
Abstract
Bacteriophage endolysins are potential alternatives to conventional antibiotics for treating multidrug-resistant gram-negative bacterial infections. However, their structure–function relationships are poorly understood, hindering their optimization and application. In this study, we focused on the individual functionality of the C-terminal muramidase domain of Gp127, a modular endolysin from E. coli O157:H7 bacteriophage PhaxI. This domain is responsible for the enzymatic activity, whereas the N-terminal domain binds to the bacterial cell wall. Through protein modeling, docking experiments, and molecular dynamics simulations, we investigated the activity, stability, and interactions of the isolated C-terminal domain with its ligand. We also assessed its expression, solubility, toxicity, and lytic activity using the experimental data. Our results revealed that the C-terminal domain exhibits high activity and toxicity when tested individually, and its expression is regulated in different hosts to prevent self-destruction. Furthermore, we validated the muralytic activity of the purified refolded protein by zymography and standardized assays. These findings challenge the need for the N-terminal binding domain to arrange the active site and adjust the gap between crucial residues for peptidoglycan cleavage. Our study shed light on the three-dimensional structure and functionality of muramidase endolysins, thereby enriching the existing knowledge pool and laying a foundation for accurate in silico modeling and the informed design of next-generation enzybiotic treatments.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Spurred by the prevalence and spread of multidrug-resistant bacteria, there has been a universal call for new classes of antibiotics over the last decade. Bacteriophages (phages) and phage-derived lytic enzymes, as natural predators of bacteria, have been regarded as promising candidates since their discovery [1]. Endolysins are phage-encoded enzymes expressed at the end of the lytic cycle to degrade the host cell wall, which subsequently causes bacterial lysis and releases newly produced phage particles [2]. Over billions of years, intimate co-evolution between bacteria and phages has resulted in extraordinarily active and specific endolysins [3]. The exogenous application of recombinant lysins as enzyme-based antibiotics (enzybiotics) has several advantages over routine antibiotics, including rapid and effective action against stationary and exponential-phase bacteria, low resistance potential, killing of targeted pathogens with low activity towards normal flora, minimal side effects [4], and biodegradability without accumulation due to their proteinic nature. Furthermore, Artilysin (artificial endolysin) technology, which involves the fusion of endolysins with outer membrane permeabilizer peptides, has provided an efficient method for killing gram-negative bacteria [5, 6]. These potential benefits have led researchers to engineer and develop endolysin-based enzybiotics against multidrug-resistant pathogens.
The architecture of endolysins varies according to their origin. Endolysins from Gram-positive-targeting bacteriophages are typically modular, consisting of one or two enzymatic active domains (EADs) and a cell-wall-binding domain (CWBD) connected by a short linker [7]. Although most endolysins encoded by Gram-negative-targeting phage have a globular structure containing a single EAD [8, 9], there are some modular structure examples with higher activity than their globular counterparts in this group [10]. The CWBD recognizes cell-wall characteristics specific to bacteria, while the EAD is responsible for breaking particular bonds within the cell-wall peptidoglycan (PG/murein) structure [7]. The PG consists of alternating residues of β-(1,4) glycosidic bonded N-acetylglucosamine (GlcNAc) and N-acetylmuramic acid (MurNAc) in which D-lactoyl group of the MurNAc residues are substituted by a pentapeptide stem [11]. The classification of endolysin is done according to the enzymatic activity type of EADs and consists of three main categories: (i) amidases which hydrolyze the amide bond between the pentapeptide stems and the sugar backbone; (ii) endopeptidases which break the bond between two amino acids; and (iii) glycosidases which target the β-(1,4) glycosidic bonds in the sugar backbone, included glucosaminidases, muramidases (lysozymes) and lytic transglycosylases. Glucosaminidases act on the reducing side of GlcNAc, whereas muramidases and lytic transglycosylases operate on the reducing side of MurNAc with the difference that they release a product including 1,6-anhydro ring at the MurNAc residue in a lytic transglycosylases reactions [2, 12].
Muramidases, also known as lysozymes, are considered to be the most widely distributed muralytic enzymes. One of their characteristics is the amino-acid sequence's incredible diversity. In most of them, two central residues are involved in catalysis: glutamic acid (Glu, E), a general-acid catalytic residue, and aspartic acid (Asp, D), a general-base catalyst residue, or cysteine (Cys, C). In addition to Glu and Asp/Cys, a third lateral and catalytically influential residue, threonine (Thr) or serine (Ser), may be involved in the catalytic reaction [13]. Goose egg-white lysozyme (GEWL) is exceptional, with only one catalytic residue – Glu [14]- characteristic of lytic transglycosylases despite the difference in cleavage products. Among endolysins with lysozyme activity, two classes have been reported so far. Muramidases from the glycoside hydrolase family 25 (with Glu and Asp residues) and muramidases targeting Gram-negative bacteria (unusual catalytic center characterized by only one/two Glu residues and no Asp) [12]. Two mechanisms have been suggested based on an inversion [15] or retention [16] of the anomeric center of the cleaved PG glycoside. Since they only cleave bacterial PG and do not catalyze the cleavage of substrate analogs, which are more manageable in studying enzymatic processes [16], determining the stereochemical products is complex, and the role of catalytic residues and the precise mechanism remains open [12].
One of the most common engineering approaches to modify endolysins, in addition to producing chimeric lytic enzymes, is the deletion of certain domains [17]. Protein truncation modifies protein properties and provides insights into their structures. This approach has been utilized to alter various aspects of endolysins, including their lytic and antibacterial activities, dependence on CWBD, specificity, plasma half-life, and thermostability [3, 17]. However, domain truncation should be carefully performed and tested for each endolysin. Deleting the CWBD, which is usually responsible for peptidoglycan binding or improving cleavage, may reduce the lytic activity of endolysins [18,19,20]. The removal of CWBD may disrupt the folding pathway of the EAD, leading to the formation of misfolded or unfolded intermediates that are prone to aggregation. Additionally, the CWBD may exert a stabilizing effect on the EAD, protecting it from denaturation and aggregation under stress conditions such as high temperature, low pH, or proteolytic degradation. The CWBD might also play a structural role in maintaining the conformation and dynamics of the EAD. However, this is not always the case, and sometimes the removal of CWBD can improve the activity or spectrum of endolysins. This depends on various factors such as the type of endolysin, catalytic mechanism, experimental conditions, and bacterial species [3, 17]. Examples of truncated endolysins with improved performance include PlyGBS [21], LysK [22,23,24], PlyLCAT [25], PlyBa04CAT [26], and CD27L [27]. The mechanisms behind the increased activity or spectrum of these truncated endolysins are not fully understood, but they might be related to the smaller size and positive charge of the catalytic domain, which could help them penetrate the peptidoglycan layer more easily [3, 17, 25]. Thus, the role of the binding domain in endolysins is complex, variable, and requires careful analysis and validation of individual endolysins.
The present study aims to investigate the structural characteristics contributing to the lytic activities of muramidase-targeting gram-negative bacteria, leading to the production of highly effective enzybiotics. To achieve this, we utilized the recombinant E. coli O157: H7 phage modular endolysin (Gp127) as a model, which is encoded by PhaxI phage previously isolated in our laboratory [28]. We hypothesized that the EAD of Gp127 even in the absence of CWBD is sufficient for its catalytic activity, and that its structure and function can be modulated by rational design for the production of effective enzybiotics. To test our hypothesis, we performed the examination of in silico tools and comparison with in vitro data by (i) protein modeling and structure validation by circular dichroism (CD) analysis, (ii) docking experiment and molecular dynamic simulation for pocket detection and stability verification, and (iii) expression, purification, and enzyme activity assay of the truncated form.
2 Materials and Methods
2.1 Sequence Mining
Sequence-based analysis of the bacteriophages, isolated and sequenced previously in our laboratory, was performed to disclosure of a muramidase-type endolysin. GP127, a hypothetical protein with NCBI Accession No. YP_007002724.1, encoded by PhaxI phage and specific to E. coli O157: H7 [28], was selected as a query for further sequence homology analysis using NCBI BLASTP [29], ExPASy ProtParam [30], and HHpred, allowing a broad search of databases [31]. To explore and map the active site, orthologous sequences of the determined catalytic domain, restricted to those corresponding to non-redundant protein sequence (nr) and Viruses (taxid:10,239), were identified and analyzed using PSI-BLAST (Position-Specific Iterative Basic Local Alignment Search Tool) [32] and PFAM (Protein family database) [33] respectively. Multiple sequence alignment (MSA) of aligned sequences was carried out using MAFFT v.7 [34] online server with 'Default' settings and the annotation of results was performed by the WebLogo server [35].
