
Abstract
Bacteriophage-derived endolysin enzymes play a critical role in disintegration of the host bacterial cell wall and hence have gained considerable attention as possible therapeutics for the treatment of drug-resistant infections. Endolysins can target both dividing and non-dividing cells and given the vital role peptidoglycan plays in bacterial survival, bacteria are less likely to modify it even if continuously exposed to lysins. Hence, probability of bacteria developing resistance to lysins appear bleak. Endolysins from mycobacteriophages offer great potential as alternative therapeutics for the drug-resistant TB. However, considering that a large number of mycobacteriophages have been discovered so far, the information on endolysins come from only a few mycobacteriophages. In this study, we report the structural and functional characterization of endolysins (LysinA and LysinB) encoded by mycobacteriophage PDRPxv which belongs to B1 sub cluster. On in silico analysis, we found LysinA to be a modular protein having peptidase domain at the N-terminal (104 aa), a central amidase domain (174 aa) and the peptidoglycan binding domain (62 aa) at the C-terminal. Additionally, ‘H-X-H’, which is a conserved motif and characteristic of peptidase domains, and the conserved residues His-His-Asp, which are characteristic of amidase domain were also observed. In LysinB enzyme, a single α/β hydrolase domain having a catalytic triad (Ser-Asp-His) and G-X-S-X-G motif, which are characteristic of the serine esterase enzymes were predicted to be present. Both the enzymes were purified as recombinant proteins and their antimycobacterial activity against M. smegmatis was demonstrated through turbidimetric experiments and biochemical assay. Interesting observation in this study is the secretory nature of LysinA evident by its periplasmic expression in E.coli, which might explain the ability of PDRPxv to lyse the bacterial host in the absence of transmembrane Holin protein.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Mycobacterium tuberculosis is responsible for about 1.5 million deaths worldwide annually. It is also estimated that 2–3% of new cases and 12–17% of reinfection cases are multi-drug resistant (MDR) (World Health Organisation 2018). India has the highest rates of occurrence of tuberculosis and the second highest MDR TB burden in the world (https://www.tbfacts.org/tb-statistics-india/). This highlights the need to devise new strategies to fight this deadly pathogen. Besides discovering and developing novel antibiotics, use of biologicals such as phages as well as their lytic enzymes (lysins), which are natural killers of bacteria are gaining attention. Phage-derived endolysins are promising because of their ability to act on cell walls even on exogenous application (Lai et al. 2015; Gerstmans et al. 2016; Love et al. 2018). Hence, their usage as potential alternative antibacterial agents and their synergistic effect with the existing antibiotics is being actively explored (Pastagia et al. 2013; Kim et al. 2018). Bacteriophages infecting Gram-positive bacteria require holin and endolysin (LysinA) to lyse the bacterial cell wall whereas phages that infect Gram-negative bacteria need to have additional proteins to disrupt the outer membrane (OM) as well (Gigante et al. 2017). This is achieved by proteins called Spanins, which mediate the fusion of the cytoplasmic membrane with the outer membrane thereby resulting in the removal of the last barrier for the phages, to release and complete the lysis of the host cell (Rajaure et al. 2015).
Mycobacterial cell exhibits a complex cell wall (envelop) consisting of a cytoplasmic membrane surrounded by a peptidoglycan layer covalently linked to the arabinogalactan-peptidoglycan (mAGP) complex and mycolic acids (Chatterjee 1997; Bugg et al. 2011). Thus, bacteriophages that infect mycobacteria have to overcome this complex barrier for the release of viral progeny after the completion of lytic cycle (Payne et al. 2009) (Fig. 1). Mycobacteriophages are the phages that infect mycobacterial species and several of them encode two endolysins, LysinA and LysinB, which act on the peptidoglycan layer and on the mycolic acid–arabinogalactan layer, respectively. To date, a large number of phages infecting Mycobacterium spp. have been reported worldwide (Hatfull 2012; Smith et al. 2013; Franceschelli et al. 2014; Pope et al. 2015), but relatively fewer studies have been carried out on the mycobacteriophage-encoded endolysins (Payne et al. 2012; Grover et al. 2014; Lai et al. 2015; Fraga et al. 2019). Amongst these, endolysins from D29 and Ms6 mycobacteriophages, which belong to A2 and F1 sub-clusters, respectively, are the most reported ones (Payne et al. 2009; Gil et al. 2010; Pohane et al. 2014).

reproduced from Gerstmans et al. (2016)
Schematic representation of the site of action of LysinA (Pacman: Pink) and LysinB (Pacman: Blue) enzymes on the mycobacterial cell wall,
In the present study, we describe cloning, overexpression, purification and functional characterization of LysinA and LysinB endolysins derived from mycobacteriophage PDRPxv, which belongs to B1 Sub-cluster (Sinha et al. 2020). To our understanding, this is the first report on Lysin enzymes derived from a B cluster mycobacteriophage.
