F = 1267.1 and an average relative error of 4.89%. The selected descriptors were then used to develop an artificial neural network (ANN) model for the activity coefficients. Findings of the study indicated that a 7–8-1 ANN trained by Levenberg–Marquardt algorithm results in the improved predictions, especially in view of a decrease as large as 47.24% in the average relative error compared to the SMLR model. The AI indices with a total contribution of 81.43% showed the dominant role of the atomic groups of the oxo compounds in determination of their activity coefficients at infinite dilution in water.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The infinite dilution activity coefficient, γ∞, is an important thermodynamic property of both practical and theoretical interest. The parameter provides insight into the kinds of physical and chemical intermolecular forces involved in the solute–solvent interactions, which is useful for estimating aqueous solubility and selecting solvents in many industrial processes including high purity extraction, azeotropic rectification, chemical separations and environmental pollution control [1]. Moreover, study of the infinite dilution activity coefficients is of great value in investigating the thermodynamic behavior of dilute aqueous solutions, developing new thermodynamic models [2] as well as calculating the solubility of solids in supercritical gases [3], excess enthalpies [4] and Henry constants [5]. Many practical implications in environmental, chemical and biochemical processes [6] and extensive applications in commercially important products like pharmaceuticals, coatings and paints [7, 8] make it essential to study γ∞ values. Gas–liquid chromatography [9], the dilutor method [10] and differential ebulliometry [11] are the most common methods of measuring γ∞ values. Nevertheless, for the reasons like safety, cost and technical availability, prediction of the infinite dilution activity coefficients by QSPR modeling [12,13,14,15] as an alternative method is of great importance.
Since 1947 when Wiener reported the first application of graph theory to QSPR modeling, many graph theoretical topological descriptors possessing high prediction potency of various physicochemical properties have been developed [16, 17]. Using the descriptors, useful information is obtained about molecular features such as size, shape, branching, symmetry, as well as the atom and bond types without the need for optimizing the geometry of molecules [18]. Atomic-level indices are highly efficient topological descriptors, which allow estimation of the individual contributions of the molecular fragments and atomic groups to the properties of chemical compounds. One of the most important descriptors of this type called atom-type-based AI indices was introduced by Ren [19]. In addition to describing the structure of a molecule at the atomic-level, the descriptors encode the structural environment of each atom-type in the molecule. The atomic-level AI indices combined with the bulk property topological descriptor of Xu [20, 21] (or mXu [22]) showed satisfactory linear correlations to various properties such as molecular total surface area, enthalpies of vaporization, Pitzer’s acentric factor, water solubility, narcosis activity, etc. [23,24,25,26]. Ren also employed the indices in linear regression modeling of the quantitative structure-retention relationship (QSRR) of aldehydes and ketones on gas chromatographic columns [27]. Moreover, Panneerselvam et al. used the atomic-level indices to predict the boiling points of trialkyl phosphates [28]. Recently, our group has successfully applied the atom-type-based topological descriptors combined with MLR technique for estimating the standard formation enthalpies of acyclic alkanes [29] and normal boiling points of esters [30]. The group has also reported the first application of ANN modeling in a QSRR study of monomethylalkanes using the topological indices [31].
In this work, the benefits of the atomic-level AI and mXu topological descriptors in QSPR modeling of the infinite dilution activity coefficients of a group of oxygen containing organic compounds in water are illustrated. The structure–property relationships were investigated by SMLR and ANN modeling techniques. Additionally, the role of atomic groups affecting the activity coefficients of the studied molecules was characterized. As far as the author is aware, this is the first report on QSPR modeling of the infinite dilution activity coefficient of organic compounds using the atomic-level AI topological indices.
2 Method
2.1 Data Set and Topological Descriptors
The experimental values of the room temperature activity coefficients at infinite dilution for 108 oxygen containing organic compounds in water were taken from the literature [32,33,34,35,36,37,38,39]. Table 1 lists the data set including C2–C18 linear and branched alcohols, ketones, ethers, esters, aldehydes and carboxylic acids with ln γ∞ values in the range of 1.32–23.34.