2.2 Protein modeling
The protein's three-dimensional structure was predicted by YASARA suit Version 20.12.24.W.64 [36]. Homology modeling parameters set on slow modeling speed, 3 PSI-BLAST iterations to template search with a maximum E-value of 0.5, 5 templates total, and up to 5 best alignments per template. Fifty confirmations were tried per loop, and a maximum number of 10 were considered in oligomerization states and termini extensions for missing residues. Extracting a PSSM, position-specific scoring matrix, from UniRef90 and matching findings by searching the PDB was done to achieve templates. To avoid misleading results, the C-terminal purification tag was excluded from the template search. The template with the top total score (the product of the scores of BAST alignment, the WHAT_CHECK [37] quality, and the target coverage) from the hits found was selected as the main template. For alignment correction and modeling of loops, the target sequence profile creation and secondary structure prediction were made by running PSI-BLAST and PSI-Pred algorithms [38], respectively. After building the three initial homology models, they were sorted by their overall Z-scores; a number indicates standard deviations in the quality of the protein model and high-resolution X-ray structures [36, 37, 39]. Furthermore, a hybrid model with a combination of the best segments of the three models was built, hoping to raise accuracy, but discarded due to low quality. For quality improvement, the final model (Gp127) was refined and energy minimized by explicit solvent molecular dynamics simulation (MDS) with the YASARA2 force field in the YASARA program [40] as well as GalaxyRefine Server [41]. The optimized model were assessed using PROCHECK [39], ProSA-web [42], and Verify3D [43] to evaluate the stereochemical quality.
2.3 Molecular Dynamic Simulation
To yield stable structures, the refined model was subjected to MDS for 150 ns (3 × 50 ns) using the Amber14 force field [44] in the YASARA program [36] with "md_analyze.mcr" macro. The pKa of side chains was predicted [45] at physiological pH 7.4, and the allotment of protonation states was conducted correspondingly. The cube simulation cell containing physiological solution (water and 0.9% NaCl) was positioned 20 angstroms away from the protein and then adjusted in size to achieve a solvent density of 0.997 g·L−1 at a temperature of 300 K. Particle mesh Ewald (PME) [46] and the cutoff radius of 8 Å for nonbonded interactions were utilized for the most accuracy. The periodic boundary for cell boundaries, constrained hydrogen atoms, four fs timestep, and constant pressure 1 bar and temperature 300 °K (NPT ensemble) [47] were considered for the main simulation. Snapshots were recorded every 100 ps and then analyzed using "md_analyze.mcr" macro for calculating the total potential energy of the system, alpha-carbon Root-Mean-Square Deviations (CA-RMSDs), and per-residue protein secondary structure as a function of simulation time. The trajectory was then analyzed in 10 blocks using the "md_analyzeblock" macro, and after elimination of equilibration time (20 ns, four first blocks), the energy-minimized structure was extracted per block. Every structure was analyzed using PROCHECK [39]. The truncated form, GP127-EAD (90–263; Muramidase domain), was stabilized as the same manner. The highest quality of every structure was selected as the receptor for the docking experiment.
2.4 Docking Experiment
Docking experiment was performed using the YASARA program [36] to determine the critical residues: (i) Library designing: Intact peptidoglycan substrate was extracted from a crystal structure (PDB ID: 2MTZ), and a tetramer AMU-NAG-AMU-NAG molecule was built from it. After hydrogen addition, optimization, and energy minimization, the structure was subjected to MDS for 300 ns using the Amber14 force field at physiological pH 7.4. As mentioned above, the trajectory was blocked, and the resulting 60 tautomers of the AMU-NAG-AMU-NAG molecule were considered as a library for the next step. (ii) Virtual screening: the protein's binding pocket was mapped using the CASTp web server [48], and then the cube cell was located 5 Å around the predicted residues. The library screening was done with the built-in "Dock_runscreening" macro for 100 docking runs for each ligand using AutoDock Vina [49], Flexible residues of the receptor as well as ligands, and clustering of poses according to 5 Å heavy atom RMSDs [36]. AMBER03 force field [50] was initially utilized to assign point charges and subsequently damped the system to simulate the less polar Gasteiger charges, which was employed to optimize the AutoDock scoring process. The results were sorted by binding energy according to YASARA report parameters (more positive energies show stronger ligand binding) [36]. (iii) Pocket confirming: The most popular complexes were subjected to MDS for 150 ns (3 × 50 ns) and analysis similar to the primary simulation process. Furthermore, Per-residue contacts with ligand, Ligand conformation RMSD after superposing on the ligand, and Ligand movement RMSD after superposing on the receptor were considered for estimating the stability of interactions and pocket confirming. The hydrogenic and hydrophobic contacts after MDS were represented by LigPlot analysis [51], predicted using the PDBSum server [52].
2.5 Recombinant Plasmid Construction
The MEGAWHOP method [53] was used for cloning constructs of both total protein (Gp127) and its truncated form (Gp127-EAD) using the pET28a ( +) Kanr (Novagen-USA) expression vector, provides a C-terminal 6-His-tag for affinity chromatography. Firstly, orf84 sequence was amplified from the PhaxI genome by Pfu DNA polymerase (Thermo Fischer Scientific) and hybrid primers, F1: CCCCTCTAGAAATAATTTTGTTTAACTTTAAGAAGGAGATATACCATGGCGATTCTAAAACTTGGAAACCGAGG, F2 (for truncated form): CCCCTCTAGAAATAATTTTGTTTAACTTTAAGAAGGAGATATACCATGGAACTGGCCAGTATGAAAGCTGTGAATCAG and R: GTGGTGGTGGTGGTGGTGCTCGAGACAGAAACTCTTGTATGCTGCCG (The underlined characters indicate sequences complementary to the pET28a plasmid). The products were then used as a mega-primer for the second round of PCR to replicate the entire destination plasmid using Phusion DNA polymerase (New England Biolabs). Finally, constructs were purified from parenteral plasmid using DpnI (Takara Bio) and subsequently verified by sequencing.
2.6 Recombinant Protein Preparation
Gp127 was produced as previously described using E. coli Bl21(DE3) pLysS (Thermo Fischer Scientific) with the same purification buffers, followed by dialysis against PBS (pH 7.4) for 16 h [10]. The protein that underwent dialysis was preserved at a temperature of 4°C without any discernible reduction in its activity, and subsequently employed for the purpose of conducting a CD analysis. Recombinant expression of Gp127-EAD was checked in 50 mL lysogeny broth (LB) using E. coli strains: Bl21(DE3) pLysS, BL21(DE3) (Thermo Fischer Scientific), Rosettagami(DE3) (Novagen), BL21-Gold(DE3) (Agilent Technologies), and C43(DE3) (Sigma-Aldrich), to explore the host with the ability to overexpress protein.
Small-scale expression of Gp127-EAD was performed in 50 mL of LB with 1% glucose using E. coli BL21-Gold(DE3) for selecting the appropriate solubilization buffer. The expression was induced at OD600nm of 0.6 using 0.25 mM IPTG, and incubation was continued for 4 h at 37°C. The cells were centrifuged at 7000 × g for 10 min at 4 °C. The resulting pellet was resuspended in lysis buffer I (100 mM NaH2PO4, 10 mM Tris–HCl, 1 mM PMSF, pH 8), disrupted by sonication (20 cycles:10s pulse and 10s rest on ice), and clarified at 12,000 × g for 15 min at 4 °C. The pellet was subjected to lysis buffer II (lysis buffer I containing 0.1% Triton X-100), and sonication and centrifugation were performed as described above. After washing and aliquoting into six fractions, the final pellet was screened for six different solubilization agents added to lysis buffer 1 at pH 8 [54, 55]: A. 8 M urea, B. 6 M GuHCl, C. 2 M urea (pH 4), D. 50 mM NaCl, 5% glycerol, 0.4% Triton (pH 7.9), E. 2 M urea, 6 M β-mercaptoethanol, F. 2 M urea, 6 M n-propanol. Suspended inclusion bodies were mixed well by vertexing and incubated for 2 h at room temperature. Both insoluble (precipitate) and soluble (supernatant) samples were loaded onto a 12% SDS-PAGE gel and subsequently, the identification of protein bands was carried out through staining with Coomassie Brilliant Blue.