In silico analysis of lysin genes was carried out to characterize their domain architecture and to identify the conserved structural motifs. The anti-mycobacterial activity of LysinA (and a mixture of LysinA & LysinB) was assessed using turbidimetry assay, and the esterase activity of LysinB was measured by in vitro assay using para-Nitrophenyl butyrate (pNPB) as the substrate. We also identified a σ70-like promoter region upstream of the LysinA gene, suggesting its putative role in expression of the endolysins during the late infection cycle.
Materials and methods
Bacteria, chemicals and plasmids
All cloning steps were performed in Escherichia coli (E. coli) DH5α strain, grown in Luria Bertani (LB) broth and agar (HiMedia, India) at 37 °C. Mycobacterium smegmatis mc2155 was grown in Middlebrook 7H9 broth supplemented with 0.02% glycerol. The pET28a vector (Novagen, USA) was a kind gift from Dr. S. Ramachandran (Institute of Genomics and Integrative Biology, New Delhi). Primers and analytical grade chemicals were purchased from Sigma-Aldrich. Ni–NTA agarose and other PCR purification/plasmid isolation kits were obtained from Qiagen (Germany). Phenylmethanesulfonylfluoride (PMSF) and other chemicals were purchased from HiMedia Laboratories (India).
In silico analysis
The putative endolysin gene sequences (gp51 & gp52) were retrieved from the genome of mycobacteriophage PDRPxv (Genbank Accession No. KR029087.1) and analysed by Blastp, NCBI-CDD Search, InterProScan and HHPred tools for homology based functional annotation (Altschul et al. 1990; Söding et al. 2005; Jones et al. 2014; Marchler-bauer et al. 2015). SignalP was used to find the presence of signal peptide (Petersen et al. 2011) and prediction for non-signal peptide triggered protein secretions was made using SecretomeP (https://www.cbs.dtu.dk/services/SecretomeP/) and ProtCompB (https://www.softberry.com). The recommended threshold value for a prokaryotic sequence to have non-signal peptide triggered protein secretion in SecretomeP is 0.5. The σ70 promoter was predicted using BPROM (https://www.softberry.com).
Cloning and expression of endolysins
Genes coding for endolysins lysinA and lysinB were amplified by polymerase chain reaction using the following primers (Eurofins, India): LysAF 5′-ATG CCG GGA TCC ATG CCC ATG CGC- 3′; LysAR 5′- GCG AAG CTT CTA CGG TTT GAG GGC G-3′; LysBF 5′- AGG GGA TCC AGC ATG ACA CAG -3′; LysBR 5′- CAC ACT CGA GCG CTA CGC GGG CAG -3′ and cloned in pMALc2x and pET28a vectors, respectively. E. coli BL21 (DE3) cells were transformed with the recombinant pET28a plasmids and the transformed cells were grown in LB media (containing 50 µg/ml kanamycin) at 37 °C overnight. Similarly, E. coli Rosetta-gami™ (DE3) pLysS cells were transformed with the recombinant plasmid pMALc2x-LysA. Isolated colonies of transformed cells were grown in LB media containing 100 µg/ml ampicillin and incubated at 37 °C overnight.
Transformed cells were grown in 5 ml LB at 37 °C for 3–4 h, till O.D.600 reached ~ 0.4–0.6. An aliquot of the uninduced E. coli culture was collected in a separate tube and to the remaining culture, inducer isopropyl-1-thio-β-d-galactopyranoside (IPTG) was added to a final concentration of 0.1 mM. The culture was incubated further at 16 °C, overnight with shaking and the cells were harvested by centrifugation at 8000 r.p.m for 10 min. The cell pellets obtained from both uninduced and induced E. coli cultures were resuspended in 1 × SDS-gel loading buffer and electrophoresed on 12% SDS-PAGE. The protein bands were visualized by staining the gel with Coomassie Brilliant Blue R250.
Purification of endolysins
Purification of LysinA
LysinA was cloned in the expression vector pMALc2x having maltose-binding protein (MBP) as the tag and expressed in E. coli Rosetta-gami™ (DE3) pLysS cells. The recombinant LysinA obtained in the soluble fraction was purified by affinity chromatography. Briefly, the amylose resin (New England Biolabs, USA) was equilibrated in the Column buffer (25 mM Tris, 0.3 M NaCl, 1 mM EDTA, pH 7.9) according to manufacturer’s instructions. The recombinant LysinA obtained in soluble form was mixed with 2 ml of amylose resin and incubated overnight at 4 °C. The protein-matrix mixture was poured onto a Poly-Prep Chromatography Column (Qiagen, Germany) and the flow through was discarded. The column was washed with the column buffer and the bound enzyme was eluted using column buffer containing maltose (25 mM Tris, 0.3 M NaCl, 10 mM maltose, pH 7.9). The eluted fractions were analyzed using 10% SDS-PAGE.