The topological indices for the studied molecules were calculated during the following steps [27]:
(i) Illustration of the hydrogen depleted structure of each molecule by the molecular graph.
(ii) Deriving the distance matrix, D = [dij]n×n, whose elements are the shortest path length between the atoms i and j in the molecular graph. Then, the sum over the column i (or row j) of the matrix was calculated to give the distance sum vector, S = [si]n×1.
(iii) Coding of the graph by the vertex degree vector, V = [vi]n×1, whose elements are the number of connections to the atom i.
(iv) Calculation of Xu index for each graph by the following equation where the sum is over all i atoms in the molecular graph.
(v) Calculation of the atomic-level AI topological descriptors for each atom i belonging to jth atom-type in the graph as follows:
where the perturbing term, ϕi(j), reflects the impact of the structural environment of the ith atom on the topological index value. The AIi(j) values of m atoms of the same type was then utilized to obtain the desired atom-type topological descriptor, AI(j), using the following equation:
To modify the Xu and AI topological descriptors for differentiation of the heteroatoms (oxygen in the work) and multiple bonds, vi values were replaced by the degree of vertex developed by Ren, vm, [19]. The parameter is defined using the number of connections of the atom (δ), valence connectivity of Kier–Hall, δv [40], and the principal quantum number of the valence shell (N).
where δ is calculated by the difference between the number of valence electrons and the number of hydrogens bonded to an atom.
2.2 Model Development
The quantitative structure–activity coefficient correlations for the studied compounds were firstly generated by SMLR using SPSS software [41]. In the modeling process, the room temperature values of ln γ∞ for the oxo compounds (dependent variable) and the calculated topological descriptors (independent variables) were mathematically correlated by the following equation:
where a0 is a constant and the parameters a1 and bj are the contribution coefficients of mXu index and jth AI index, respectively. The oxo compounds was randomly split into a training set with 86 molecules and a prediction set with 22 molecules, and the corresponding ln γ∞ values were utilized for developing the model and model validation, respectively. To select the best linear model, the coefficient of multiple determination (R2), adjusted correlation coefficient (\({\text{R}}_{{{\text{adj}}}}^{2}\)), Fisher-ratio (F) and standard error (SE) of the generated equations were compared. Moreover, the values of t-scores for the model coefficients indicating the level of significance of the topological indices in the model, along with the statistics of standard error for the coefficients, were evaluated to choose a high quality subset of the indices.
In the next step, a feed-forward back propagation ANN algorithm was written in MATLAB [42] to develop the nonlinear QSPR model. The theoretical explanations for the modeling technique can be found in the literature [43]. The nonlinear model was generated using inputs and targets, normalized in the range of − 1 to + 1 to achieve the minimum computational errors. The network included an input layer with Ni neurons equal to the number of topological indices selected by SMLR, a hidden layer with Nh neurons, and an output layer with 1 neuron representing the targets, i.e. the ln γ∞ values. The neurons were intercorrelated by the connections called weight (W) and bias (b) whose values were modified during the network training. In this work, the Levenberg–Marquardt algorithm was utilized to train the network because of its robustness and accuracy [44] and the performance function of root mean squared error (RMSE) was employed based on the following definition:
where the subscripts exp and pred refer to the experimental and predicted values of the activity coefficients for jth molecule of N model compounds, respectively. Optimum Nh value was also determined during the training stage of ANN through search within the range defined by the criterion, ρ, (Eq. 7) and observing the variations in RMSE.
where the lower limit of ρ value is adjusted at 1 to avoid memorizing the data by the neural network, and the upper limit should not exceed 3 due to the inability of ANN to generalize [45].
The data used as the training and prediction sets for ANN modeling were the same as those employed for developing SMLR model. However, the training set was randomly divided into two sets with 64 and 22 data as the training and validation sets, respectively, to reveal the overtraining of ANN by tracking RMSE values as a function of epoch number. Additionally, different combinations of the linear, logarithmic sigmoid and hyperbolic tangent sigmoid transfer functions were utilized for the hidden and output layers to achieve the best architecture for the neural network.
