
Abstract
Solvent basicity is recognized as playing a major role in solvation and is included, through empirical basicity parameters, in linear free energy relationships that account for the effects of changes in solvent on chemical reactions. It is reasonable to postulate that the basicity of a solvent molecule reflects some combination of its molecular properties. In the present study, density functional calculations using the B3LYP functional, and Hartree–Fock calculations have been used to calculate the partial atomic charges (using the Hirshfeld and CM5 models), orbital energies, polarizabilities, dipole moments and quadrupolar amplitudes for over one hundred molecules for which there are experimental values for two basicity parameters, Kamlet and Taft’s hydrogen bond acceptor strength, β, and Gutmann’s donor number, DN, a measure of Lewis basicity. Regression of the experimental β and DN values against molecular descriptors reflecting the above molecular properties yields a remarkably consistent picture. For both parameters the values for alcohols and amines lie systematically off of the regression lines through the remaining compounds, which include alkanes, aromatics, halogenated alkanes and aromatics, esters, carbonates, carboxylic acids, ketones, ethers, nitriles, phosphates, sulfides and sulfates. Independent of the calculation method or method of estimating the partial atomic charges, both experimental β and DN are essentially determined by two molecular properties: the charge on the most negative atom of the molecule and the molecular orbital from which charge donation would occur. The regression results using any of the fours sets of descriptors (reflecting the two calculation methods and two methods of charge estimation) are remarkably similar for β and DN supporting the view that these are measures of the same “basicity”.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Basicity and acidity are among the core concepts in solution chemistry. The earliest approaches to acidity and basicity, due to Arrhenius [1] and to Brønsted [2] and Lowry [3], focused on the proton acidity. Lewis [4, 5] subsequently broadened the idea of acid–base chemistry to include, for example, the binding of ligands, as Lewis bases, to metal ions, acting as Lewis acids. While proton basicity, as reflected in pKa values and Lewis basicity as reflected in the binding constants of metal–ligand complexes are different, it is likely that they both reflect some fundamental “basicity” of the moiety.
The general importance of basicity in solutions is widely recognized and it is one of the properties universally included in linear free energy type equations, such as those due to Kamlett and Taft [6], for the correlation of chemical parameters with molecular properties. Thus, a number of experimental solvent parameters relating to solvent basicity have been developed [7, 8] of which Gutmann’s donor number [9], DN, and Kamlet and Taft’s hydrogen bond acceptor basicity [10], β, are commonly used.
The development of computational methods and the availability of increasing computer power have opened the possibility of calculating, as opposed to measuring, values of these basicity parameters or replacing them with molecular descriptors generated computationally. A number of computational studies have been reported, commonly these have involved the calculation of pKa values of acids [11,12,13,14,15,16,17] or the correlation (and prediction) of experimental basicity parameters with computationally derived molecular descriptors [18,19,20]. In principle, either approach can provide insight into the origins of a moiety’s basicity, from the relative contributions of the different descriptors used.
The present study involves the correlation of experimental basicity parameters with computationally derived molecular descriptors; however, it is not directed to the prediction of the solubility parameters but, rather, to explore which molecular properties lead to a moiety’s basicity. The experimental parameters considered are Kamlet and Taft’s hydrogen bond acceptor parameter, β, and Gutmann’s donor number, DN.
2 Basicity Parameters
It is worthwhile considering the basicity scales and whether they measure the same “basicity” of a molecule.
Gutmann’s DN was originally defined as the absolute value of the molar enthalpy of interaction of the base with antimony pentoxide, a very strong Lewis acid, with the reaction carried out in dilute solutions with dichloroethane as solvent. In effect it was intended as a measure of the Lewis basicity of an isolated or very weakly solvated, molecule of the base [9].
In contrast, Kamlet and Taft’s β was defined as the difference between the shifts in the absorbance maximum of two dyes, which differed in that one had an acidic proton [10]. In this case the measurements were carried out on dilute solutions of the dye with the base as solvent. Thus, β was intended to be a measure of the hydrogen bond basicity, or hydrogen bond acceptor strength, of the base as solvent.
So the scales differ in two ways, the DN scale being a measure of Lewis basicity while β was intended to be a measure of hydrogen bond acceptor strength. The two scales also differ in that the DN was measured for an isolated base molecule in a weakly interacting solvent while β was a property of the bulk liquid.