Purification of LysinB
The recombinant plasmid pET28a-LysinB was expressed in E. coli BL21 (DE3) cells; His-tagged LysinB was obtained in the soluble fraction and purified by affinity chromatography. Ni–NTA matrix (Ni–NTA agarose, Qiagen, Germany) was equilibrated in the binding buffer (25 mM Tris, 0.5 M NaCl, pH 7.9) according to manufacturer’s instructions. The soluble fraction containing recombinant LysinB was mixed with 2 ml of Ni–NTA matrix, incubated overnight at 4ºC and the enzyme-matrix mixture was poured onto a Poly-Prep Chromatography Column (Qiagen, Germany). After discarding the flow through, column was washed with wash buffer I (25 mM Tris, 0.5 M NaCl, 20 mM imidazole, pH 7.9) followed by wash buffer II (25 mM Tris, 0.5 M NaCl, 40 mM imidazole, pH 7.9). Finally, the bound protein was eluted using elution buffer (25 mM Tris, 0.5 M NaCl, 200 mM imidazole, pH 7.9). The eluted fractions were analyzed using 12% SDS-PAGE.
Turbidimetric assay for the anti-M. smegmatis activity of LysinA and LysinB
M. smegmatis cells (O.D.600 ~ 0.6) were resuspended in 7H9 media and incubated with LysinA and LysinB individually or in combination, and with BSA (negative control); experiment was done in duplicate at a fixed concentration of 100 µg/ml for each protein. Treated M. smegmatis cell suspensions were incubated at 37 °C (with gentle shaking) for 24 h in a 96 well microtiter plate. At the end of the incubation period, O.D. was measured at 600 nm.
Assay for the esterase activity of LysinB
LysinB activity was assayed as described by Gilham and Lehner (2005), by measuring the release of pNP (p-Nitrophenyl) using p-Nitrophenyl Butyrate (pNPB) as the substrate. The reaction mixture containing 20 mM Tris–HCl pH 8.0, 100 mM NaCl, 0.1% Triton X-100, pNPB (50 μM) and 2 μg of LysinB enzyme (in a final volume of 100 μl) was incubated at room temperature for 30 min. pNP was determined by measuring absorbance at 420 nm and the amount released was calculated using a standard curve. In the reaction kept as control, all the components except LysinB were included.
Periplasmic expression of PDRPxv LysinA
The periplasmic expression of LysinA was analyzed using the Ueguchi and Ito (1990) method with slight modifications. E. coli Rosetta-gami™ (DE3) pLysS cells containing pMALc2x-LysinA recombinant plasmid were grown in 5 ml of LB media containing ampicillin (50 µg/ml) and chloramphenicol (34 µg/ml) to an O.D600 of 0.6. Cells were induced with 0.3 mM IPTG and grown for another 18 h at 16 °C. To recover proteins secreted in the E. coli periplasm, the cell pellet was resuspended in 2 ml of buffer, containing 20% sucrose and 30 mM Tris–HCl. To this, 1 mM EDTA was added and the cell suspension was vortexed for 5–10 min at room temperature. Cells were then centrifuged at 10,000×g for 10 min and the pellet obtained was resuspended in 2 ml of ice-cold 5 mM MgSO4, incubated in ice for 10 min and centrifuged again to collect the supernatant containing periplasmic fraction.
Western blotting of periplasmic fraction
Proteins present in the periplasmic fraction were separated by 12% SDS–PAGE, transferred to a PVDF membrane and blocked overnight at 4 °C with 5% BSA. Membrane was incubated with mouse anti-MBP antiserum (Santacruz Biotech, USA) and anti-mouse IgG HRP-conjugated antibody for the identification of LysinA-MBP fusion protein in the periplasmic fraction. Blots were developed using H2O2 as the substrate and 3,3′-diaminobenzidine as the chromophore.
Cloning of putative promoter sequence present in proximity to endolysin genes
To experimentally test the activity of gp49 proximal to lysinA (gp51) and lysinB (gp52) in PDRPxv genome and identified as putative promoter region by using BPROM tool, gp49 (564 bp) was cloned upstream to lacZ gene in pSD5B, a promoter less shuttle vector kindly given by Prof. Vani Brahmachari (Dr. Ambedkar Center for Biomedical Research, University of Delhi). Oligonucleotide pair (Eurofins, India) Gp49 F: 5′- CGC TCT AGA AGC TCA CCA CCC GGC CTT G - 3′ and Gp49 R: 5′ - CGC GCA TGC ATG GCC ACC CCG TTC GAC – 3′ was used for the amplification of the identified region. The amplicon and the vector were digested with XbaI and SphI restriction enzymes and ligated using T4 DNA ligase. Ligated product was transformed into E. coli DH5α cells. The transformants were screened by colony PCR and recombinant plasmid containing Gp49 was confirmed by sequencing and was designated as pPrLys.