3 Results and Discussion
3.1 Quantitative Structure–Activity Coefficient Relationships
As mentioned, the mXu index in combination with AI topological indices was firstly correlated to ln γ∞ data using SMLR modeling technique. The best SMLR model obtained for the activity coefficients is as follows:
The statistics of R2 show that the developed model explains ~ 99% of the variances in the activity coefficients. Moreover, \({\text{R}}_{{{\text{adj}}}}^{2}\) equals R2 indicating a high significance level of the model and the F-value implies that the relationship described by SMLR equation is significant with a certainty of 99.99%. Values of the t-scores for the model coefficients are 18.291, − 2.225, 7.658, 3.615, − 7.872, − 3.241 and − 3.951, respectively, proving that all the selected topological descriptors are significant to the model developed for ln γ∞ data. Investigation of the prediction power of the linear model was graphically done using the residual plot shown in Fig. 1. As illustrated, the residuals with a range of − 1.36 to + 1.11, did not follow a normal distribution around the average error of zero. Moreover, the average relative deviation (ARD) obtained for the model was 4.89%, suggesting that the present model can not make sufficiently accurate predictions for the desired data. Values of mXu and AI indices entered in the SMLR model and the predicted activity coefficients are listed in Table 1.

The plot of the residuals resulted from MLR model vs. experimental ln γ∞ values of the oxygen containing organic compounds
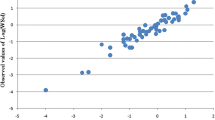
In order to achieve a more accurate model, ANN modeling of the ln γ∞ values was also examined. Therefore, the topological descriptors were used as inputs for developing the nonlinear model. To find the best ANN architecture, different combinations of the transfer functions were applied and the optimum Nh value of the generated networks was sought within the range of 3–9, based on Eq. 7. By comparing the results (not shown), the best statistical quality was found to belong to an ANN with 7–8-1 topology and the transfer functions of tansig-linear for the hidden-output layers. Table 2 gives the characteristics of the best ANN model found for predicting ln γ∞ values of the studied compounds. As shown, the nonlinear model could predict ln γ∞ values for the three sets of training, validation and prediction with R2 > 0.99 and RMSE < 0.3. Optimum values of the weights and biases for the proposed ANN model are presented in Table 3. Additionally, Fig. 2 indicates the values of mean squared error (MSE) against the epoch number for the training, validation and prediction sets. Obviously, the best performance of the ANN was achieved at epoch 26 with a MSE value of 0.0718. The ANN predicted ln γ∞ values for the model molecules are given in Table 1, and the quality of the predictions is graphically illustrated in Figs. 3 and 4. According to Fig. 3, the nonlinear model offers satisfactory efficiency to correlate the activity coefficients to the topological indices as judged from the good agreement between the data points and the straight line indicating perfect predictions. Moreover, the residual plot shown in Fig. 4 shows that there is no systematic error in the developed model, and the residuals ranging from − 0.63 to + 0.71, are considerably smaller than those obtained by SMLR. To further illustrate the efficiency of proposed ANN model for prediction of ln γ∞ values, the residuals reported by He and Zhong [46] as well as Estrada et al. [47] for the same compounds are also shown in the figure. The data clearly indicate the narrower range of the residuals resulted from the ANN model compared to those previously obtained by the researchers. It was also found that the average relative deviation of the nonlinear model (2.58%) was not only 47.24% lower than the developed SMLR model, but also 33.16% lower than ADR reported by He and Zhong, and 65.08% lower than the report of Estrada et al. Relatively small deviations of ANN predicted activity coefficients from the experimental values prove the superiority of the nonlinear model over the previous regression models developed for ln γ∞ of the oxo compounds.