In carrying out this work we have used the DN and β values listed in Marcus’s review [7]. This review presents data for around one hundred solvents where both β and DN values are available and we restricted this study to those compounds. To bring the scales into approximate correspondence we have divided the DN values by 37, which makes the DN* for hexamethylphosphoramide equal to its β value (1.05).
It is worth noting at the outset that both the β and DN* values show essentially no evidence of steric hindrance. Thus, β and DN* depend essentially on the principal functional group of the base. If steric effects were important than one would observe a systematic decrease in the basicity measures as larger side-groups are introduced. This is generally not the case, as is clear from Table 1 where the β and DN* values for different groups of molecules, with varying sizes of side-groups, are listed.
It is also clear from Table 1 that the β and DN* values are in remarkably good agreement except for several amines, where the DN* values are systematically higher. This is confirmed by Fig. 1 where the DN* values are plotted against the β values. Marcus reports a value 0.76 for the correlation coefficient (R2) of this plot for the 110 compounds for which he reports values [7]; however, if the values for simple alkyl amines (butyl-, diethyl-, triethyl and tributyl-amine) are excluded, R2 for the remaining 106 compounds is 0.86 and removing all of the amines, anilines and pyridines from the correlation raises R2 to 0.92 (97 data points).

Plot of DN* against β: light blue circles, alkanes and aromatics; dark blue squares, halogenated alkanes and aromatics; orange circles, esters and carbonates; green circles, carboxylic acids; purple circles, ketones; black diamonds, ethers; red triangles, nitriles; dark green squares, RS, RS = O, RP = O; dark blue circles, amides; light brown triangles, water alcohols and polyols; grey triangles, pyridines; brown squares, amines and anilines (Color figure online)
Thus, despite their different formulations, β and DN (or DN*) appear to be measures of the same “basicity” of solvents; the only clear exception being nitrogen bases.
3 Computational Details
Since the focus of this work is to understand the relationship between the measures of basicity and molecular properties of the bases, calculations were carried out on isolated molecules rather than in the presence of a reaction field.
It seemed important to assess the reliability of the calculated molecular properties and so calculations were carried out using Hartree–Fock and density functional methods (using the B3LYP functional). All calculations were carried out using the Gaussian 09 suite of programs [21]. The structures of the molecules were optimized using both methods and the molecular properties recovered for the minimized structures.
Initially calculations were calculated using the 6–31G(d,p) and 6–311G(2d,2p) basis sets and Mulliken partial charges were used. However, it is clear the Mulliken charges are highly sensitive to the basis set used and so don’t provide a suitable basis for analyses of the solvent parameters. Thus, the calculations were repeated and Hirshfeld and CM5 partial charges were recovered.
To observe the effect of basis set on the molecular properties, density functional calculations were carried out for DMSO using the B3LYP functional and nine basis sets ranging from 6–31G(d,p) to 6–311++(3df,2p) and including Dunning’s aug-CC-pVTZ and aug-cc-pVQZ. These calculations show that the Mulliken partial charges vary widely and non-monotonically with changes in basis set (partial charges on the S=O oxygen atom are − 0.699, − 0.463 and − 1.121 a.u. for the 6–31G(d,p), 6–311++(3df,2p) and aug-cc-pVQZ basis sets, respectively). In contrast, the Hirshfeld and CM5 partial charges are relatively insensitive to basis set and show reasonable convergence for the more complete basis sets (Hirshfeld partial charges for the S=O oxygen atom are − 0.401, − 0.385, − 0.385, − 0.392 and − 0.389 a.u. for the 6–31(d,p), 6–311+(3df,2p), 6–311++(3df,2p), aug-CC-pVTZ and aug-cc-pVQZ basis sets, respectively).
To ensure that the results were general, rather than specific to DMSO, calculations were carried out using both the density functional (B3LYP functional) and Hartree–Fock methods and the 6–311+G(3df,2p) and aug-cc-pVTZ basis sets for n-hexane, m-xylene, ethanol, DMSO and N,N-dimethyl formamide, DMF. This showed that the Hirshfeld and CM5 partial charges were method dependent but not basis set dependent, at least for these basis sets. Thus, calculations were carried out using both density functional and Hartree–Fock methods but only using the 6–311G+(3df,2p) basis set, which was computationally less demanding than the aug-cc-pVTZ basis set.