Larger scale production of Gp127-EAD was performed in 200 mL media, as mentioned above. The final pellet was resuspended in the selected solubilization buffer (100 mM NaH2PO4, 10 mM Tris–HCl, 1 mM PMSF, 5 M GuHCl, pH 8) and applied to a HiTrap™ IMAC HP 1 ml column (GE Healthcare Life Science) charged with 0.1 M NiSO4. The column was washed with wash buffer (100 mM NaH2PO4, 10mM Tris–HCl, 20mM imidazole, and 10% glycerol) and then proteins were eluted with elution buffer (100mM NaH2PO4, 10mM Tris–HCl, 5 M GuHCl, and 300 mM imidazole). Refolding of denatured forms were performed using the dilution method [54, 56] (final GuHCl concentration: 0.1 M and 0.5M in Phosphate Buffered Saline (PBS). The effects of different additives were screened to prevent aggregate formation and increase the refolding yield, according to a previously described approach [56]. Flash dilution was carried out in a 96-well plate by quickly adding 10 µL of solubilized protein to 190 µL of different refolding solutions:1. 1.4 M Acetone, 2. 1.4 M DMSO, 3. 1.4 M Ethanol, 4. 1.4 M Urea, 5. 1.4 M Triton-X100, 6. 0.05% (w/v) PEG [57], 7. 1mM β-mercaptoethanol, 8. 10% Sucrose + 1mM EDTA [54], 9. 10% Glycerol, 10. 10% Sorbitol, 11. 10% Trehalose [56], 12. 50 mM Glycine. The final protein concentration was 16 µg/ml using the Bradford assay. The absorbance at 320 nm was measured after 60 min, and the sample with the lowest turbidity was chosen as the refolding solution. The purified denatured Gp127-EAD stocks were preserved at 4°C and refolded immediately before the in vitro assay.
2.7 Muralytic Activity
For the muralytic activity of the enzybiotics, outer membrane (OM) permeabilized E. coli ATCC 8739 bacteria was prepared as the substrate [58]. Briefly, the mid-exponential growth phase was centrifuged and incubated in a chloroform-saturated 50 mM Tris–HCl buffer pH 7.7 for 45 min. Next, the permeabilized cells were washed in PBS buffer pH 7.4 and resuspended to an OD600 of 1.5. A zymogram qualitative assay was performed using 12% SDS-PAGE, in which 0.2% (wt/vol) OM permeabilized E. coli ATCC 8739 was incorporated into the separating gel before polymerization. After electrophoresis, the gel was incubated in PBS (pH 7.4) with 1% Triton X-100 for 16 h at 37°C to renature the proteins for peptidoglycan hydrolysis. After 0.5 h staining with 0.1% methylene blue in 0.01% KOH and washing, the muralytic activity of the individual protein band was observed as a clear zone [59]. Lytic activity was quantified using the ActivityCalculator tool and the standardized method (https://www.biw.kuleuven.be/logt/ActivityCalculator.htm) [60]. Thirty microliters of the desired enzybiotic was added to 270 µL of OM-permeabilized E. coli ATCC 8739 cells in a 96-well plate. The turbidity reduction assay was performed using OD655 spectrophotometrically decreasing over time in a Synergy HTX multimode plate reader.
2.8 Circular Dichroism Spectroscopy
The recording of CD spectra of GP127 was conducted using the Jasco J-810 Spectropolarimeter. Far-UV measurement was taken using a quartz cuvette with a path length of 1 mm. The 0.2 mg/ml protein solution was scanned within the range of 195 to 250 nm. The protein's CD spectrum was adjusted by subtracting the spectrum of the blank PBS solution (pH 7.4) to ensure accurate measurements of the protein's spectral properties.
3 Results
3.1 Highlighting Sequence Features
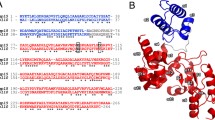
As regards analysis results of the E. coli phage PhaxI genome, a 264-aa protein (GP127, an inferred molecular mass of 28.8 kDa and theoretical pI of 8.99) predicted to act as a modular endolysin (Fig. 1, A), with an N-terminal CWBD (9–64; PG_binding_1 domain; pfam01471) and a C-terminal EAD (90–263; Muramidase domain; pfam11860) [61]. Muramidase (lysozyme) domain founded in bacteria (mainly Proteobacteria) and phage (Caudovirales morphotype) is responsible for cleavage the β -(1,4) glycosidic bond in the backbone of the peptidoglycan. Pfam's lytic enzymes that contain a muramidase catalytic domain exhibit a variety of architectures, ranging from a solitary muramidase catalytic domain to domains featuring muramidase alongside recognized CWBDs (with three alpha-helices) at the N- or C-terminus. To identify the evolutionary conservation pattern, the muramidase domain of PhaxI endolysin was compared against viruses using PSI-BLAST and 222 aligned sequences (E value < 0.01, available at the time of the study) were then submitted to MAFFT server. WebLogo analyses of the catalytic domain orthologs showed 21 identical amino acids (Fig. 1, C). Although GP127 has 100% sequence similarity to one of the muramidase family members, the characterized Salmonella phage 10 endolysin [10], the structure of the protein was not reported at the time of our study. HHpred search indicated that there was 46% identity to Burkholderia AP3 phage endolysin, PDB ID: 5NM7 [62], with an E-value of 7.8e-40 and a probability of 100% (Fig. 1, B). A BLAST search restricted to the Protein Data Bank (PDB) identified four proteins (5NM7, 4LPQ, 1LBU, 4FET), and only one of them, 5NM7, covered the muramidase domain of GP127.

Overview of the GP127 sequence. A. Domain architecture of the modular GP127 endolysin, the N-terminal CWBD (blue: residues Arg9 to Ile64); the hinge region (residues Arg65 to Val89); the C-terminal EAD (red: residues Glu90 to Phe263). B. Homology structure detection of GP127 sequence by HHpred analysis and HMM-HMM comparison. C. Logo representation of the EAD orthologous sequences
3.2 Three-Dimensional Structures
The lack of an experimentally resolved structure prompted us to generate a 3D model for GP127 using the YASARA program, which runs three PSI-BLAST iterations (to adopt PSSM from UniRef90) and PDB searches (to find a match) to identify possible templates (Table S1). Three models were predicted for GP127 based on the PDB structure 5NM7 (chain G), the best template with the highest percentage of BLAST E-value, alignment score, and coverage. According to the related alignment of the selected model, 256 of 272 target residues (94.1%) are aligned to template residues where the sequence identity and similarity are 46.1% and 60.2% respectively ('similar' means that the BLOSUM62 score is > 0). Improving the quality of the model using a hybrid construction (a combination of the best parts of the three models) failed, and the initial structure was considered as the final model. For quality improvement, the structure was refined and the energy minimized using GalaxyRefine Server and the YASARA program. The optimized model was assessed using Ramachandran analysis, which showed 96.7% of residues in the most favored regions without any residues in generously allowed and disallowed regions (Figure S1, A). The overall G-factors was 0.29. Among the main chain stereochemical parameters, the overall G-factor and omega angle standard deviation were better than the ideal values, whereas the other four properties were within acceptable ranges (Table 1; Figure S2). Regarding the side-chain stereochemical parameters, the Chi-1 gauche minus standard deviation was within the suitable regions, and all other parameters showed better quality than the ideal values (Table 2; Figure S3). Good and reliable structural quality was further confirmed with ProSA-web and Verify3D scores of -6.65 and 98.53, respectively (Figure S1, B-C).
To yield stable structures, refined GP127 and its truncated form (GP127-EAD) were subjected to MDS for 150 ns using the YASARA program. The average values for the total potential energy of GP127 and GP127-EAD were -1,036,747 and 912,452 kJ mol −1 with SDR of 0.08% and 0.09% respectively (Table S2, A). CA-RMSD indicated an average value of 0.38 ± 0.05 nm and 0.27 ± 0.03 nm for GP127 and GP 127-EAD structures, respectively (Table S2, B). The energy and CA-RMSD stabilized over the course of the simulation period (Figure S4). The last snapshot of each simulation was saved using the "md_analyze.mcr" macro. The last GP127 structure revealed a total of fifteen helices: α 1 (G10- G24), α 2 (G34-A48), α 3 (G57-N66), α 4 (E77-E86), α 5 (E90-S102), α 6 (E118-F130), α 7 (Q132-Q140), α 8 (D157-L169), α 9 (E171-Y175), α 10 (N189-C193), α 11 (N 197-F 205), α 12 (E 209-K 221), α 13 (D223-N232), α 14 (N234-N243), and α 15 (Q251-K261) (Fig. 2, A). Far-UV CD data were acquired for GP127 (Figure S5), further validating the in silico model. The predicted secondary structure content of the computational model estimated using YASARA was found to be 59.2% helix, 4% β-sheet, 8.1% turn, and 28.7% coil, similar to those obtained from the GP127 CD spectrum analyzed with the BeStSel server [63] except for some minor discrepancies (56.6% helix, 5.2% β-sheet, 8.6% turn, and 29.6% coil). The good stereochemical quality of the last GP127-EAD structure was assessed by Ramachandran analysis (95.8% of residues in the most favored regions with an overall G-factor of 0.33) and further confirmed with Verify3D and ProSA-web scores of 98.53 and -6.65, respectively (Figure S6), which revealed a secondary structure similar to the EAD of GP127 except one extra β-sheet with 61.4% helix, 8.2% β-sheet, 9.2% turn, and 21.2% coil. The secondary structure composition of both GP127 and GP127-EAD residues did not undergo significant changes during the simulations (Fig. 3, A). The average percentage of GP127 secondary structure was 57.6% ± 0.65 helix, 5.3% ± 0.12 β-sheet, 9.9% ± 0.61 turn, and 26.5% ± 0.78 coil. GP127-EAD had an average of 57.2% ± 0.5 helix, 6.2% ± 0.98 β-sheet, 11.2% ± 0.41 turn, and 23.9% ± 1.2 coil during simulation. The observed alterations are consistent with the forecasts and marginal conformational transitions were observed at the junctions of alpha helices and/or beta strands with coils in the N-terminus or C-terminus.

