
Abstract
Bone morphogenic protein-2 (BMP-2) is the most documented member of BMP family and plays a crucial role in bone formation and growth. In this study, we systematically analyze and compare the complex crystal structures and interaction properties of BMP-2 with its cognate receptors BMPR-I/BMPR-II and with its natural antagonist crossveinless-2 (CV-2) using an integrated in silico-in vitro strategy. It is found that the antagonist-binding site is not fully overlapped with the two receptor-binding sites on BMP-2 surface; the antagonist can competitively disrupt BMP-2–BMPR-II interaction using a blocking-out-of-site manner, but has no substantial influence on BMP-2–BMPR-I interaction. Here, the antagonist-binding site is assigned as a new functional epitope armpit to differ from the traditional conformational epitope wrist and linear epitope knuckle at receptor-binding sites. Structural analysis reveals that the armpit comprises three sequentially discontinuous, structurally vicinal peptide segments, separately corresponding to a loop region and two β-strands crawling on the protein surface. The three segments cannot work independently when splitting from the protein context, but can restore binding capability to CV-2 if they are connected to a single peptide. A systematic combination of different-length polyglycine linkers between these segments obtains a series of designed single peptides, from which several peptides that can potently interact with the armpit-recognition site of CV-2 with high affinity and specificity are identified using energetic analysis and fluorescence assay; they are expected to target BMP-2–CV-2 interaction in a self-inhibitory manner.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Bone regeneration is a complex, well-orchestrated physiological process of bone formation and growth, which can be observed in continuous remodeling throughout adult life and is a necessary phenomenon in bone healing such as fracture due to osteoporosis and surgery [1]. Bone morphogenetic proteins (BMPs) are known to play a central role in bone formation, growth and regeneration [2], which belong to the transforming growth factor β (TGF-β) family and regulate bone remodeling via a variety of signaling pathways [3]; activation of these BMP signaling has been found to be involved in the development of osteoblasts and their inhibition may represent one of the attractive strategies for certain bone disorders such as osteoproliferation and osteoarthritis [4]. BMP-2 is the most documented member of BMP family, which is known to have two functional epitopes referred to wrist and knuckle [5]; the former is a highly discontinuous conformational epitope that comprises different elements of both BMP-2 monomers and binds to type-I receptor (BMPR-I), whereas the latter is a continuous linear peptide epitope located at the β7-strand of one monomer surface and binds to type-II receptor (BMPR-II) [6]. Recently, the recognition site of TGF-β1 by its receptor proteins was revealed. The TGF-β1 is structurally homologous with BMP-2; both belong to the same superfamily. Therefore, the recognition site can be considered as a new epitope, which, however, is considerably different to wrist and knuckle [7].
Over the past decades, a number of natural antagonists such as noggin, chordin, gremlin, crossveinless, USAG-1 and follistatin have been uncovered to inhibit the biological activity of BMP-2 via competitively blocking the protein interaction with its cognate receptors [8]. Previously, we successfully designed a number of BMPR-I based cyclic peptides to target BMP-2 using an integrated strategy [9, 10]. Later, the strategy was used to rationally derive osteogenic peptides from BMP-2 knuckle epitope region [11]. Here, we further employed the strategy to systematically investigate the intermolecular recognition and interaction of BMP-2 with its antagonist crossveinless-2 (CV-2) at structural level. By comparing the crystal complex structures of BMP-2 with CV-2 and with its receptors BMPR-I/BMPR-II, we found that the BMP-2 antagonist-binding site is overlapped with neither wrist epitope nor knuckle epitope. Instead, the BMP-2 adopts a moderate surface region to interact with CV-2, which, in addition to wrist and knuckle epitopes, was assigned as a new functional epitope termed armpit. The armpit epitope was investigated systematically based on BMP-2–CV-2 complex crystal structure, which contains three discrete peptide segments that are close to each other on the protein surface. We also split these peptides from full-length BMP-2 protein context and then redesigned them to derive a number of new peptides, which exhibited self-inhibitory capability against the native BMP-2–CV-2 interaction by competing with BMP-2 for CV-2.