The molecular properties recovered were the Hirshfeld and CM5 partial charges on the most negative and most positive atoms, the polarizability, dipole moment, quadrupolar amplitudeFootnote 1 of the molecule and the energies of the filled, donor, and unfilled, acceptor, orbitals. Plots of the polarizabilities, dipole moments and quadrupolar amplitudes from the Hartree–Fock calculations (6–311G+(3df,2p) basis set) against those from the density functional calculations are linear so that, for these, the choice of calculation method in immaterial. This is not the case for the orbital energies, shown for the occupied donor orbitals in Fig. 2, where the values from the Hartree–Fock calculations generally fall on two lines when plotted against the density functional results, one line through the nitriles, alkanes and aromatic compounds (including halogenated compounds) and the second line passing through the values for most of the other compounds.

Plot of the donating orbital energies E(OMO) calculated using the Hartree–Fock method against those from the density functional method: light blue circles, alkanes and aromatics; dark blue squares, halogenated alkanes and aromatics; orange circles, esters and carbonates; green circles, carboxylic acids; purple circles, ketones; black diamonds, ethers; red triangles, nitriles; dark green squares, RS, RS = O, RP = O; blue circles, amides; light brown triangles, water alcohols and polyols; grey triangles, pyridines; brown squares, amines and anilines (Color figure online)
Similar plots of the Hirshfeld derived partial charges calculated using the Hartree–Fock method against those from the density functional method are also linear, while those for the CM5 derived charges show small deviations for some classes of compounds. However, when CM5 derived charges are plotted against Hirshfeld derived charges, as in Fig. 3 where the negative atom partial charges from the density functional calculations are plotted, it is found that the data lie on several lines, depending on the functional group.

Plot of the CM5 derived partial charges on the most negative atom against the Hirshfeld derived values, calculated using the density functional method; light blue circles, alkanes and aromatics; dark blue squares, halogenated alkanes and aromatics; orange circles, esters and carbonates; green circles, carboxylic acids; purple circles, ketones; black diamonds, ethers; red triangles, nitriles; dark green squares, RS, RS = O, RP = O; blue circles, amides; light brown triangles, water alcohols and polyols; grey triangles, pyridines; brown squares, amines and anilines (Color figure online)
For each molecular property a molecular descriptor, QX, was constructed as:
where X represents the molecular property and the subscripts max and min refer to the maximum and minimum calculated values of X (note that for the negative charge Xmax is the largest negative charge, for example). This gives a series of descriptors that vary between zero and unity, making comparison of their relative importance straightforward.
Because of the differences in the E(OMO) values calculated using the Hartree–Fock and density functional methods and those between the Hirshfeld and CM5 based partial atomic charges, both the β and DN* values were analysed using descriptors derived from both computational methods and from both the Hirshfeld and CM5 based partial atomic charges.
All of the molecular properties recovered from the quantum mechanical calculations and the experimental β and DN* values are provided in the supplementary material. The molecular descriptors recovered from the DF calculations using the CM5 derived charges are also listed there.
4 Analysis of β and DN*
The approach adopted assumes linear relationships between the basicity parameters and the molecular descriptors; thus we write:
where QX are molecular descriptors and the aX and bX are the related coefficients.
The molecular properties recovered for the minimized chemical structures are largely straightforward; thus, the molecular polarizability, dipole moment and quadrupolar amplitude are unambiguous.
In considering the partial charges on atoms the situation is simple except in the cases of alkanes where the most negative atoms are the carbon atoms and so are not directly available to neighbouring molecules; in these cases, the net charge on the CHn moiety was taken (these are reported by Gaussian). For aromatic compounds this wasn’t necessary, presumably because the carbon atom partial charges are available from above or below the ring.
The question of which orbital’s energy is taken is more complex. As an example, the nitriles all have very similar β of DN* values and for the simple alkyl nitriles the highest occupied molecular orbital (HOMO) is the CN π-bonding orbital but in benzyl cyanide and benzonitrile the first and second HOMOs are the benzene ring π-bonding orbitals and the third HOMO, which is the CN π-bonding orbital, was taken as the donating orbital. Similar situations arise in a few other cases; thus, the HOMO of chloroethanol is associated with the Cl atom and the second HOMO with the O atom and the HOMO of tetramethyl urea is associated with the N atoms and the second HOMO, which is on the C=O, was taken as the donating orbital.