Three-dimensional models of GP127 (I) and GP127_EAD (II) after MDS. A. The predicted chains of GP127 [N-terminal CWBD (red), C-terminal EAD (cyan), hinge region (yellow)] and GP127_EAD. B. Amino acid conservation according to the ConSurf score. C. Amino acid solubility according to the CamSol method

Per-residue protein secondary structure of GP127 (I) and GP127_EAD (II) without (A) or with (B) ligand during MDS. [Helix (blue), sheet (red), turn (green), coil (cyan), helix 3 10 (yellow) and helix Pi (orange)]. The plots were generated by YASARA
Further structural analysis was performed to visualize the conserved and soluble spots on the surface of the predicted 3D structures using the ConSurf [64] and CamSol [65] servers, respectively. Conservation analysis revealed remarkably conserved residues that are situated in close proximity to the main groove of the protein. It is highly plausible that these residues comprise the PG binding area of the enzyme. (Fig. 2, B). The CamSol method identified highly soluble spots in CWBD (Fig. 2, C).
At the end of our in silico study, the crystal structure (resolution: 2.99 Å) of an E. coli O157: H7 phage endolysin, LysT84 (PDB ID:7RUM), was reported with 98.5% sequence identity to GP127 and an RMSD of 1.5 [66], which strongly verified the quality of our proposed 3D structure using the YASARA program (Fig. 4, A, I). Therefore, we updated our HHpred analysis with the GP127-EAD sequence to identify new crystal structures with similar 3D structures to this muramidase domain (Table 3). Three structures were identified with a probability of 100%. LysT84 and AP3gp15 (our template for GP127 homology modeling) are modular endolysins, but Pae87 lacks the CWBD present in the N-terminal. This recently reported crystal structure of Pseudomonas aeruginosa phage endolysin (PDB ID: 7Q4T, resolution: 1.27 Å) [67] exhibits the typical lysozyme-like alpha/beta fold and has two putative glutamate catalytic residues, Glu29 and Glu46. The distance between delta carbons of the two Glu residues is 16.21 Å. The structural alignment between GP127-EAD and Pae87 using YASARA showed an RMSD of 1.925 A over 151 aligned residues with 54.97% sequence identity (Fig. 4, A, II), and Glu 101 of GP127-EAD, positioned similarly to Glu29 (Fig. 4, B, I). On the other hand, Glu 118 approximately positioned to Glu 46 but 18.79 Å away from Glu 101. Another recently published result concerning muramidase AbLys1 (PDB ID:8APP), belonging to glycoside hydrolase family 24, which contains a small antenna-like N-terminal domain and a larger C-terminal domain with six α-helices and a β-hairpin, has indicated the involvement of the conserved catalytic triad (Glu52, Asp61, and Thr67) using bioinformatic analysis [68]. Interestingly, although no significant sequence similarity to GP127-EAD was found using BLASTP analysis, Glu52 and Thr67 showed positional similarity to Glu101 and Ser179 of GP127-EAD, respectively (Fig. 4, B, II). It is important to note that Glu118 of GP127-EAD is located further away from Asp61. The distance between the delta carbon of Glu52 and the gamma carbon of Asp61 is 12.46 Å, which is smaller than the corresponding groups observed in both GP127-EAD and Pae87.

Structural alignment of the predicted structures. A. Superimposition of the GP127 on: I. LysT84 (green), and II. Pae87 (purple). [N-terminal CWBD (red), C-terminal EAD (cyan), hinge region (yellow)]. B. Comparison of the putative catalytic center of the GP127-EAD with: I. Pae87 (purple) and II. AbLys1(orange); putative catalytic residues are shown as the sticks
3.3 Substrate Binding Region
To determine the critical binding residues, refine, and estimate the stability of the ligand interactions as a factor for activity verification of GP127-EAD, a three-step experiment was performed using the YASARA program. Previous reports indicate that variations exist in the binding affinity between tautomers and that preferential binding and stability of a particular tautomer form over the other results in a shift in the equilibrium of tautomeric forms [69, 70]. The binding conformations of 60 tautomers of the AMU-NAG-AMU-NAG molecule within the predicted active site of both GP127 and GP127-EAD were screened to select the correct tautomer. The best complex was selected based on the binding energy and interaction of the ligand with Glu 101, formerly reported as a critical residue, and then subjected to MDS to investigate the pattern of receptor flexibility in ligand binding (Fig. 5). The GP127 and GP127-EAD complexes had average potential energies of -1,008,309 and -1,020,838 kJ mol − 1, respectively, with an SDR of 0.09% (Table S3, A). The average value of CA-RMSD was 0.23 ± 0.04 nm and 0.22 ± 0.03 nm for GP127 and GP127-EAD structures, respectively (Table S3, B), indicating low structural changes in the presence of ligand. The RMSD of the heavy atoms of the ligand over time was measured after superposing the receptor onto its reference structure. This procedure provides insights into the motion of the ligand in its binding pocket, which is stable for both GP127-EAD and GP127. In addition, the RMSD of the ligand atoms over time was measured after superposing on the reference structure of the ligand. The acquired data summarized the conformational changes of the ligand, indicating the stable conformation of the selected tautomer for both GP127 and GP127-EAD during the simulation. Furthermore, the per-residue number of contacts (Fig. 6) and the secondary structure composition (Fig. 3, B) of both structures interacting with the ligand did not change significantly compared to that without the ligand during the simulation time. The average of binding energy was -312.81 and -332.92 for GP127 and GP127-EAD, respectively. The types of contacts made with the ligand as a function of simulation time (Fig. 7) indicate that GP127_EAD established a higher number of interactions with the ligand (Fig. 8; Table 4). The most critical binding residues were Val100, Glu101, Gln184, Asn189, and Asn250 which were present in both last structures and their conservation was verified according to both ConSurf (Fig. 2, B) and Weblogo analysis (Fig. 1, C), which emphasizes their importance in the PG interaction.

Stability comparison of GP127 (I) and GP127_EAD (II) interacted with ligand during MDS. A. Total potential energy. B. CA-RMSD values. C. Ligand movement after superposing on the receptor. D. Ligand conformation after superposing on the ligand. [n = 3 × 50 ns, nm: nanometer, ns: nanosecond]

Per-residue number of contacts of GP127 (I) and GP127_EAD (II) without (A) or with (B) ligand during MDS. (See plot legend). The plots were generated by YASARA

Per-residue contacts of GP127 (I) and GP127_EAD (II) with ligand during MDS. [hydrogen bonds (red), hydrophobic contacts (green), ionic interactions (blue), mixed contacts (plot legend)]. Plots were generated using YASARA software

Docking analyses of GP127 (I) and GP127_EAD (II) interacting with the ligand. A. 3D representation of the last structure using YASARA [hydrogen bonds: broken yellow lines; hydrophobic contacts: green lines]. B. 2D Ligplot interaction diagrams [hydrogen bonds (broken green lines) and hydrophobic contacts (brick-red spoked arcs)]
3.4 In Vitro Activity
For in vitro validation of GP127-EAD activity, recombinant protein expression was first examined using E. coli Bl21(DE3) pLysS as it was done with GP127. In contrast to GP127, which was overexpressed in soluble form (Figure S7, I), GP127-EAD was undetectable in SDS-PAGE analysis (Figure S7, II) under the different IPTG concentrations, temperatures and media conditions (data not shown). The growth of E. coli Bl21(DE3) pLysS cells transformed with either the empty vector, GP127, or GP127-EAD was monitored by measuring the optical density at 600 nm. It was found that GP127 was toxic to the cells, as indicated by the lower optical density values; however, cell growth was not significantly affected by GP127-EAD compared with the empty vector (Fig. 9, A). Therefore, expression was tested in four other E. coli strains, and SDS-PAGE analysis showed that E. coli BL21-Gold (DE3) cells are able to overexpress GP127-EAD, but as inclusion bodies (IBs). GP127 was successfully overexpressed in the E. coli BL21-Gold (DE3) cells in soluble form, similar to E. coli Bl21(DE3) pLysS. The optical density of the culture containing the GP127-EAD construct showed an upward trend in contrast to soluble GP127, indicating no GP127-EAD activity in the inclusion body form (Fig. 9, B). The increase in optical density may indicate cell proliferation and production of inactive proteins after IPTG induction. However, it is important to consider another possible cause for this observation, which could be light scattering from inclusions rather than increased growth. Various potential solutions were explored to address the inclusion body formation of GP12-EAD during the expression process. These included incubation at low temperature, induction with lower IPTG concentrations, supplementation of growth media with ethanol, glycerol, or glucose, and alteration of temperature to induce heat shock protein expression. However, none of these approaches resulted in the conversion of GP12-EAD IBs into a soluble form (data not shown). To determine the optimal buffer for solubilization of IBs, various conditions were investigated. The results showed that lysis buffer B, consisting of 5 and 6 M guanidine hydrochloride (GuHCl) at a pH of 8, yielded the highest amount of solubilized protein (Figure S8). The purified protein was highly pure as demonstrated by SDS-PAGE (Figure S9. I). Due to the precipitates that occurred during refolding on the column, the flash dilution method was used to facilitate the refolding process of the purified protein. Different additives were screened to prevent aggregate formation and increase refolding yield (Fig. 10). The lowest turbidity was observed in the presence of 1.4 M urea. The predicted activity of GP127-EAD was checked in vitro, using OM-permeabilized E. coli ATCC 8739 as a substrate. A qualitative zymogram assay confirmed the activity of GP127-EAD in the absence of CWBD (Figure S9. II). The muralytic activity of GP127-EAD was quantified under substrate saturating conditions using the Activity Calculator in PBS containing the selective additive. It was determined to be 6143 U/nM, as calculated from the slope of the linear regression of the corresponding dose-dependent saturation curve.