Mean squared errors of the training, validation and prediction sets versus epoch numbers for the best ANN topology found for estimation of ln γ∞ values

Predicted versus experimental ln γ∞ values based on the developed ANN model

3.2 Structural Interpretation of the Infinite Dilution Activity Coefficients
To assess the role of structural characteristics of the studied compounds which determine the ln γ∞ values, the relative importance of each topological descriptor (IMj) was calculated using the connection weights of the developed ANN by the following equation [48]:
where the superscripts i, h and o for the weights refer to the input, hidden and output layers of the network, and the superscripts k, m and n refer to the corresponding neurons, respectively. Calculated values of IMj for the topological indices are presented in Fig. 5. Obviously, the mXu index characterizing the molecular size [20] had the maximum contribution indicating the dominant role of the molecular size in determining the ln γ∞ values. The atomic-level AI topological indices with an overall contribution of 81.43% showed highly considerable role in determination of the activity coefficients of the oxo compound. Among the indices, AI (–CH3) had a contribution of 17.59% indicating that the degree of branching was nearly as effective as bulkiness of the molecule in determining the ln γ∞ values. The functional groups of –OH and = O as an indication of the molecular polarity were in the next ranks in view of contribution to the activity coefficients. Moreover, relatively large IMj values for AI (> CH–) and AI (–CH2–) proved the dominant role of the position of branching in the molecule as well as the hydrophobic interactions in determining ln γ∞ values, respectively. The descriptor of AI (> C =) with IMj = 9.90% was also important in determination of the activity coefficients for the studied oxo compounds. According to the results, the topological indices entered in the proposed ANN model allowed to achieve beneficial insights about the contribution and role of the structural characteristics affecting ln γ∞ of the oxygen containing organic compounds.

Relative importance of the topological descriptors entered in the generated ANN model
4 Conclusion
In the study, the atomic-level AI topological indices combined with mXu index were employed for SMLR and ANN modeling of the room temperature activity coefficients at infinite dilution for a group of oxygen containing organic compounds. The results showed that a 7–8-1 ANN was superior over the linear model in predicting the activity coefficients. Obtaining an average relative deviation of 2.58%, which is significantly lower than those previously reported using the regression models, validated the prediction power of the nonlinear ANN model generated for the desired data. Among the topological indices, mXu and AI (–CH3) were found to be the most important descriptors affecting ln γ∞ values indicating the major role of the molecular bulkiness and degree of molecular branching in determining the activity coefficient values. The findings of the study suggest the atomic-level topological indices combined with the neural network modeling as a promising choice to achieve improved prediction results in QSPR study of the infinite dilution activity coefficients of the oxo compounds in water at 298.15 K.
References
Hsieh, C.M., Lin, S.T.: Prediction of 1-octanol–water partition coefficient and infinite dilution activity coefficient in water from the PR+COSMOSAC model. Fluid Phase Equilib. 285, 8–14 (2009)
Kojima, K., Zhang, S., Hiaki, T.: Measuring methods of infinite dilution activity coefficients and a database for systems including water. Fluid Phase Equilib. 131, 145–179 (1997)
Cheng, J.S., Tang, M., Chen, Y.P.: Correlation of solid solubility for biological compounds in supercritical carbon dioxide: comparative study using solution model and other approaches. Fluid Phase Equilib. 194–197, 483–491 (2002)