In general, use of the ring π-bonding orbitals of aromatic compounds led to calculated β and DN* that were much too large and, in all cases, the highest non-ring orbital was used. This result seems counter-intuitive but may reflect the fact that the ring π-bonding orbitals are diffuse, with the electrons bound by several C nuclei. This is likely specific to this case, which involves the basicity of molecules with hard donor atoms.
The procedure adopted to analyze the β and DN* data was to begin by carrying out a multiple regression of all of the values, fitting them to all seven of the molecular descriptors. The regression was repeated after the removal of descriptors for which the p-values of the coefficients indicated that they were statistically insignificant. This is shown in Tables 2 and 3 which give the results of the regression all of the β and DN* values (106 data) against all seven descriptors and against those with statistically significant coefficients, respectively.
There is a difference between the regressions using the CM5 and Hirshfeld based partial atomic charges. Thus, for the β values, regressions using the CM5 partial charges return only two significant descriptors, Qq−, based on the partial charge of the most negative atom, and QE(OMO), based on the energy of the donating orbital. The regressions of the DN* values return these descriptors as significant but also return significance for QP, based on the molecular polarizability, for the density functional derived values and for QQP, based on the molecular dipoles, for the Hartree–Fock calculations.
In contrast, regressions of the β values using Hirshfeld charges, in addition to the dependences on Qq− and QE(OMO), show statistically significant coefficients for Qq+, based on the partial charge of the most positive atom, and for QDP. In the case of DN*, the regressions show statistically significant coefficients for Qq+ and, for the density functional results, the dependence on Qq− is lost (p = 0.3).
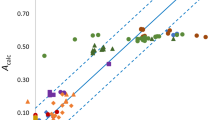
When the β values calculated using the solvent descriptors and the coefficients in Table 3 are plotted against the experimental values it becomes clear that the calculated values for the alcohols and primary or secondary amines lie on lines quite different from that through the values for the other compounds. This is shown in Fig. 4, which compares the experimental β values with those calculated from the descriptors recovered from the density functional calculations and the CM5 based partial charges using the coefficients in Table 3.

Plot of calculated against experimental β values, with βcalc values calculated using the coefficients in Table 3 based on density functional calculations and CM5 derived partial charges. Symbols: light blue circles, alkanes and aromatics; dark blue squares, halogenated alkanes and aromatics; orange circles, esters and carbonates; green circles, carboxylic acids; purple circles, ketones; black diamonds, ethers; red triangles, nitriles; dark green squares, RS, RS = O, RP = O; blue circles, amides; light brown triangles, water alcohols and polyols; grey triangles, pyridines; brown squares, amines and anilines; the solid red line represents perfect agreement (Color figure online)
The plots of values of β calculated using the other coefficients in Table 3 show similar patterns, with the calculated values for the alcohols and aniline or amines with NH hydrogens are systematically out of agreement with the experimental values.
In the case of the DN* values the same pattern is observed except that, in these cases, the values for the amines, anilines and pyridines lie off the line through the data for the other systems. Given the fact that, in contrast to most compounds, the DN* values of several of these nitrogen bases are significantly higher than their β values (see Fig. 1), it is perhaps not surprising that there isn’t good agreement between the calculated and experimental DN* values.
In view of the consistency of these results the β and DN* values were regressed with the values for the alcohols and NH hydrogen bases removed from the β data set and those of the alcohols and amines, anilines and pyridines removed from the DN* data set. The results are shown in Tables 4 and 5 which give the results of the regressions of the β and DN* values (84 and 77 data, respectively) against all seven descriptors and against those with statistically significant coefficients, respectively.
The results shown in Tables 4 and 5 are clear and essentially consistent. Thus, for the β values, all four regressions indicate that only the dependences on Qq− and QE(OMO) are statistically significant. This is also true for the DN* values, except for the regression using the Hartree–Fock and CM5 charge derived molecular descriptors, where the dependences on QP, QDP and Qq+ are marginally significant, with p values of 0.01, 0.03 and 0.01, respectively, as compared to Qq− and QE(OMO) for which the p values are 9 × 10−20 and 2 × 10−12, respectively.