2 Materials and Methods
2.1 Complex Crystal Structures of BMP-2 Dimer with Its Natural Antagonist CV-2 and Its Cognate Receptors BMPR-I/BMPR-II
The biologically active BMP-2 protein exists in a C2-symmetrical homodimer and exposes functional sites at each of monomers to interact its cognate receptors as well as natural antagonists. The receptors include type-I (BMPR-I) and type-II (BMPR-II), and their ternary complex crystal structures with BMP-2 dimer was downloaded from the protein data bank (PDB) [11] with id 2H62. In addition, the receptor-mediated signaling can be antagonized by antagonist protein CV2 through blocking the binding of BMPR-I and BMPR-II to BMP-2 [12]; their complex crystal structure was obtained with PDB id 3BK3. Here, the cocrystallized water molecules, ions and other cofactors were manually removed from the raw crystal structures [13]; hydrogen atoms and protonation state were automatically assigned for the protein systems using REDUCE program [14] and H++ server [15], respectively.
2.2 Molecular Dynamics Simulation
The complex systems of BMP-2 dimer with receptor, antagonist and armpit epitope-derived peptides were analyzed using molecular dynamics (MD) simulations. The complexes were solvated in a TIP3P water box and subjected to 100-ps pre-simulations with gradual temperature increase from 0 to 300 K [16, 17]. Next, hundreds-nanosecond MD simulations with 2 fs time step were performed for each complex system under the isothermal-isobaric ensemble, with water, protein and peptide coupled separately to a heat bath with T = 298 K and a time constant τT = 0.1 ps using weak temperature coupling, and atmospheric pressure was maintained at 1 bar using weak semi-isotropic pressure coupling with compressibility κxyz = 5 × 10−5 bar−1 and time constant τP = 1 ps [18]. The particle-mesh-Ewald (PME) [19] and LINCS [20] were used to treat electrostatic interactions and hydrogen-involving covalent bonds, respectively.
The complex binding energetics were characterized using molecular mechanics/Poisson-Boltzmann surface area (MM/PBSA) [21], which calculated the complex interaction energy ΔEint and the desolvation free energy ΔGdsol upon the complex binding using molecular mechanics (MM) approach and finite-difference solution of implicit solvent model (PBSA), respectively [22]. The solvent dielectric constant was assigned to 80. If the complex is formed by BMP-2 dimer with an armpit epitope-derived peptide, conformational flexibility of the peptide ligand was dissected with Quasiharmonic approach [23] to estimate entropy penalty − TΔS upon the peptide binding. Consequently, the total binding free energy ΔGttl can be written as follows [9]:
or
where < ··· > represents average over the conformational snapshots collected over a single simulation trajectory and i corresponds to the ith snapshot of the complex.
2.3 Fluorescence-Based Analysis
The fluorescence polarization (FP) assays were performed at 298 K following a protocol modified from our previous works [9, 10]. Briefly, the conjugated fluorescein (FITC) was used to label synthetic peptides. Titrations were conducted by monitoring FP as a function of increasing BMP-2 protein concentration added to 10 μM FITC-peptides in a buffer containing 50 mM Tris–HCl, 100 mM NaCl and 5 mM EDTA. Each assay was performed in triplicate. The dissociation constant (Kd) was determined by fitting titration curves to the equation:
where the [P] is the BMP-2 protein concentration, F is the polarization value at a measured concentration, and F0 and F∞ are the polarization values of BMP-2-free and BMP-2-saturated peptides, respectively.
3 Results and Discussion
3.1 Structure-Based Identification of BMP-2 Armpit Epitope
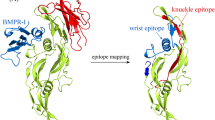
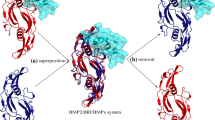
Traditionally, two regions on BMP-2 protein surface, referred to wrist and knuckle epitopes, are known as the functional binding sites of its receptor proteins BMPR-I and BMPR-II, respectively [24]. Here, the two complex crystal structures of BMP-2 dimer with its cognate receptors BMPR-1/BMPR-2 (PDB: 2H62) and BMP-2 dimer with its natural antagonist CV-2 (PDB: 3BK3) were superposed onto each other in Fig. 1. As can be seen, the BMP-2 dimer is structurally similar in the two complex crystal structures, with a small backbone-atom RMSD = 1.85 Å between the two BMP-2 dimers separately in crystal structures 2H62 and 3BK3. This is expected since the dimer structure architecture is rigid and well folded, which is not affected significantly by its receptor or antagonist binding.