Analysis of protein toxicity in expression hosts. A. E. coli BL21(DE3) pLysS. B. E. coli BL21-Gold(DE3). Cultures were induced with 0.25 mM IPTG at an OD600nm of 0.6

Effect of additives on the prevent aggregate formation of GP127-EAD. [N1. No additive, N2. 1.4 M Acetone, N3. 1.4 M DMSO, N4. 1.4 M Ethanol, N5. 1.4 M Urea, N6. 10 mM Triton-X100, N7. 0.05% (w/v) PEG, N8. 1mM β-mercaptoethanol, N9. 10% Sucrose + 1mM EDTA, N10. 10% Glycerol, N11. 10% Sorbitol, N12. 10% Trehalose, N13. 50 mM Glycine]
4 Discussion
The antimicrobial potential of modular gram-negative phage endolysins remains largely unexplored, in part due to limited structural and biochemical characterization. The classification of enzymes based only on amino acid sequence similarity leads to false classifications [62]. The complexity of determining the stereochemical products and understanding the role of catalytic residues in enzymes that exclusively cleave bacterial PG is well recognized. The study of their precise mechanism is particularly challenging because of their inability to catalyze the cleavage of substrate analogs that are easily manageable. In addition, the presence of flexible linkers in modular proteins leads to the hardening of crystallography process, resulting in low-resolution structures in the existing limited structures [66]. The prediction of the 3D model and revision of critical residues using docking and molecular dynamic simulation studies can provide information on the structural characteristics and details of the mechanism of action as a crucial step towards unexplored potential and further engineering.
This study investigated the structural characteristics of the individual muramidase domain using E. coli O157:H7 endolysin as a model for the examination of in silico tools and comparison with in vitro data on the stability and activity of the recombinant enzybiotic. This can simplify the identification of the catalytic residue, putative mechanism, and subsequent engineering approaches of the muramidase family. The CD spectrum analysis, as well as the structural homology and low RMSD between our proposed model and two recently resolved crystal structures [66, 67], indicate the reliability of homology-based 3D structure prediction by YASARA when the percentage of template identity is not very high (46% in our case). A good complementary tool for this program was the GalaxyRefine Server, which was able to completely refine the errors in the structure and improve the stereochemical parameters.
To assess the stability in silico, MDS was firstly performed for GP127. The MDS results demonstrated the stable structure of GP127, as indicated by the RMSD and Rg measurements. The secondary structure composition of GP127 residues remained almost consistent throughout the simulations and matched the data obtained from the far-UV CD experiments, further validating the in silico analysis. Subsequently, the per-residue secondary structures of GP127 and GP127-EAD were compared using MDS data. The results suggest that the removal of CWBD does not significantly affect the secondary structure of the EAD, as the truncated form (GP127-EAD) exhibits a secondary structure composition similar to that of the EAD in GP127. Moreover, the per-residue number of contacts of GP127 and GP127-EAD was analyzed using MDS, and the results suggest that the stability and folding of the EAD are not compromised by the removal of CWBD. In the next step, CamSol analysis was employed to predict the protein solubility of GP127 and GP127-EAD. The results suggest that CWBD truncation may lead to an increase in the aggregation form of EAD, as CWBD contains highly soluble spots. These in silico predictions were substantiated by in vitro data, which highlighted the reduced solubility and increased aggregation tendency of the EAD. We attributed the high risk of aggregation of EAD to two possible reasons: the deletion of CWBD, which may act as a solubility-enhancing tag for EAD, and/or the inclusion body refolding process that may produce misfolded or unfolded intermediates that are prone to aggregation. A variety of solubilization agents were employed to preserve the secondary and tertiary structures, reducing aggregation during the refolding process and enhancing the recovery of functional GP127-EAD from IBs. However, only the buffer containing 6 M GuHCl could solubilize the IBs, which is a stronger denaturant usually associated with the complete denaturation of proteins. Certain IBs can be rendered soluble at low concentrations of denaturants, thereby preserving their partial structure and facilitating efficient refolding. Therefore, it is important to determine the lowest possible denaturant concentration for solubilization. Among the different GuHCl concentrations, only 5 and 6 M solubilized the protein, highlighting the limited solubility of GP127-EAD. In the next step, the gradual removal of the denaturant during on-column refolding was failed because of on-column precipitation, which strongly suggests a propensity for protein aggregation. Many proteins form conformational intermediates at moderately denaturing concentrations, which results in irreversible precipitation. In this case, rapid reduction of the denaturant with dilution can decrease the loss caused by aggregate formation, particularly through the incorporation of suitable additives into the refolding buffer [71]. Dilution was performed at 25 °C, and the samples were allowed to stand for 1 h to complete the refolding procedure. The temperature used was low enough to avoid damaging the protein and high enough to help the thermal motion of the molecule reach its native shape by melting any transient misfolded conformations. Chaotropic agents are employed in protein refolding solutions to dissolve the aggregates formed by partially folded intermediates. The inclusion of a minimal concentration of a chaotrope, like 1 M urea or 0.5 M GuHCl, can effectively accomplish this objective without destabilizing the stable native structures [56]. Since aggregation formation was detected in the dilution of about 50-fold, a concentration of 0.5 M GuHCl was chosen as a final denaturant concentration (tenfold dilution) that is presumably enough to cause ‘‘salting in’’ and to enhance the solubility of the native protein. The effectivity of different additives aimed at reducing aggregation and stabilizing the native fold was screened. The addition of 1.4 M urea prevented protein aggregation, leading to improved refolding yields. This may be due to the similarity of the molecular structure of urea to that of GuHCl. A better effect of urea compared with stabilizers was previously observed in the refolding of denatured lysozyme with GuHCl [57]. The final buffer with the selective additive was considered for the refolding process before activity assay.
The in silico activity of Gp127-EAD was confirmed based on the design of the tautomer library, virtual screening around the mapped binding region, and confirmation of the pocket based on ligand stability during MDS. It was shown that the proposed ligand conformation did not undergo any significant jump during MDS, contrary to what was previously observed for the MDS of an endolysin interacting with a carbohydrate analog [70]. This stability in the substrate-binding region and a higher number of interactions with the ligand could be a sign of GP127-EAD being as active as GP127. The similarity of GP127-EAD secondary structure profile with Pae87, which does not have CWBD, further validated the probability of GP127-EAD activity without CWBD. The in vitro data supported these modeling results, and GP127-EAD exhibited individual activity.