Analysis of promoter strength
Recombinant plasmid pPrLys and the vector pSD5B were electroporated separately in M. smegmatis mc2155 and plated in M7H10 agar media containing 25 μg/ml kanamycin. M. smegmatis cells transformed with pPrLys/pSD5B were grown in 7H9 broth and O-Nitrophenyl-β-d-galactopyranosidase (ONPG) assay was performed according to Miller (1972) with some modifications. M. smegmatis cells transformed with pSD5B were taken as the negative control. Cell pellets obtained from 100 ml of M. smegmatis culture were resuspended in 10 ml of assay buffer (0.1 M sodium phosphate buffer, pH 7.0, 10 mM KCl, 1 mM MgSO4, 50 mM β-mercaptoethanol). Cells were lysed by sonication in ice (30 s on/30 s off mode) for 15 cycles, centrifuged at 12,000 rpm and the supernatant was collected in a fresh centrifuge tube. After estimating the total protein present in the supernatant fraction by Bradford method, 200 μl of ONPG (4 mg/ml) was added to the supernatant and incubated at 37 °C till the development of yellow color. Reaction was stopped by adding 1 M Na2CO3 and the release of o-Nitrophenol was measured at A420. The assay was performed in duplicate. Promoter activity was calculated using the equation (Pathak et al. 2015):
Results
Location of endolysin genes on PDRPxv genome
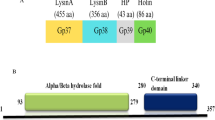
LysinA (gp51) and lysinB (gp52) sequences (Fig. 2A, B) were retrieved from PDRPxv genome (Genbank Accession No. KR029087.1). While SignalP prediction indicated absence of a signal peptide in both the enzymes, SecratomeP analysis gave a score of 0.601 for LysinA and 0.308 for LysinB. Next, to identify the domains and their organization in LysinA, full-length protein sequence of Gp51 (429 amino acids) was analyzed using Blastp and NCBI-CDD, which showed the enzyme to be modular in structure with an N-terminal Peptidase domain (21–125 aa), a central N-acetylmuramoyl-l-alanine amidase domain (159–333 aa) and a C-terminal peptidoglycan recognition protein (PGRP) conserved domain (cd06583), localized between amino acid residues 367 and 429. On comparing PDRPxv LysinA with its experimentally verified counterpart in D29, we found domains at N- and C-terminal to be similar in both the enzymes but the central domain in D29 is a lysozyme like domain (Pohane et al. 2014) (Fig. 2B). The conserved H-X-H motif, which is a characteristic of the metal-dependent endopeptidase family of enzymes/proteins was also observed in the N-terminal region of LysinA enzyme (Fig. 3A).

A Predicted domains in LysinA enzyme of PDRPxv. Modular structure of LysinA enzyme from D29 and Ms6 mycobacteriophages. B Predicted domain architecture of LysinB enzyme from PDRPxv, D29 and Ms6 mycobacteriophages

Sequence alignment of LysinA enzymes of mycobacteriophages belonging to B1 sub-cluster (including PDRPxv) with Ms6 and D29. A Sequence alignment of residues predicted to be present in the peptidase domain and B Sequence alignment of residues predicted to be present in the amidase domain of LysinA enzymes. Protein sequence of LysinA from the mycobacteriophages belonging to B1 sub cluster were compared with D29 and Ms6 LysinA protein sequences. Conserved residues are marked with dots (filled circle) and residues involved in the enzyme activity are marked with an asterisk (*)
LysinB enzyme is a mycolylarabinogalactan esterase that cleaves the ester linkage joining the mycolic acid-rich outer membrane to arabinogalactan, releasing free mycolic acids in mycobacterial species (Payne et al. 2009). Upon analyzing the protein sequence of PDRPxv LysinB enzyme (451 amino acids), a single α/β hydrolase domain, similar to that present in LysinB from D29 and Ms6 was observed (Fig. 2B). The conserved G-X-S-X-G motif (Ser177), which is a characteristic of serine esterase enzymes and is considered to be important for the activity of LysinB enzymes was also observed. Besides, other catalytic residues such as aspartic acid (Asp267) and histidine (His419) of the catalytic triad (Ser-Asp-His) that have been reported in the serine esterases (Fig. 4) were also identified. Notably, gene coding for holin was not predicted either in the vicinity of lysinA and lysinB genes or anywhere else in the PDPRxv genome.

Sequence alignment of residues predicted to be present in the α/β hydrolase domain of LysinB genes of B1 sub-cluster mycobacteriophages (including PDRPxv) with those of Ms6 and D29. Sequence alignment showed residues involved in the esterase activity of LysinB enzymes to be conserved. Conserved residues are marked with dots (filled circle) and residues involved in the enzyme activity are marked with an asterisk (*)
Purification and functional characterization of endolysins
LysinA gene was cloned in pMALc2x expression vector and lysinB in pET28a. Both, the MBP-tagged LysinA and the His-tagged LysinB (expressed in E. coli Rosetta-gami™ (DE3) pLysS and BL21 (DE3), respectively) yielded the proteins in the soluble fraction. Purity of each recombinant enzyme was above (90%) as analyzed by SDS-PAGE (Fig. 5).