The β and DN* values calculated using the four sets of descriptors and the coefficients in Table 5 are compared to the experimental values in Fig. 5.

Plots of calculated against experimental β values, with βcalc values calculated using the coefficients in Table 5. Symbols: light blue circles, alkanes and aromatics; dark blue squares, halogenated alkanes and aromatics; orange circles, esters and carbonates; green circles, carboxylic acids; purple circles, ketones; black diamonds, ethers; red triangles, nitriles; dark green squares, RS, RS = O, RP = O; blue circles, amides; the solid red line represents perfect agreement. a Density functional calculations and CM5 derived partial charges. b Hartree–Fock calculations and CM5 derived partial charges. c Density functional calculations and Hirshfeld derived partial charges. d Hartree–Fock calculations and Hirshfeld derived partial charges (Color figure online)
5 Discussion
It is clear from Fig. 1 and Table 1 that β or DN* are closely aligned, indicating that, in these cases at least, the hydrogen and Lewis basicities are very similar. The nitrogen bases may be an exception to this, since there is poor agreement between β or DN* for these.
This is borne out by the results of the regressions, which show that, in both cases, only the descriptors reflecting the partial charge on the most negative atom, Qq−, and that reflecting the energy of the charge donating orbital, QE(OMO), have meaningful statistical significance.
As can be seen from Table 5, the results are surprisingly consistent over all of the regressions. Thus, the average value of the intercept is 0.28 with those for the β and DN* values being 0.31 and 0.26, respectively, with relatively little dependence on the calculation method or the method used to estimate the partial atomic charges.
Perhaps surprisingly, given the differences in the orbital energies from the density functional and Hartree–Fock calculations (see Fig. 2), the coefficients recovered for QE(OMO) from the regressions are also substantially independent of the calculation method. The average coefficient recovered for QE(OMO) is 0.56 with the values for the for the β and DN* values being 0.59 and 0.54, respectively.
There are differences for the coefficients recovered for Qq−, reflecting the differences in the Hirshfeld and CM5 based partial charges (see Fig. 3); however, there is consistency between the values recovered from the regressions of the β and DN* data with these averaging 0.94 and 0.62 for descriptors based on the CM5 and Hirshfeld charge models, respectively; again, these are substantially independent of the calculation method.
The fact that the alcohols and nitrogen bases don’t fit on the regression line through the other classes of compounds indicates that there are factors not accounted for in the present treatment. The liquid alcohols and amines are hydrogen bonded liquids, which could impinge on their β values but, in principal, the DN* values are measured for individual molecules where hydrogen bonding shouldn’t be a factor. In practice, this distinction between β and DN* is commonly lost and donor numbers are determined using proxy experiments where, for example, the NMR [22] or visible [23] spectra of a probe molecule are measured in different liquid solvents and the donor number is estimated from the variation of the measured property with donor numbers for other solvents. In these cases, the values determined are properties of the liquid solvent and need not coincide with the property of the isolated molecule.
The nitrogen bases have previously been recognized as differing from the other, commonly oxygen, bases. For example, Popov pointed out very early that pyridine behaved as a weaker base than is indicated by its donor number [24]. This is clearly reflected in Fig. 1, where the DN* values of several nitrogen bases are far higher than would be expected from their β values.
We currently have no explanation for why these compounds differ so strongly from the others for which data are available and will reserve further consideration of this to a later paper.
For the remaining compounds, including nitriles, a number of conclusions can be drawn. Thus, the β and DN* values are essentially different measures of the same molecular property and are substantially determined by the magnitude of the negative charge on the most negative atom in the molecule and the energy of the orbital from which charge donation will occur. Perhaps surprisingly, given what one commonly reads, there is no detectable influence of the dipole moment of the solvent molecule nor of its polarizability on β and DN*.
The relative importances of Qq− and QE(OMO) are made ambiguous by the differences of the partial atomic charges recovered using the Hirshfeld and CM5 methods (see Table 5 for example). Thus, with Qq− based on CM5 charges the contribution of QE(OMO) is around half of that from Qq− while the contributions seem to be approximately equal when Qq− values are calculated using Hirshfeld charges.