Superposition between the complex crystal structures of BMP-2 dimer with its cognate receptors BMPR-1/BMPR-2 (PDB: 2H62) and BMP-2 dimer with its natural antagonist CV-2 (PDB: 3BK3). The backbone-atom RMSD value between the two BMP-2 dimers separately in crystal structures 2H62 and 3BK3 is 1.85 Å
The complex crystal structure of BMP-2 dimer with its cognate receptors is shown in Fig. 2a, where the BMPR-I and BMPR-II is bound to the wrist and knuckle epitopes on BMP-2 surface, respectively. The wrist is a conformational epitope that spans across different regions in both two subunits of the dimer, whereas the knuckle epitope is a linear epitope that crawls along a β-strand in one subunit. The complex crystal structure of BMP-2 dimer with its natural antagonist CV-2 is shown in Fig. 2b, which exhibits a distinct binding mode as compared to BMPR-I and BMPR-II. The binding site of CV-2 is separated from BMPR-I binding site, while it partially overlaps with BMPR-II binding site. Therefore, CV-2 is thought not to influence the BMPR-I recognition by BMP-2. Instead, it may competitively disrupt BMP-2–BMPR-II interaction using a blocking-out-of-site manner, that is, some protein components that are out of CV-2 binding site can sterically block the proper location of BMPR-II on BMP-2 surface. Therefore, the CV-2 can be regarded as a BMPR-II specific inhibitor. However, the CV-2 binding site is considerably different to BMPR-II binding site; the latter only contains one continuous β-strand, whereas the former comprises of three discontinuous regions (partially including the β-strand). Here, we called the CV-2 binding site of BMP-2 as armpit epitope to distinguish it with wrist epitope representing BMPR-I binding site and knuckle epitope representing BMPR-II binding site. Here, the three epitopes are mapped onto the protein surface of BMP-2 dimer in Fig. 2c. It is evident that they have different sizes and presents distinct shapes. Considering that the solvent accessible surface area (ASA) described by Lee and Richards [25] would help to analyze and compare BMP-2 interaction interfaces representing the three epitopes, we computed the interface area (IA) of BMP-2 complexes with BMPR-1, BMPR-2 and CV-2 via the ASA approach using PISA server [26]. Consequently, the IA values of BMP-2–BMPR-I (wrist epitope), BMP-2–BMPR-II (knuckle epitope) and BMP-2–CV-2 (armpit epitope) complexes were calculated as 749.6, 407.6 and 585.2 Å2, respectively; they are different significantly, suggesting that the armpit epitope is not consistent with traditional wrist and knuckle epitopes.

a Crystal complex structure of BMP-2 dimer with the wrist epitope and knuckle epitope of its cognate receptors BMPR-I and BMPR-II, respectively (PDB: 2H62). b Crystal complex structure of BMP-2 dimer with the armpit epitope of its natural antagonist CV-2 (PDB: 3BK3). c Mapping of wrist, knuckle and armpit epitopes onto the protein surface of BMP-2 dimer
3.2 Sequence, Structural and Energetic Analysis of BMP-2 armpit Epitope
The residue importance in BMP-2–CV-2 binding was investigated using alanine scanning strategy [27], which virtually mutated each BMP-2 residue to alanine and calculated total binding energy change upon the mutation using MM/PBSA method. The strategy has recently been successfully used to computationally identify the core recognition epitope of TGF-β1 by its receptor proteins [7]. Consequently, the resulting energy changes can be used to characterize the relative contribution of each BMP-2 residue to the binding, which are visualized as a histogram plot in Fig. 3. If a mutation causes energy loss (> 0) it is positive contribution residue and should be important for the binding; vice versa. Generally, the wild-type BMP-2 residues can positively contribute to the binding, and only very few are negative, although most mutations can only affect BMP-2–CV-2 binding moderately or modestly, This is expected if considering that the sequence and structure of BMP-2 protein has been evolutionally compatible with its natural antagonist CV-2; the residue mutation would impair the compatibility and therefore cause unfavorable effect on the binding. However, there are also few residues that seem to play an important role in the binding, with energy change > 0.8 kcal/mol. Interestingly, these important residues are not distributed evenly, which can be clustered into three discrete regions in BMP-2 protein, namely segment 1 (31WIVAPP36), segment 2 (87ISMLML92) and segment 3 (98VVLKNYQ104); they come together to define the armpit epitope.