Expression and purification of LysinA and LysinB from PDRPxv mycobacteriophage. A Agarose gel analysis of PCR amplified lysinA and lysinB genes. Lane M: DNA marker; Lane 1: lysinA (1290 bp) and Lane 2: lysinB (1356 bp). B, C SDS-PAGE analysis of affinity purified recombinant LysinA and LysinB enzymes, respectively. Lane M: Protein molecular weight markers; Lane 1: Purified fraction. Molecular weight (90 kDa) of the purified protein seen in B is inclusive of MBP tag (42.5 kDa) and LysinA (47.5 kDa)
LysinA activity was studied using turbidimetry assay. The effect of exogenous addition of LysinA (100 µg) to the exponentially growing M. smegmatis cells was measured by recording O.D.600. LysinA enzyme alone showed 23% cell lysis whereas in combination with LysinB enzyme (100 µg each), 43% cell lysis was observed when compared to the control. The esterase activity of LysinB enzyme was analysed using pNPB as the substrate, where release of pNP product was measured at 420 nm. Specific activity of LysinB was found to be 0.56 U mg−1 (Fig. 6).

Enzyme activity of PDRPxv encoded LysinA and LysinB. A The activity of LysinA enzyme was studied by turbidimetric assay. LysinA enzyme alone or in combination with LysinB was added to M. smegmatis cells (~ 1 × 106 cells) and O.D.600 was measured after 48 h of incubation. LysinA showed 23% cell lysis; LysinB showed 34% lysis, and LysinA+LysinB showed 43% cell lysis, B Specific activity (U mg−1) of LysinB enzymes from PDRPxv (0.72), D29 (0.56) and Ms6 (0.12) depicted. Experiments were done in duplicate
Periplasmic expression of LysinA enzyme
To validate the secretory nature of LysinA as predicted by SecretomeP, periplasmic fraction of E. coli cells expressing LysinA was extracted and analyzed by Western blotting, using anti-MBP antibody. As evident in Fig. 7, the presence of LysinA enzyme in the periplasmic fraction was confirmed.

Periplasmic expression of LysinA. a SDS-PAGE analysis of periplasmic fraction isolated from E. coli Rosetta-gami™ (DE3) pLysS expressing pMALc2x-LysinA. Lane 1: Unstained protein marker; Lane 2: Periplasmic fraction. b Western blot analysis of periplasmic fraction. Lane 1: Pre-stained protein marker: Lane 2: Periplasmic fraction containing LysinA enzyme identified using anti-MBP antiserum
Cloning and in vivo functional assay of promoter identified in proximity to endolysins
Analysis of regions flanking the endolysins in PDRPxv genome had conveyed the presence of a putative σ70-like promoter sequence upstream of lysinA gene (within the gp49 ORF), where − 10 region consisting of GGCCATCAT consensus sequences was found 200 bp upstream of lysinA gene and − 35 region TCGGCA was found 223 bp upstream to the gene. To verify the predicted promoter region, this 564 bp (gp49) upstream region of the lysinA gene was cloned as transcriptional lacZ fusion in a promoter probe vector (pSD5B), resulting in the plasmid ‘pPrLys’. On conducting ONPG assay, PrLys was found to induce β-galactosidase activity in M. smegmatis with 73.3 miller units (Fig. 8).

ONPG Assay for measuring the activity of the putative promoter (gp49). The x-axis represents the control (M. smegmatis-pSD5B) and M. smegmatis-pPrLys promoter and the y-axis represents promoter activity in Miller units. The experiments were done in duplicate
Discussion
Mycobacterial infections are proving to be increasingly difficult to treat with the growing incidence of multidrug-resistant strains and due to an excruciatingly slow pace of discovery of new TB drugs. In this context, bacteriophage-derived endolysins are gaining notice as promising alternative/adjuncts to the antibiotics. Owing to their crucial role in breakdown of the cell wall for releasing the phage progeny (Borysowski et al. 2006), endolysins cause lysis of the host bacteria at the end of the infection cycle. The presence of mycolic acid in the mycobacterial cell wall poses an additional barrier for the phages to escape the infected cell and therefore in addition to LysinA, mycobacteriophages also have LysinB enzyme. In this study, we performed functional analysis of LysinA and LysinB encoded by mycobacteriophage PDRPxv, which is a double-stranded lytic DNA mycobacteriophage, belonging to B1 sub-cluster of Siphoviridae family (Sinha et al. 2020). Its genome size is 69,171 bp with a GC content of 66%.