Shen, S., Nagata, I.: Prediction of excess enthalpies of ketone–alkane systems from infinite dilution activity coefficients. Themochim. Acta 258, 19–31 (1995)
Morton, D.W., Young, C.L.: Henry's law constants and infinite dilution activity coefficients of C2–C8 hydrocarbons in phenylalkanes. J. Chem. Thermodyn. 28, 895–904 (1996)
Sandler, S.I.: Infinite dilution activity coefficients in chemical, environmental and biochemical engineering. Fluid Phase Equilib. 116, 343–353 (1996)
Lindvig, T., Hestkjær, L.L., Hansen, A.F., Michelsen, M.L., Kontogeorgis, G.M.: Phase equilibria for complex polymer solutions. Fluid Phase Equilib. 194–197, 663–673 (2002)
Kolář, P., Shen, J.W., Tsuboi, A., Ishikawa, T.: Solvent selection for pharmaceuticals. Fluid Phase Equilib. 194–197, 771–782 (2002)
Dohnal, V., Ondo, D.: Refined non-steady-state gas–liquid chromatography for accurate determination of limiting activity coefficients of volatile organic compounds in water: application to C1–C5 alkanols. J. Chromatogr. A 1097, 157–164 (2005)
Krummen, M., Gmehling, J.: Measurement of activity coefficients at infinite dilution in N-methyl-2-pyrrolidone and N-formylmorpholine and their mixtures with water using the dilutor technique. Fluid Phase Equilib. 215, 283–294 (2004)
Dallinga, L., Schiller, M., Gmehling, J.: Measurement of activity coefficients at infinite dilution using differential ebulliometry and non-steady-state gas-liquid chromatography. J. Chem. Eng. Data 38, 147–155 (1993)
Xu, J., Zhang, H., Wang, L., Ye, W., Xu, W., Li, Z.: QSPR analysis of infinite dilution activity coefficients of chlorinated organic compounds in water. Fluid Phase Equilib. 291, 111–116 (2010)
Atabati, M., Zarei, K., Borhani, A.: Predicting infinite dilution activity coefficients of hydrocarbons in water using ant colony optimization. Fluid Phase Equilib. 293, 219–224 (2010)
Xu, J., Wang, L., Wang, L., Zhang, H., Xu, W.: Predicting infinite dilution activity coefficients of chlorinated organic compounds in aqueous solution based on three-dimensional WHIM and GETAWAY descriptors. J. Solution Chem. 40, 118–130 (2011)
Zarei, K., Atabati, M.: Prediction of infinite dilution activity coefficients of halogenated hydrocarbons in water using classification and regression tree analysis and adaptive neuro-fuzzy inference systems. J. Solution Chem. 42, 516–525 (2013)
Xiao, F., Peng, G., Nie, C., Wu, Y., Dai, Y.: Quantum topological method studies on the thermodynamic properties of polychlorinated phenoxazines. J. Mol. Struct. 1074, 679–686 (2014)
Atabati, M., Emamalizadeh, R.: A Quantitative structure property relationship for prediction of flash point of alkanes using molecular connectivity indices. Chin. J. Chem. Eng. 21, 420–426 (2013)
Mamy, L., Patureau, D., Barriuso, E., Bedos, C., Bessac, F., Louchart, X., Martin-Laurent, F., Miege, C., Benoit, P.: Prediction of the fate of organic compounds in the environment from their molecular properties: a review. Crit. Rev. Environ. Sci. Technol. 45, 1277–1377 (2015)
Ren, B.: Novel atom-type AI indices for QSPR studies of alcohols. Comput. Chem. 26, 223–235 (2002)
Ren, B., Chen, G., Xu, Y.: A novel topological index for QSPR/QSAR study of organic compounds. Acta Chim. Sin. 57, 563–571 (1999). (in Chinese)
Ren, B., Xu, Y., Chen, G.: Estimation of heat capacity of complex organic compounds by a novel topological index. J. Chem. Eng. China 50, 280–286 (1999). (in Chinese)
Ren, B.: Novel atomic-level-based AI topological descriptors: application to QSPR/QSAR modeling. J. Chem. Inf. Comput. Sci. 42, 858–868 (2002)
Ren, B.: Application of novel atom-type AI topological indices in the structure-property correlations. J. Mol. Struct. (THEOCHEM) 586, 137–148 (2002)
Ren, B.: Atom-type-based AI topological descriptors: application in structure-boiling point correlations of oxo organic compounds. J. Chem. Inf. Comput. Sci. 43, 1121–1131 (2003)
Ren, B.: Atomic-level-based AI topological descriptors for structure–property correlations. J. Chem. Inf. Comput. Sci. 43, 161–169 (2003)