Figure 5a–d compare the β values calculated using the four sets of descriptors with the experimental data; the corresponding graphs comparing the calculated DN* with the experimental values are essentially similar and are provided in the supplementary material.
Consideration of Table 5 suggests that there is no clear “best method” for calculating the molecular properties, with the R2 and F values being fairly consistent for the β and DN* values, except for the DN* values calculated using the Hartree–Fock method and CM5 derived charges, where the mild significances of the quadrupole amplitude, charge on the most positive atom and energy of the accepting (unoccupied) orbital likely affect the statistical results.
It is clear from Fig. 5a–d that the values calculated for the nitriles (red triangles) are sensitive to both the calculation method and model used to derive the partial atomic charges, lying on the line through the other values for the DF calculations and CM5 charges but clustering below the line with DF calculations and/or Hirshfeld derived charges. Of course, the values for the other groups of compounds are also shifted by changes in the calculation method or source of partial charges, resulting in the relatively small variations in R2 and F.
The present results don’t allow the choice of either a preferred calculation method or preferred model for estimating the partial atomic charges.
6 Conclusions
Regression of the experimental β and DN* values against the molecular descriptors recovered from the density functional and Hartree–Fock calculations, with the partial atomic charges estimated using the Hirshfeld and CM5 models yield a remarkably consistent picture.
In both cases the calculated values for the alcohols and some nitrogen bases lie systematically off of the regression line through the alkanes, aromatics, halogenated alkanes and aromatics, esters, carbonates, carboxylic acids, ketones, ethers, nitriles, phosphates, sulfides and sulfates. The reason for the differences between the results for the alcohols and amines and the other solvents isn’t clear.
Both β and DN* are substantially determined by two molecular properties: the charge on the most negative atom of the molecule and the molecular orbital from which charge donation would occur. The apparent relative importance of these properties differs, depending on the method of charge estimation, but is consistent between β and DN* for either method. The last point strongly indicates that β and DN* are essentially measures of the same basicity, despite the differences in their initial formulation.
Notes
The quadrupolar amplitude is calculated as \( A = \sqrt {\sum {q_{ij} q_{ij}}} \quad i = x,y,z\quad j = x,y,z \) where the qij are the components of the traceless quadrupole.
References
Svante Arrhenius, S.A.: Recherches sur la Conductivité Galvanique des Électrolytes. Doctoral Dissertation, Stockholm, Royal Publishing House, P.A. Norstedt and Söner (1884)
Brönsted, J.N.: Einige bemerkungen über den begriff der säuren und basen. Rec. des Trav. Chim. Pays-Bas 42, 718–728 (1923)
Lowry, T.M.: The uniqueness of hydrogen. J. Soc. Chem. Ind. 42, 43–47 (1923)
Lewis, G.N.: Valence and the Structure of Atoms and Molecules. American Chemical Society Monograph Series, vol. 14. The Chemical Catalog Co., Inc., New York (1923)
Jensen, W.B.: The Lewis acid–base definitions: a status report. Chem. Rev. 78, 1–22 (1978)
Kamlet, M.J., Taft, R.W.: Linear solvation energy relationships–local empirical rules or fundamental laws of chemistry—a reply to the chemometricians. Acta Chem. Scand. 39, 611–628 (1985)
Marcus, Y.: The properties of organic liquids that are relevant to their use as solvating solvents. Chem. Soc. Rev. 22, 409–416 (1993)
Sandström, M., Persson, I., Person, P.: A study of solvent electron-pair donor ability and Lewis basicity scales. Acta Chem. Scand. 44, 653–675 (1990)