Despite varying IPTG concentrations, temperatures, and media conditions, none of the five E. coli strains tested could overexpress GP127-EAD in a soluble form, even when employing E. coli Bl21(DE3) pLysS, selected for its ability to soluble overexpress T7 lysozyme, the complete endolysin GP127, and Salmonella Duf3380 endolysin [10], as well as E. coli C43(DE3), which is resilient to toxic protein expression. This could suggest a potential blockage in protein expression due to a defense mechanism against highly toxic proteins. Only E. coli BL21-Gold(DE3) cells were able to overexpress GP127-EAD; however, the resulting protein aggregated into IBs without displaying any activity. This was confirmed through SDS-PAGE analysis of both soluble and insoluble fractions, and monitoring the growth of the cells, respectively. Several factors, such as expression level, solubility, folding, and stability, can contribute to a protein adopting this form. Nevertheless, toxicity often plays a crucial role in IB formation, as some toxic proteins are better expressed in this state due to their biological inertness [72]. Furthermore, various methods failed to solubilize IBs during the expression process. These findings suggest that GP127_EAD might possess high toxicity towards E. coli cells, potentially leading to the inhibition or misfolding of its expression under different conditions as a means to protect the cells' viability. The muralytic activity of the purified refolded protein was confirmed by qualitative zymography and quantitative standardized methods. However, it is important to note that the yield of inclusion body refolding directly affected the amount of activity and risk of aggregation [57]. Moreover, the differences in expression conditions for the full enzyme versus its truncated variant cast doubt on the validity of any comparative analysis regarding their activity and stability.
Muramidases have a diverse amino acid sequence and typically use a general-acid catalytic residue, Glu, as well as a general-base catalyst residue, Asp, or Cys, while additionally, Thr or Ser are sometimes involved in the third lateral. Goose egg-white lysozyme is unusual because it uses only Glu [14]. A few muramidases targeting gram-negative bacteria have been characterized, including Gp110 [10], AP3gp15 (PDB ID:5 NM7) [62], LysT84 (PDB ID:7RUM) [66], and Pae87 (PDB ID:7q4t) [67]. LysT84 and Pae87 have recently been released after our in silico studies. Rodrïguez-Rubio et al. (2016) [10] have showed that based on sequence conservation, Glu101 of Gp110 is a catalytic residue. Maciejewska et al. (2017) [62] reported that a Glu101Ala mutation in AP3gp15 causes catalytic deactivation. The second putative catalytic residue, Asp154, is located 19.5 Å away from Glu101, which is too far for an inversion mechanism to occur since it requires a distance of 9 Å. This suggests that the PF11860 domain family, without any catalytic aspartate residues, has low homology to known lysozymes and, similar to lytic transglycosylases, has only one catalytic residue, Glu. Recently, Love et al. (2022) [66] proposed a new mechanism for this subfamily that involves two key Glu residues, Glu 101 and Glu 118, located on the opposite side of the active site cavity, and Ser as a third lateral residue. A combination of structural analysis, mutagenesis, and molecular dynamics simulations confirmed the necessity of three critical residues for catalysis. In inverting β-glycosidases, the average distance of 2 oxygen atoms of the inter-carboxylic is recorded to be 8.5 ± 2.0 A˚, whereas in those that use the retaining mechanism, it is shorter at 6.4 ± 0.6 Å or 4.8 ± 0.3 Å [73]. Although the distance between Glu101 and Glu118 in LysT84 and AP3gp15 is 15.1 and 13.6 Å in the crystal structures, Love et al. indicated the closer position in LysT84 during MDS is around 9–10 A˚. However, the average distance was 15.1 A˚ and the shorter reported distance was related to the first 40 ns when the system had not yet reached equilibrium. Their hypothesis was that upon substrate binding, a significant conformational shift is required to increase flexibility and position them closer to PG cleavage. Our results indicate that the distance between the carbon delta of Glu 101 and Glu 118 of both GP127 and GP127-EAD interacting with the ligand was still too large to carry out the proposed mechanism (Table S4). This can be further confirmed based on structural alignment analysis of GP127-EAD, Pae87 [67], and AbLys1 [68] revealing the spatial arrangement of substrate binding region and potential similarities of GP127-EAD to other related enzymes within the glycoside hydrolase family. Glu 101 of GP127-EAD, formerly reported as a critical residue [62], was positioned similarly to Glu29 of Pae87 and Glu52 of AbLys1, while Ser179 of GP127-EAD, the third recently proposed critical residue [66], was located similarly to Thr67 of AbLys1 indicating the important roles of Glu101 and Ser179 in the catalytic process. The smaller distance between Glu29 and Glu46 in Pae87, and Glu52 and Asp61 in AbLys1 compared to their counterparts in GP127-EAD (Glu 101 and Glu 118) can suggest differences in substrate specificity and catalytic efficiency among these enzymes. However, it may also confirm that Glu118 is too far away to catalyze the substrate through the proposed mechanism. On the other hand, although the proposed mechanism suggested that CWBD properly organizes the active site and the distance between the critical residues for PG cleavage, our in silico and in vitro data have shown that EAD with the positive charge has high toxicity and can be active individually. Further investigations are required to confirm the essential features of the catalytic processes of these muramidase families for the development of effective enzymes.
The difference in murein composition between gram-positive and gram-negative bacteria influences their susceptibility to phage-derived lytic enzymes. Most endolysins derived from phages specific to gram-negative bacteria are unable to lyse intact gram-negative cells without the use of outer membrane permeabilizers, or binding to specific cationic peptides [6, 62, 74, 75]. Furthermore, these enzymes exhibit a broad spectrum of activity against gram-negative strains but lack peptidoglycan-degrading activity against gram-positive bacteria [62, 74, 76,77,78,79]. As previously reported, enzymes with this muramidase domain demonstrate potent lytic activity against gram-negative Burkholderia, Escherichia coli, Pseudomonas, Salmonella, and Klebsiella strains [10, 62, 66] but fail to break down the cell wall of gram-positive Staphylococcus strain [62]. The thick peptidoglycan layer of gram-positive bacteria exhibits significant variations in peptide composition, crosslinks, and glycan chain modifications, while gram-negative bacteria have more conserved peptidoglycan with 1–3 layers of the A1γ chemotype. However, in our initial antibacterial test, we used E. coli O157:H7 as a representative gram-negative bacterium and S. aureus (ATCC 10917) as a representative gram-positive bacterium with and without EDTA to determine the appropriate substrate (Figure S10). As anticipated, the antibacterial activity of Gp127 alone was negligible against both strains (a log reduction of 0.11 ± 0.05 and 0.076 ± 0.03 for E. coli and S. cereus, respectively). However, the addition of 0.5 mM EDTA resulted in a reduction of 2.54 ± 0.14 log units in viable E. coli cells, indicating nearly complete elimination. In the case of S. cereus, a log reduction of 0.2 ± 0.11 was observed. Considering the high toxicity of the EAD as well as the necessity of CWBD at an optimal distance to maintain stability, we suggested that the synthetic recombination of endolysin domains from different sources, along with the addition or swapping of CWBD [3], can be a suitable approach resulting in potent chimeric enzymes with enhanced stability and efficacy against both gram-negative and gram-positive strains. For this engineering approach, the selected CWBDs should exhibit high solubility, as indicated in Fig. 2 C, and be connected to the EAD via a flexible linker. This design ensures an optimal distance between the domains to minimize aggregation and promote effective interaction between EAD and CWBD.
In summary, our study contributes to the essential structural and biochemical understanding of modular gram-negative phage endolysins, which are promising candidates for antimicrobial therapy and enzybiotic development. We explored the individual functionality of EAD from PhaxI endolysin and assessed the predictability of enzyme characteristics using in silico methods within the muramidase family. The role of CWBD in endolysins is complex and variable, necessitating careful analysis and validation. Our findings challenge the current view that CWBD is essential for active site organization and efficient peptidoglycan cleavage, as we demonstrated that its removal does not significantly impact the structure or dynamics and EAD maintains its individual activity. We also found a possible discrepancy between the proposed catalytic mechanism and the actual spatial arrangement of the key residues, suggesting the need for further research to clarify the catalytic mechanisms and role of the other critical residues through mutagenesis experiments. Additionally, we recognized that CWBD might act as a solubility-enhancing tag for the protein, and its removal could expose the hydrophobic regions, leading to the protein aggregation. To overcome recombinant expression challenges in E. coli, which can also lead to misfolded or unfolded intermediates prone to aggregation, we primarily recommend assessing the activity and stability of chemically synthesized EAD. Furthermore, repositioning the cleavage site may improve solubility, although careful consideration must be given to the potential effects of the surrounding sequence and structure on folding, function, and stability. A comprehensive exploration of these variations, particularly the influence of specific amino acids on the overall protein structure, is crucial for gaining a thorough understanding of the muramidase domain. Moreover, we propose the use of EAD as a potential building block to create chimeric enzybiotics by integrating diverse binding domains with the incorporation of a flexible linker to ensure adequate spacing between the EAD and the different CWBD, which probably acts as a protective cap, preventing EAD aggregation. This approach has the potential to significantly enhance the efficacy and longevity of therapeutic agents. Our research not only enriches the existing knowledge base but also lays the foundation for more accurate predictions and strategic designs of potent enzybiotic-based treatments in the future.