Ren, B.: Application of novel atom-type AI topological indices to QSPR studies of alkanes. Comput. Chem. 26, 357–369 (2002)
Ren, B.: Atom-type-based AI topological descriptors for quantitative structure–retention index correlations of aldehydes and ketones. Chemom. Intell. Lab. Syst. 66, 29–39 (2003)
Panneerselvam, K., Antony, M.P., Srinivasan, T.G., Rao, P.R.V.: Estimation of normal boiling points of trialkyl phosphates using retention indices by gas chromatography. Thermochim. Acta 511, 107–111 (2010)
Safa, F., Yekta, M.: Quantitative structure–property relationship study of standard formation enthalpies of acyclic alkanes using atom-type-based AI topological indices. Arab. J. Chem. 10, 439–447 (2017)
Osaghi, B., Safa, F.: QSPR study on the boiling points of aliphatic esters using the atom-type-based AI topological indices. Rev. Roum. Chim. 64, 183–189 (2019)
Safdel, F., Safa, F.: Atom-type-based AI topological indices for artificial neural network modeling of retention indices of monomethylalkanes. J. Chromatogr. Sci. 57, 1–8 (2019)
Sutter, J.M., Jurs, P.C.: Adoption of Simulated Annealing to Chemical Optimization Problems. In: J.H. Kalivas (ed.), Elsevier, Amsterdam (1995)
Mitchell, B.E., Jurs, P.C.: Prediction of infinite dilution activity coefficients of organic compounds in aqueous solution from molecular structure. J. Chem. Inf. Comput. Sci. 38, 200–209 (1998)
Dallas, A.J.: Fundamental solvatochromic and thermodynamic studies of complex chromatographic media. PhD Thesis. University of Minnesota, Minneapolis, MN (1995)
Li, J.: Solvatochromic and thermodynamic studies of retention in gas chromatography and gas-liquid equilibria. PhD Thesis. University of Minnesota, Minneapolis, MN (1992)
Sutter, J.M., Dixon, S.L., Jurs, P.C.: Automated descriptor selection for quantitative structure-activity relationships using generalized simulated annealing. J. Chem. Inf. Comput. Sci. 35, 77–84 (1995)
Li, J., Dallas, A.J., Eikens, D.I., Carr, P.W., Bergmann, D.L., Hait, M.J., Eckert, C.A.: Measurement of large infinite dilution activity coefficients of nonelectrolytes in water by inert gas stripping and gas chromatography. Anal. Chem. 65, 3212–3218 (1993)
Wessel, M.D.: PhD Thesis, Pennsylvania State University, University Park, PA (1996)
Yaws, C.L., Yang, H., Hopper, J.R., Hansen, K.C.: Organic chemicals: water solubility data. Chem. Eng. 97, 115–116 (1990)
Kier, L.B., Hall, L.H.: Molecular connectivity in structure–activity studies. Research Studies Press, Letchworth (1986)
SPSS for Windows.: Statistical package for IBM PC, Release 20.0, SPSS Inc., https://www.spss.com.
MATLAB R2013a, The Math Works Inc., https://www.mathworks.com
Agatonovic-Kustrin, S., Beresford, R.: Basic concepts of artificial neural network (ANN) modeling and its application in pharmaceutical research. J. Pharm. Biomed. Anal. 22, 717–727 (2000)
Marquardt, D.: An algorithm for least-squares estimation of nonlinear parameters. J. Soc. Ind. Appl. Math. 11, 431–441 (1963)
Andrea, T.A., Kalayeh, H.: Applications of neural networks in quantitative structure-activity relationships of dihydrofolate reductase inhibitors. J. Med. Chem. 34, 2824–2836 (1991)
He, J., Zhong, Ch.: A QSPR study of infinite dilution activity coefficients of organic compounds in aqueous solutions. Fluid Phase Equilib. 205, 303–316 (2003)
Estrada, E., Díaz, G.A., Delgado, E.J.: Predicting infinite dilution activity coefficients of organic compounds in water by quantum-connectivity descriptors. J. Comput. Aided Mol. Des. 20, 539–548 (2006)
Aleboyeh, A., Kasiri, M.B., Olya, M.E., Aleboyeh, H.: Prediction of azo dye decolorization by UV/H2O2 using artificial neural networks. Dyes Pigm. 77, 288–294 (2008)
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Safa, F. Atomic-Level Topological Indices for Prediction of the Infinite Dilution Activity Coefficients of Oxo Compounds in Water. J Solution Chem 49, 222–238 (2020). https://doi.org/10.1007/s10953-020-00954-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10953-020-00954-8