The contribution of each BMP-2 residue to CV-2 binding. The three hotspot segments 1, 2 and 3 of BMP-2 representing armpit epitope are identified
The three segments at BMP-2–CV-2 complex interface are highlighted in Fig. 4. They are sequentially discontinuous but spatially vicinal, roughly parallel with each other. The segment 1 is a loop region, and the segments 2 and 3 are two β-strands of BMP-2 protein; they can directly contact and interact with CV-2, thus contributing significantly to the complex binding. Subsequently, the three segments were derived from BMP-2 surface to obtain three corresponding peptides, and their binding energies to CV-2 were calculated and decomposed using MM/PBSA method and Quasiharmonic approach (Table 1). The binding energetic components of BMP-2 protein and armpit epitope to CV-2 were also analyzed and compared with the three segment peptides. It is seen that the BMP-2 exhibits a high binding potency to CV-2 with total binding energy ΔGttl = − 21.8 kcal/mol, which is a compromise between negative interaction ΔEint = − 174.2 kcal/mol and positive desolvation effect (ΔGsol = 152.4 kcal/mol). The binding of armpit epitope to CV-2 also shows a similar energetic profile with BMP-2 protein, although the energetic components of armpit epitope seem to be moderately degraded relative to the full-length BMP-2 protein. This is not unexpected since the protein context has recently been found to confer affinity and specificity to peptide-mediated protein interactions, although the context does not directly participate in the interaction, which can exerts indirectly effects such as conformational selection and flexibility constraint to help the interactions [28]. In addition, the three segment peptides have only a moderate or modest binding capability to CV-2, with ΔGttl ranging between − 2.3 and − 6.9 kcal/mol, suggesting that each of the three independent peptides of armpit epitope cannot be recognized and bound effectively by CV-2, although they can work together as a complete armpit epitope in BMP-2 protein context to interact tightly with CV-2. In particular, the three independent peptides were speculated to incur a large entropy penalty upon binding to CV-2, separately with − TΔS = 21.6, 27.4 and 29.2 kcal/mol, indicating that they possess large intrinsic disorder when splitting from protein context to solvent.

Highlight of armpit epitope at the complex interface of BMP-2 dimer with its antagonist CV-2. The epitope consists of three sequentially discontinuous but structurally vicinal segments 1, 2 and 3. The segment 1 is a loop region, and the segments 2 and 3 are two β-strands
3.3 Molecular Design of BMP-2 Armpit-Derived Self-Inhibitory Peptides
The BMP-2 armpit epitope was found to cover three protein segments; they are discontinuous in sequence but vicinal in structure. Therefore, we considered to artificially combine them together to obtain a series of single peptides that mimic the native conformation of armpit epitope on BMP-2 protein surface to target CV-2 binding site, thus competitively disrupting BMP-2–CV-2 interaction. Such peptides are known as self-inhibitory peptides that are a kind of small protein segments that are split from the complex interface of protein–protein interactions but restore the potential capability to rebind at the interface as a general mechanism to inhibit their cognate interactions [29]. Here, the gap between armpit segments 1 and 2 is assigned as break 1, and the gap between segments 2 and 3 is break 2. Two flexible polyglycine linkers poly-(Gly)n and poly-(Gly)m were added to breaks 1 and 2 to connect the three segments into a single peptide, respectively. The linker lengths n and m at breaks 1 and 2 range from 0 to 4, respectively, and their systematic combination between the two breaks can totally generate 5 × 5 = 25 single peptides. Subsequently, the complex structures of CV-2 with the 25 designed peptides were modeled manually and equilibrated with MD simulations, based on which their binding energies were calculated using energetic analysis. The peptide binding energies associated with the 5 × 5 systematic combination are visualized as a heatmap in Fig. 5, which straightforwardly characterizes the relative binding potency of the 25 designed peptides to CV-2. As can be seen, the binding potency of these peptides is distributed unevenly, which is improved consistently with the increases of linker lengths n and m. Obviously, a high-potency region with n ≥ 3 and m ≥ 1 are revealed from the heatmap, which is termed as hot block and represents eight potent binders of CV-2, suggesting that long linker for break 1 and moderate linker for break 2 are good choices to confer high affinity to designed single peptides.