Lysis cassettes in phages are generally situated at different chromosomal locations (Payne et al, 2009) with LysinB gene being present downstream to LysinA in all the cases. Both the genes are invariably linked and present either towards left side of the structural genes (like in D29 and Bxz2 mycobacteriophages) or on the right side (as seen in PDRPxv in this study) (Fig. S1). In PDRPxv genome, gp51 and gp52 genes code for endolysins lysinA and lysinB, respectively. While the gene size of lysinA (1290 bp) from PDRPxv was comparable to D29 (1482 bp) and Ms6 (1155 bp) mycobacteriophages, lysinB (1355 bp) gene was considerably larger than its counterpart in D29 (765 bp) and Ms6 (999 bp) (Payne et al. 2009; Pohane et al. 2014). We also observed a stretch of glycine-rich region towards the C-terminal of LysinB. To understand if this feature is observed in other phages too, we examined the reported LysinB protein sequences and found glycine-rich region to be a common feature in phages belonging to B1 sub cluster. This is an interesting observation, though we don’t yet understand its significance. While the domains in endolysins are usually conserved (Drulis-Kawa et al. 2015), the lysin enzymes are known to exhibit considerable divergence in their nucleotide and protein sequences. The analysis of PDRPxv LysinB shows a single α/β hydrolase domain similar to that present in LysinB from D29 and Ms6 (Fig. 3B). Also, by multiple sequence alignments, we found it to show less than 20% identity with D29 and Ms6 LysinB protein sequence; though, the G-X-S-X-G motif (Fig. 4), which is a characteristic of serine esterase enzymes (Gil et al. 2010; Henry et al. 2011) was present in all the three enzymes.
In LysinA enzymes, essentially four types of activities have been reported, based upon the bond in the peptidoglycan layer they act upon. These are: (i) lysozymes (ii) transglycosidases (iii) amidases and (iv) endopeptidases (Oliveira et al. 2013; Love et al. 2018; Catalão and Pimentel 2018). Protein sequence analysis of LysinA from PDRPxv showed the presence of the conserved H-X-H motif, which is a characteristic of the metal-dependent endopeptidase family of enzymes/proteins. The presence of His179, His301, and Asp313, which appear to be the conserved residues in amidase domain (Fig. 3A and Fig. 3B) were also observed. On comparing PDRPxv LysinA from those from Ms6 and D29, we found that while LysinA in PDRPxv and D29 are modular, their counterpart in Ms6 has a single domain which possesses both N-acetylmuramoyl-l-alanine amidase/PGRP domain in its central region (Fig. 2A). Although the predicted variations in the domain architecture of LysinA enzymes from the three mycobacteriophages are noteworthy, however the fact that these phages belong to different clusters perhaps also needs to be considered before the significance of their structural variability could be assessed.
To experimentally validate the results obtained from computational analysis, LysinA and LysinB genes were cloned in expression vectors and the predicted promoter region was cloned in a promoter probe vector (pSD5B). The esterase activity of recombinant LysinB enzyme (cloned in pET28a vector) showed a specific activity of 0.56 U mg−1, which is comparable to the activity of LysinB observed in D29 (0.72 U mg−1) and Ms6 (0.12 U mg−1). When LysinA was cloned in pET28a vector, it was consistently obtained in insoluble form even under varied conditions of incubation temperature, concentration of inducer etc. (unpublished data). Therefore, LysinA gene was cloned in expression vector pMALc2x, which yielded the recombinant protein in soluble form. Interestingly, we observed the expressed recombinant LysinA to cause partial lysis of E. coli cells when expression was induced using IPTG. Though we did not find the signal peptide in LysinA, a score of 0.610 in SecretomeP (higher than the recommended threshold value of 0.5 for a prokaryotic sequence to have non-signal peptide-triggered protein secretion) indicated secretory nature of the protein. The periplasmic expression of LysinA was confirmed by Western blotting (Fig. 7) which explain its access to peptidoglycan and thus the ability to cause damage to the bacterial cell wall. This also expounds how intracellular LysinA translocate in the absence of holin in PDRPxv and act upon the peptidoglycan.
Finally, the purified recombinant LysinA and LysinB were tested for their ability to kill M. smegmatis cells. We found that the combination of two enzymes was more effective than LysinA alone, thus confirming that the enzymes work together towards the lysis of mycobacterium for the release of phage progeny. Similar observation has been made earlier (Payne et al. 2009), where co-treatment with LysinA and LysinB proteins showed improved cell lysis. It is pertinent that regulation of the synthesis of endolysins should be stringent since they are critical for determining the time of bacterial cell lysis (Garcia et al. 2002). While in L5 mycobacteriophage, genes that encode ‘host lysis’ and the ‘structural proteins’ are transcribed as a single operon, in Ms6 mycobacteriophage, they are not (Brown et al. 1997). In PDRPxv, we predicted a putative σ70-like promoter sequence upstream to lysinA and experimentally demonstrated it to have regulatory activity through the expression of the β-galactosidase reporter gene. The proximal location and the demonstrated activity (Fig. 8) suggest that the promoter sequence might be playing a role in the expression of Lysin enzymes, though more experimental data would be required for it to be certain.