Abbreviations
- EAD:
-
Enzymatic active domain
- CWBD:
-
Cell-wall-binding domain
- MDS :
-
Molecular dynamic simulation
References
Abdelkader K, Gerstmans H, Saafan A, Dishisha T, Briers Y (2019) The preclinical and clinical progress of bacteriophages and their lytic enzymes: the parts are easier than the whole. Viruses 11:96
Young R (1992) Bacteriophage lysis: mechanism and regulation. Microbiol Rev 56:430–481
Gerstmans H, Criel B, Briers Y (2018) Synthetic biology of modular endolysins. Biotechnol Adv 36:624–640
Pastagia M, Schuch R, Fischetti VA, Huang DB (2013) Lysins: the arrival of pathogen-directed anti-infectives. J Med Microbiol 62:1506–1516
Briers Y, Walmagh M, Grymonprez B, Biebl M, Pirnay J-P, Defraine V, Michiels J, Cenens W, Aertsen A, Miller S (2014) Art-175 is a highly efficient antibacterial against multidrug-resistant strains and persisters of Pseudomonas aeruginosa. Antimicrob Agents Chemother 58:3774–3784
Briers Y, Walmagh M, Van Puyenbroeck V, Cornelissen A, Cenens W, Aertsen A, Oliveira H, Azeredo J, Verween G, Pirnay JP et al (2014) Engineered endolysin-based “Artilysins” to combat multidrug-resistant gram-negative pathogens. mBio 5:10–1128
Nelson DC, Schmelcher M, Rodriguez-Rubio L, Klumpp J, Pritchard DG, Dong S, Donovan DMJ (2012) Endolysins as antimicrobials. Adv Virus Res 83:299–365
Briers Y, Lavigne RJFM (2015) Breaking barriers: expansion of the use of endolysins as novel antibacterials against Gram-negative bacteria. Future Microbiol 10:377–390
Oliveira H, Melo LD, Santos SB, Nobrega FL, Ferreira EC, Cerca N, Azeredo J, Kluskens LD (2013) Molecular aspects and comparative genomics of bacteriophage endolysins. J Virol 87:4558–4570
Rodríguez-Rubio L, Gerstmans H, Thorpe S, Mesnage S, Lavigne R, Briers Y (2016) DUF3380 domain from a Salmonella phage endolysin shows potent N-acetylmuramidase activity. Appl Environ Microbiol 82:4975–4981
Vollmer W, Blanot D, De Pedro M (2008) Peptidoglycan structure and architecture. FEMS Microbiol Rev 32:149–167
Love MJ, Abeysekera GS, Muscroft-Taylor AC, Billington C, Dobson RCJ (2020) On the catalytic mechanism of bacteriophage endolysins: Opportunities for engineering. Biochim Biophys Acta Proteins Proteom 1868:140302
Wohlkonig A, Huet J, Looze Y, Wintjens R (2010) Structural relationships in the lysozyme superfamily: significant evidence for glycoside hydrolase signature motifs. PLoS ONE 5:e15388
Weaver L, Grütter M, Matthews BW (1995) The refined structures of goose lysozyme and its complex with a bound trisaccharide show that the “Goose-type” ly’sozymes lack a catalytic aspartate residue. J Mol Biol 245:54–68
Hermoso JA, Monterroso B, Albert A, Galan B, Ahrazem O, Garcia P, Martinez-Ripoll M, Garcia JL, Menendez M (2003) Structural basis for selective recognition of pneumococcal cell wall by modular endolysin from phage Cp-1. Structure 11:1239–1249
Martinez-Fleites C, Korczynska JE, Davies GJ, Cope MJ, Turkenburg JP, Taylor EJ (2009) The crystal structure of a family GH25 lysozyme from Bacillus anthracis implies a neighboring-group catalytic mechanism with retention of anomeric configuration. Carbohydr Res 344:1753–1757
São-José C (2018) Engineering of phage-derived lytic enzymes: Improving their potential as antimicrobials. Antibiotics 7:29
Loessner MJ, Kramer K, Ebel F, Scherer S (2002) C-terminal domains of Listeria monocytogenes bacteriophage murein hydrolases determine specific recognition and high-affinity binding to bacterial cell wall carbohydrates. Mol Microbiol 44:335–349
Porter CJ, Schuch R, Pelzek AJ, Buckle AM, McGowan S, Wilce MC, Rossjohn J, Russell R, Nelson D, Fischetti VA (2007) The 1.6 Å crystal structure of the catalytic domain of PlyB, a bacteriophage lysin active against Bacillus anthracis. J Mol Biol 366:540–550
Becker SC, Swift S, Korobova O, Schischkova N, Kopylov P, Donovan DM, Abaev IJ (2015) Lytic activity of the staphylolytic Twort phage endolysin CHAP domain is enhanced by the SH3b cell wall binding domain. FEMS Microbiol Lett 362:1
Cheng Q, Fischetti VA (2007) Mutagenesis of a bacteriophage lytic enzyme PlyGBS significantly increases its antibacterial activity against group B streptococci. Appl Microbiol Biotechnol 74:1284–1291
Horgan M, O’Flynn G, Garry J, Cooney J, Coffey A, Fitzgerald GF, Ross RP, McAuliffe OJA, Microbiology e (2009) Phage lysin LysK can be truncated to its CHAP domain and retain lytic activity against live antibiotic-resistant staphylococci. Appl Environ Microbiol 75:872–874
Fenton M, Keary R, McAuliffe O, Ross RP, O’Mahony J, Coffey A (2013) Bacteriophage-Derived Peptidase CHAP(K) Eliminates and Prevents Staphylococcal Biofilms. Int J Microbiol 2013:625341
Fenton M, Casey PG, Hill C, Gahan CG, Ross RP, McAuliffe O, O’Mahony J, Maher F, Coffey A (2010) The truncated phage lysin CHAP(k) eliminates Staphylococcus aureus in the nares of mice. Bioeng Bugs 1:404–407
Low LY, Yang C, Perego M, Osterman A, Liddington R (2011) Role of net charge on catalytic domain and influence of cell wall binding domain on bactericidal activity, specificity, and host range of phage lysins. J Biol Chem 286:34391–34403
Low LY, Yang C, Perego M, Osterman A, Liddington RC (2005) Structure and lytic activity of a Bacillus anthracis prophage endolysin. J Biol Chem 280:35433–35439
Mayer MJ, Garefalaki V, Spoerl R, Narbad A, Meijers R (2011) Structure-based modification of a Clostridium difficile-targeting endolysin affects activity and host range. J Bacteriol 193:5477–5486
Shahrbabak SS, Khodabandehlou Z, Shahverdi AR, Skurnik M, Ackermann HW, Varjosalo M, Yazdi MT, Sepehrizadeh Z (2013) Isolation, characterization and complete genome sequence of PhaxI: a phage of Escherichia coli O157: H7. Microbiology (Reading) 159:1629–1638
Johnson M, Zaretskaya I, Raytselis Y, Merezhuk Y, McGinnis S, Madden TL (2008) NCBI BLAST: a better web interface. Nucleic Acids Res 36:W5-9
Gasteiger E, Hoogland C, Gattiker A, Wilkins MR, Appel RD, Bairoch A (2005) Protein identification and analysis tools on the ExPASy server. The proteomics protocols handbook Springer, 571–607
Soding J, Biegert A, Lupas AN (2005) The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res 33:W244-248
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J et al (2014) Pfam: the protein families database. Nucleic Acids Res 42:D222-230
Katoh K, Rozewicki J, Yamada KD (2019) MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform 20:1160–1166
Crooks GE, Hon G, Chandonia JM, Brenner SE (2004) WebLogo: a sequence logo generator. Genome Res 14:1188–1190
Krieger E, Vriend GJB (2014) YASARA View—molecular graphics for all devices—from smartphones to workstations. Bioinformatics 30:2981–2982
Hooft RW, Vriend G, Sander C, Abola EE (1996) Errors in protein structures. Nature 381:272
Jones DT (1999) Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol 292:195–202
Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26:283–291
Krieger E, Joo K, Lee J, Lee J, Raman S, Thompson J, Tyka M, Baker D, Karplus KJPS, Function B (2009) Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins: Structure. Function, and Bioinformatics 77:114–122
Heo L, Park H, Seok C (2013) GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res 41:W384-388
Wiederstein M, Sippl MJ (2007) ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res 35:W407-410
Luthy R, Bowie JU, Eisenberg D (1992) Assessment of protein models with three-dimensional profiles. Nature 356:83–85
Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C (2006) Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins: Structure. Function, and Bioinformatics 65:712–725
Krieger E, Nielsen JE, Spronk CA, Vriend G (2006) Fast empirical pKa prediction by Ewald summation. J Mol Graph Model 25:481–486
Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG (1995) A smooth particle mesh Ewald method. J Chem Phys 103:8577–8593