Heatmap of the systematic combination effect between the different lengths of poly-(Gly)n linker at break 1 and poly-(Gly)m linker at break 2. A hot block represents the potent combination region is highlighted
The binding energetic components of eight designed peptides in the hot block to CV-2 were decomposed and listed in Table 2. It is evident that the total binding energy of these connected peptides is improved relative to that of three independent segments, although their flexibility and entropy penalty also increase upon the connection. This is expected since the intrinsic disorder of peptides is roughly proportional to peptide length [30], combination of three independent segments into a single peptide would significantly increase peptide length, which causes high entropy cost upon the peptide binding. However, the unfavorable entropy effect could be largely counteracted by favorable interaction energy incoming from peptide length increase, thus exhibiting an improved total binding energy. In order to substantiate the computational design, four out of the eight designed peptides were selected, namely, pGly(3–2), pGly(3–3), pGly(4–2), pGly(4–3) and pGly(4–4), which were predicted to have the highest binding potency to CV-2, with ΔGttl > − 10 kcal/mol; their binding affinities were measured using fluorescence-based assays. Titration of CV-2 to peptide buffer can moderately and considerably shift the fluorescence polarization curves shown in Fig. 6, indicating that, as expected, these peptides can effectively bind to CV-2 protein in solvent. The Kd values can be readily derived from these polarization curves and provided in Table 1. As might be expected, a good consistence between the calculated ΔGttl and experimental Kd can be observed, with a significant correlation R = 0.73. The four tested peptides were determined to have high or moderate affinity, with Kd ranging between 18.2 and 114.2 μM, in which the pGly(3–2) peptide exhibited the highest affinity (Kd = 18.2 μM).

Fluorescence polarization curves of five designed armpit-derived peptides binding to CV-2
4 Conclusion
The intermolecular interaction between BMP-2 protein and its natural antagonist CV-2 was investigated systematically at structural, energetic and dynamic levels. It is revealed that the antagonist binding site is not fully overlapped with receptor binding sites on BMP-2 surface, thus assigned as a new epitope armpit, which is considerably different to the traditional receptor recognition epitopes wrist and knuckle in shape and size. The armpit epitope consists of three sequentially discontinuous, structurally vicinal peptide segments, separately representing a loop region and two β-strands of BMP-2; they cannot work effectively in independent manner to recognize and bind CV-2 if lack of protein context support. However, connection of the three segments into a single peptide can partially restore the epitope binding capability. By systematically optimizing the combination of two polyglycine linker lengths between these segments obtained a number of designed peptides that can potently bind to CV-2 with high affinity, which can be exploited as lead molecular entities to further develop new competitively inhibitory peptides disrupting BMP-2–CV-2 interaction.