To conclude, we have highlighted some of the unique properties of PDRPxv mycobacteriophage-derived endolysins, and made a broad comparison with the endolysins previously described from other mycobacteriophages. In addition, experimental validation by demonstration of biochemical and antibacterial activity of both LysinA and LysinB further support the opinion that co-treatment with endolysins could be a possible option for combating mycobacterial pathogens.
References
Altschul SF, Gish W, Miller W et al (1990) Basic local alignment search tool. J Mol Biol. https://doi.org/10.1016/S0022-2836(05)80360-2
Borysowski J, Weber-Dabrowska B, Górski A (2006) Bacteriophage endolysins as a novel class of antibacterial agents. Exp Biol Med (Maywood) 231:366–377. https://doi.org/10.1177/153537020623100402
Brown KL, Sarkis GJ, Wadsworth C, Hatfull GF (1997) Transcriptional silencing by the mycobacteriophage L5 repressor. EMBO J 16:5914–5921. https://doi.org/10.1093/emboj/16.19.5914
Bugg TDH, Braddick D, Dowson CG, Roper DI (2011) Bacterial cell wall assembly: still an attractive antibacterial target. Trends Biotechnol 29:167–173. https://doi.org/10.1016/j.tibtech.2010.12.006
Catalão M, Pimentel M (2018) Mycobacteriophage lysis enzymes: targeting the mycobacterial cell envelope. Viruses 10:428. https://doi.org/10.3390/v10080428
Chatterjee D (1997) The mycobacterial cell wall: structure, biosynthesis and sites of drug action. Curr Opin Chem Biol 1:579–588. https://doi.org/10.1016/S1367-5931(97)80055-5
Drulis-Kawa Z, Majkowska-Skrobek G, Maciejewska B (2015) Bacteriophages and phage-derived proteins—application approaches. Curr Med Chem 22:1757–1773. https://doi.org/10.2174/0929867322666150209152851
Fraga AG, Trigo G, Murthy RK et al (2019) Antimicrobial activity of mycobacteriophage D29 lysin B during Mycobacterium ulcerans infection. PLoS Negl Trop Dis 13:e0007113. https://doi.org/10.1371/journal.pntd.0007113
Franceschelli JJ, Suarez A, Terán L et al (2014) Complete genome sequences of nine mycobacteriophages. Genome Announc 2:2013–2014. https://doi.org/10.1128/genomeA.00181-14.Copyright
Garcia M, Pimentel M, Moniz-Pereira J (2002) Expression of mycobacteriophage Ms6 lysis genes is driven by two 70-like promoters and is dependent on a transcription termination signal present in the leader RNA. J Bacteriol 184:3034–3043. https://doi.org/10.1128/JB.184.11.3034-3043.2002
Gerstmans H, Rodriguez-Rubio L, Lavigne R, Briers Y (2016) From endolysins to Artilysin(R)s: novel enzyme-based approaches to kill drug-resistant bacteria. Biochem Soc Trans 44:123–128. https://doi.org/10.1042/BST20150192
Gigante A, Hampton C, Dillard R et al (2017) The Ms6 mycolyl-arabinogalactan esterase LysB is essential for an efficient mycobacteriophage-induced lysis. Viruses 9:343. https://doi.org/10.3390/v9110343
Gil F, Grzegorzewicz AE, Catalao MJ et al (2010) Mycobacteriophage Ms6 LysB specifically targets the outer membrane of Mycobacterium smegmatis. Microbiology 156:1497–1504. https://doi.org/10.1099/mic.0.032821-0
Gilham D, Lehner R (2005) Techniques to measure lipase and esterase activity in vitro. Methods 36:139–147. https://doi.org/10.1016/j.ymeth.2004.11.003
Grover N, Paskaleva EE, Mehta KK et al (2014) Growth inhibition of Mycobacterium smegmatis by mycobacteriophage-derived enzymes. Enzyme Microb Technol 63:1–6. https://doi.org/10.1016/j.enzmictec.2014.04.018
Hatfull GF (2012) Complete genome sequences of 138 mycobacteriophages. J Virol 86:2382–2384. https://doi.org/10.1128/JVI.06870-11
Henry M, Coffey A, O’Mahony J, Sleator RD (2011) Comparative modelling of LysB from the mycobacterial bacteriophage Ardmore. Bioeng Bugs 2:88–95. https://doi.org/10.4161/bbug.2.2.14138
Jones P, Binns D, Chang H et al (2014) Sequence analysis InterProScan 5: genome-scale protein function classification. Bioinformatics 30:1236–1240. https://doi.org/10.1093/bioinformatics/btu031
Kim N-H, Park WB, Cho JE et al (2018) Effects of phage endolysin SAL200 combined with antibiotics on Staphylococcus aureus infection. Antimicrob Agents Chemother. https://doi.org/10.1128/AAC.00731-18
Lai MJ, Liu CC, Jiang SJ et al (2015) Antimycobacterial activities of endolysins derived from a mycobacteriophage, BTCU-1. Molecules 20:19277–19290. https://doi.org/10.3390/molecules201019277