Gutmann, V.: Empirical parameters for donor and acceptor properties of solvents. Electrochim. Acta 21, 661–670 (1976)
Kamlet, M.J., Taft, R.W.: The solvatochromic comparison method. I. The β-scale of solvent hydrogen-bond acceptor (HBA) basicities. J. Am. Chem. Soc. 98, 377–383 (1976)
Eckert, F., Clampt, A.: Accurate prediction of basicity in aqueous solution with COSMO-RS. J. Compd. Chem. 27, 11–19 (2006)
Schwöbel, J., Ebert, R.-U., Kühne, R., Schüürmann, G.: Predictioon of the intrinsic hydrogen bond acceptor strength of organic compounds by local molecular parameters. J. Chem. Inf. Model. 49, 956–962 (2009)
Green, A.J., Popelier, P.L.A.: Theoretical prediction of hydrogen-bond basicity pK BHX using quantum chemical topology descriptors. J. Chem. Inf. Model. 54, 553–561 (2014)
Kaupmees, K., Trummal, A., Leito, I.: Basicities of strong bases in water: a computational study. Croat. Chem. Acta 87, 385–395 (2014)
Le Questel, J.-Y., Berthelot, M., Laurence, C.: Can semi-empirical calculations yield reasonable estimates of hydrogen-bonding basicity? The case of nitriles. J. Chem. Soc. Perkin Trans 2, 711–719 (1997)
Seybold, P.G., Kreye, W.C.: Theoretical estimation of the acidities of alcohols and azoles in gas phase, DMSO and water. Int. J. Quant. Chem. 112, 3769–3776 (2012)
Safi, Z.S., Omar, S.: Proton affinity and molecular basicity of m- and p-supstituted benzamieds in gas phase and solution: a theoretical study. Chem. Phys. Lett. 611–612, 321–330 (2014)
Katritzky, A.R., Fara, D.C., Yang, H., Tamm, K., Tamm, T., Karelson, M.: Quantitative measures of solvent polarity. Chem. Rev. 104, 175–198 (2004)
Devereux, M., Popelier, P.L.A., McLay, I.M.: A refined model for prediction of hydrogen bond acidity and basicity parameters from quantum mechanical descriptors. Phys. Chem. Chem. Phys. 11, 1595–1603 (2009)
Caclelli, I., Campanile, S., Giolitti, A., Molin, D.: Theoretical prediction of Abraham hydrogen bond acidity and basicity factors from a reaction field method. J. Chem. Inf. Model. 45, 3237–3333 (2005)
Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Scalmani, G., Barone, V., Mennucci, B., Petersson, G.A., Nakatsuji, H., Caricato, M., Li, X., Hratchian, H.P., Izmaylov, A.F., Bloino, J., Zheng, G., Sonnenberg, J.L., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Montgomery, J.A., Peralta, J.E., Ogliaro, F., Bearpark, M., Heyd, J.J., Brothers, E., Kudin, K.N., Staroverov, V.N., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A., Burant, J.C., Iyengar, S.S., Tomasi, J., Cossi, M., Rega, N., Millam, J.M., Klene, M., Knox, J.E., Cross, J.B., Bakken, V., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R. E., Yazyev, O., Austin, A.J., Cammi, R., Pomelli, C., Ochterski, J.W., Martin, E.L., Morokuma, K., Zakrzewski, V.G., Voth, G.A., Salvador, P., Dannenberg, J.J., Dapprich, S., Daniels, A.D., Farkas, O., Foresman, J.B., Ortiz, J.V., Cioslowski, J., Fox, D.J.: Gaussian 09 ed. Gaussian, Inc., Wallingford, CT (2009)
Hahn, S., Miller, W.M., Lichtehthaler, R.N., Prausnitz, J.M.: Donor number estimation for oxygen- and nitrogen-containing solvents via proton NMR shift of chloroform. J. Solution Chem. 14, 129–137 (1985)
Rzeszotarska, J., Ranachowski, P., Kalinowski, M.K.: Donor numbers in binary mixtures of dimethyl sulfoxide with dipolar aprotic solvents. Collect. Czech. Chem. Commun. 59, 2201–2208 (1994)
Greenberg, M.S., Popov, A.I.: Spectroscopic studies of ionic solvation. 17. Studies of preferential solvation of sodium ion in non-aqueous mixed solvent by 23Na nuclear magnetic resonance. Spectrochim. Acta A 31, 697–705 (1975)
Acknowledgements
This study was supported by University College Dublin.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Earle Waghorne, W., O’Farrell, C. Solvent Basicity, A Study of Kamlet–Taft β and Gutmann DN Values Using Computationally Derived Molecular Properties. J Solution Chem 47, 1609–1625 (2018). https://doi.org/10.1007/s10953-018-0791-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10953-018-0791-3