Krieger E, Vriend G (2015) New ways to boost molecular dynamics simulations. J Comput Chem 36:996–1007
Tian W, Chen C, Lei X, Zhao J, Liang J (2018) CASTp 3.0: computed atlas of surface topography of proteins. Nucleic Acids Res 46:W363–W367
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31:455–461
Duan Y, Wu C, Chowdhury S, Lee MC, Xiong G, Zhang W, Yang R, Cieplak P, Luo R, Lee T et al (2003) A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J Comput Chem 24:1999–2012
Laskowski RA, Swindells MB (2011) LigPlot+: multiple ligand–protein interaction diagrams for drug discovery. ACS Publications
Laskowski RA, Jablonska J, Pravda L, Varekova RS, Thornton JM (2018) PDBsum: Structural summaries of PDB entries. Protein Sci 27:129–134
Miyazaki K (2011) MEGAWHOP cloning: a method of creating random mutagenesis libraries via megaprimer PCR of whole plasmids. Methods Enzymol Elsevier, 399–406
Singh A, Upadhyay V, Panda AK (2015) Solubilization and refolding of inclusion body proteins. Methods Mol Biol 1258:283–291
Nazari M, Zarnani AH, Ghods R, Emamzadeh R, Najafzadeh S, Minai-Tehrani A, Mahmoudian J, Yousefi M, Vafaei S, Massahi S, Nejadmoghaddam MR (2017) Optimized protocol for soluble prokaryotic expression, purification and structural analysis of human placenta specific-1(PLAC1). Protein Expr Purif 133:139–151
Burgess RR (2009) Refolding solubilized inclusion body proteins. Methods Enzymol 463:259–282
Yasuda M, Murakami Y, Sowa A, Ogino H, Ishikawa H (1998) Effect of additives on refolding of a denatured protein. Biotechnol Prog 14:601–606
Lavigne R, Briers Y, Hertveldt K, Robben J, Volckaert G (2004) Identification and characterization of a highly thermostable bacteriophage lysozyme. Cell Mol Life Sci 61:2753–2759
Plotka M, Sancho-Vaello E, Dorawa S, Kaczorowska AK, Kozlowski LP, Kaczorowski T, Zeth K (2019) Structure and function of the Ts2631 endolysin of Thermus scotoductus phage vB_Tsc2631 with unique N-terminal extension used for peptidoglycan binding. Sci Rep 9:1261
Briers Y, Lavigne R, Volckaert G, Hertveldt K (2007) A standardized approach for accurate quantification of murein hydrolase activity in high-throughput assays. J Biochem Biophys Methods 70:531–533
Lu S, Wang J, Chitsaz F, Derbyshire MK, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Marchler GH, Song JS et al (2020) CDD/SPARCLE: the conserved domain database in 2020. Nucleic Acids Res 48:D265–D268
Maciejewska B, Zrubek K, Espaillat A, Wisniewska M, Rembacz KP, Cava F, Dubin G, Drulis-Kawa Z (2017) Modular endolysin of Burkholderia AP3 phage has the largest lysozyme-like catalytic subunit discovered to date and no catalytic aspartate residue. Sci Rep 7:14501
Micsonai A, Wien F, Bulyaki E, Kun J, Moussong E, Lee YH, Goto Y, Refregiers M, Kardos J (2018) BeStSel: a web server for accurate protein secondary structure prediction and fold recognition from the circular dichroism spectra. Nucleic Acids Res 46:W315–W322
Ashkenazy H, Abadi S, Martz E, Chay O, Mayrose I, Pupko T, Ben-Tal N (2016) ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res 44:W344-350
Sormanni P, Aprile FA, Vendruscolo M (2015) The CamSol method of rational design of protein mutants with enhanced solubility. J Mol Biol 427:478–490
Love MJ, Coombes D, Ismail S, Billington C, Dobson RCJ (2022) The structure and function of modular Escherichia coli O157:H7 bacteriophage FTBEc1 endolysin, LysT84: defining a new endolysin catalytic subfamily. Biochem J 479:207–223
Vazquez R, Seoane-Blanco M, Rivero-Buceta V, Ruiz S, van Raaij MJ, Garcia P (2022) Monomodular Pseudomonas aeruginosa phage JG004 lysozyme (Pae87) contains a bacterial surface-active antimicrobial peptide-like region and a possible substrate-binding subdomain. Acta Crystallogr D Struct Biol 78:435–454
Premetis GE, Stathi A, Papageorgiou AC, Labrou NEJTFJ (2023) Characterization of a glycoside hydrolase endolysin from Acinetobacter baumannii phage Ab TZA1 with high antibacterial potency and novel structural features. FEBS J 290:2146–2164
Sarvagalla S, Singh VK, Ke Y-Y, Shiao H-Y, Lin W-H, Hsieh H-P, Hsu JT, Coumar MSJJoc-amd (2015) Identification of ligand efficient, fragment-like hits from an hts library: Structure-based virtual screening and docking investigations of 2 h-and 3 h-pyrazolo tautomers for aurora kinase a selectivity. J Comput Aided Mol Des 29:89–100
Saadhali SA, Hassan S, Hanna LE, Ranganathan UD, Kumar V (2016) Homology modeling, substrate docking, and molecular simulation studies of mycobacteriophage Che12 lysin A. J Mol Model 22:180
Clark EDB, Schwarz E, Rudolph R (1999) [15] Inhibition of aggregation side reactions during in vitro protein folding. Methods Enzymol Elsevier, 217–236
Singhvi P, Saneja A, Srichandan S, Panda AK (2020) Bacterial Inclusion Bodies: A Treasure Trove of Bioactive Proteins. Trends Biotechnol 38:474–486
Mhlongo NN, Skelton AA, Kruger G, Soliman ME, Williams IHJPS, Function B (2014) A critical survey of average distances between catalytic carboxyl groups in glycoside hydrolases. Proteins: Structure. Function, and Bioinformatics 82:1747–1755
Oliveira H, Thiagarajan V, Walmagh M, Sillankorva S, Lavigne R, Neves-Petersen MT, Kluskens LD, Azeredo J (2014) A thermostable Salmonella phage endolysin, Lys68, with broad bactericidal properties against gram-negative pathogens in presence of weak acids. PLoS ONE 9:e108376
Yang H, Wang M, Yu J, Wei H (2015) Antibacterial Activity of a Novel Peptide-Modified Lysin Against Acinetobacter baumannii and Pseudomonas aeruginosa. Front Microbiol 6:1471
Walmagh M, Briers Y, dos Santos SB, Azeredo J, Lavigne R (2012) Characterization of modular bacteriophage endolysins from Myoviridae phages OBP, 201phi2-1 and PVP-SE1. PLoS ONE 7:e36991
Park Y, Lim JA, Kong M, Ryu S, Rhee S (2014) Structure of bacteriophage SPN 1 S endolysin reveals an unusual two-module fold for the peptidoglycan lytic and binding activity. Mol Microbiol 92:316–325
Maciejewska B, Roszniowski B, Espaillat A, Kesik-Szeloch A, Majkowska-Skrobek G, Kropinski AM, Briers Y, Cava F, Lavigne R, Drulis-Kawa Z (2017) Klebsiella phages representing a novel clade of viruses with an unknown DNA modification and biotechnologically interesting enzymes. Appl Microbiol Biotechnol 101:673–684
Oliveira H, Vilas Boas D, Mesnage S, Kluskens LD, Lavigne R, Sillankorva S, Secundo F, Azeredo J (2016) Structural and Enzymatic Characterization of ABgp46, a Novel Phage Endolysin with Broad Anti-Gram-Negative Bacterial Activity. Front Microbiol 7:208
Acknowledgements
We are grateful to Dr. Mahboobeh Nazari for her expert guidance on protein purification experiments, which greatly improved the quality and direction of this article.
This work was funded by the Deputy of Research, Tehran University of Medical Sciences (research code 99-2-412-48440).
Author information
Authors and Affiliations
Contributions
The project has been conceived by MJ, ZS and AE-H. The experiments and data gathering have been carried out by MJ. AG has helped in part of the experiments and analysis of the results. The project results have been supervised by ZS and AE-H and further discussed with MJ and the other authors. Obtained data and analyzes have been critically verified by ARS and AAMM. The manuscript draft has been written by MJ and revised with insights from the other authors. All authors have approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Conflict of Interests
All authors declare to have no conflict of interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Javid, M., Shahverdi, A.R., Ghasemi, A. et al. Decoding the Structure–Function Relationship of the Muramidase Domain in E. coli O157.H7 Bacteriophage Endolysin: A Potential Building Block for Chimeric Enzybiotics. Protein J 43, 522–543 (2024). https://doi.org/10.1007/s10930-024-10195-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10930-024-10195-z