Love M, Bhandari D, Dobson R, Billington C (2018) Potential for bacteriophage endolysins to supplement or replace antibiotics in food production and clinical care. Antibiotics 7:17. https://doi.org/10.3390/antibiotics7010017
Marchler-bauer A, Derbyshire MK, Gonzales NR et al (2015) CDD: NCBI’ s conserved domain database. Nucleic Acids Res 43:222–226. https://doi.org/10.1093/nar/gku1221
Miller JH (1972) Experiments in molecular genetics. Cold Spring Harb Lab Press, Cold Spring Harb NY
Oliveira H, Melo LDR, Santos SB et al (2013) Molecular aspects and comparative genomics of bacteriophage endolysins. J Virol 87:4558–4570. https://doi.org/10.1128/JVI.03277-12
Pastagia M, Schuch R, Fischetti VA, Huang DB (2013) Lysins: the arrival of pathogen-directed anti-infectives. J Med Microbiol 62:1506–1516. https://doi.org/10.1099/jmm.0.061028-0
Pathak R, Rathor N, Garima K et al (2015) lspA gene of Mycobacterium tuberculosis co-transcribes with Rv1540 and induced by surface and acidic stress. Gene 560:57–62. https://doi.org/10.1016/j.gene.2015.01.061
Payne K, Sun Q, Sacchettini J, Hatfull GF (2009) Mycobacteriophage Lysin B is a novel mycolylarabinogalactan esterase. Mol Microbiol 73:367–381. https://doi.org/10.1111/j.1365-2958.2009.06775.x
Payne KM, Hatfull GF, Nigou J (2012) Mycobacteriophage endolysins: diverse and modular enzymes with multiple catalytic activities. PLoS ONE 7(3):e34052
Petersen TN, Brunak S, von Heijne G, Nielsen H (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8:785–786. https://doi.org/10.1038/nmeth.1701
Pohane AA, Joshi H, Jain V (2014) Molecular dissection of phage endolysin aninterdomain interaction confers host specificity in lysin a of mycobacterium phage D29. J Biol Chem 289:12085–12095. https://doi.org/10.1074/jbc.M113.529594
Pope WH, Bowman CA, Russell DA et al (2015) Whole genome comparison of a large collection of mycobacteriophages reveals a continuum of phage genetic diversity. Elife 4:e06416. https://doi.org/10.7554/eLife.06416
Rajaure M, Berry J, Kongari R et al (2015) Membrane fusion during phage lysis. Proc Natl Acad Sci 112:5497–5502. https://doi.org/10.1073/pnas.1420588112
Sinha A, Eniyan K, Manohar P et al (2020) Characterization and genome analysis of B1 sub-cluster mycobacteriophage PDRPxv. Virus Res 279:197884. https://doi.org/10.1016/j.virusres.2020.197884
Smith KC, Castro-Nallar E, Fisher JN et al (2013) Phage cluster relationships identified through single gene analysis. BMC Genomics 14:410. https://doi.org/10.1186/1471-2164-14-410
Söding J, Biegert A, Lupas AN (2005) The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. https://doi.org/10.1093/nar/gki408
Ueguchi C, Ito K (1990) Escherichia coli sec mutants accumulate a processed immature form of maltose-binding protein (MBP), a late-phase intermediate in MBP export. J Bacteriol 172:5643–5649. https://doi.org/10.1128/jb.172.10.5643-5649.1990
World Health Organisation (2018) Global health TB report 2018. World Health Organization, Geneva
Acknowledgements
We thank Innovation and entrepreneurship development center (IEDC) cell at Acharya Narendra Dev College, funded by the National Science & Technology Entrepreneurship Development Board (NSTEDB), Department of Science and Technology (DST), India for supporting the project. We thank TATA-CSIR-OSDD fellowship program for providing fellowship (TCOF 21) to Ms. Avni Sinha and IEDC for provision of fellowship for undergraduate students Mr. Shazeb Ahmad (co-author) and Ms. Simran Virdi (we acknowledge her contribution in turbidimetric assay). We express our gratitude to Dr. Pawan Sharma, former Senior Scientist at the International Centre for Genetic Engineering and Biotechnology (ICGEB), India for proofreading the manuscript. We thank the Principal, Acharya Narendra Dev College for the infrastructural support and research facilities.
Funding
Innovation and entrepreneurship development center (IEDC) cell at ANDC is funded by the National Science & Technology Entrepreneurship Development Board (NSTEDB), Department of Science and Technology (DST), India.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Eniyan, K., Sinha, A., Ahmad, S. et al. Functional characterization of the endolysins derived from mycobacteriophage PDRPxv. World J Microbiol Biotechnol 36, 83 (2020). https://doi.org/10.1007/s11274-020-02858-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11274-020-02858-7