References
Dimitriou R, Jones E, McGonagle D, Giannoudis PV (2011) Bone regeneration: current concepts and future directions. BMC Med 9:66
Chen D, Zhao M, Mundy GR (2004) Bone morphogenetic proteins. Growth Factors 22:233–241
Luo K (2017) Signaling cross talk between TGF-β/Smad and other signaling pathways. Cold Spring Harb Perspect Biol 9:a022137
Rosen V (2006) BMP and BMP inhibitors in bone. Ann N Y Acad Sci 1068:19–25
Kirsch T, Nickel J, Sebald W (2000) BMP-2 antagonists emerge from alterations in the low-affinity binding epitope for receptor BMPR-II. EMBO J 19:3314–3324
Saito A, Suzuki Y, Ogata S, Ohtsuki C, Tanihara M (2003) Activation of osteo-progenitor cells by a novel synthetic peptide derived from the bone morphogenetic protein-2 knuckle epitope. Biochim Biophys Acta 1651:60–67
Chen X, Wang H, Yang S, Zheng J, Liu X, Mao G (2020) Structure-based discovery and redesign of TGF-β1 elbow epitope recognition by its type-II receptor in hypertrophic scarring biotherapy. J Mol Recog. https://doi.org/10.1002/jmr.2881
Ali IHA, Brazil DP (2014) Bone morphogenetic proteins and their antagonists: current and emerging clinical uses. Br J Pharmacol 171:3620–3632
Fan X, Xia H, Liu X, Li B, Fang J (2019) Rational design of type-IA receptor-derived cyclic peptides to target human bone morphogenic protein 2. J Biosci 44:130
Zhang A, Chen Z, Yu X, Zhang L, Song Q (2020) Rational derivation of osteogenic peptides from bone morphogenetic protein-2 knuckle epitope by integrating in silico analysis and in vitro assay. Int J Pept Res Ther. https://doi.org/10.1007/s10989-020-10058-y
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) The protein data bank. Nucleic Acids Res 28:235–242
Zhang JL, Qiu LY, Kotzsch A, Weidauer S, Patterson L, Hammerschmidt M, Sebald W, Mueller TD (2008) Crystal structure analysis reveals how the Chordin family member crossveinless 2 blocks BMP-2 receptor binding. Dev Cell 14:739–750
Zhou P, Yang C, Ren Y, Wang C, Tian F (2013) What are the ideal properties for functional food peptides with antihypertensive effect? A computational peptidology approach. Food Chem 141:2967–2973
Word JM, Lovell SC, Richardson JS, Richardson DC (1999) Asparagine and glutamine: using hydrogen atom contacts in the choice of side-chain amide orientation. J Mol Biol 285:1735–1747
Anandakrishnan R, Aguilar B, Onufriev AV (2012) H++ 3.0: automating pKa prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res 40:W537–W541
Yang C, Wang C, Zhang S, Huang J, Zhou P (2015) Structural and energetic insights into the intermolecular interaction among human leukocyte antigens, clinical hypersensitive drugs and antigenic peptides. Mol Simul 41:741–751
Zhou P, Zhang S, Wang Y, Yang C, Huang J (2016) Structural modeling of HLA-B*1502 peptide carbamazepine T-cell receptor complex architecture: implication for the molecular mechanism of carbamazepine-induced Stevens-Johnson syndrome toxic epidermal necrolysis. J Biomol Struct Dyn 34:1806–1817
Gao S, Wang Y, Ji L (2020) Rational design and chemical modification of TEAD coactivator peptides to target hippo signaling pathway against gastrointestinal cancers. J Recept Signal Transduct Res 10:1–8
Darden T, York D, Pedersen L (1993) Particle mesh Ewald: and N.log(N) method for Ewald sums in large systems. J Chem Phys 98:10089–10092
Hess B, Bekker H, Berendsen HJC, Fraaije JGEM (1997) LINCS: a linear constraint solver for molecular simulations. J Comput Chem 18:1463–1472
Homeyer N, Gohlke H (2012) Free energy calculations by the molecular mechanics Poisson-Boltzmann surface area method. Mol Inf 31:114–122
Zhou P, Wang C, Tian F, Ren Y, Yang C, Huang J (2013) Biomacromolecular quantitative structure-activity relationship (BioQSAR): a proof-of-concept study on the modeling, prediction and interpretation of protein-protein binding affinity. J Comput Aided Mol Des 27:67–78
Yu H, Zhou P, Deng M, Shang Z (2014) Indirect readout in protein-peptide recognition: a different story from classical biomolecular recognition. J Chem Inf Model 54:2022–2032
Miyazono K, Kamiya Y, Morikawa M (2010) Bone morphogenetic protein receptors and signal transduction. J Biochem 147:35–51
Lee B, Richards FM (1971) The interpretation of protein structures: estimation of static accessibility. J Mol Biol 55:379–400
Krissinel E, Henrick K (2007) Inference of macromolecular assemblies from crystalline state. J Mol Biol 372:774–797
Kortemme T, Kim DE, Baker D (2004) Computational alanine scanning of protein-protein interfaces. Sci STKE 2004:pl2
Zhou P, Miao Q, Yan F, Li Z, Jiang Q, Wen L, Meng Y (2019) Is protein context responsible for peptide-mediated interactions? Mol Omics 15:280–295
London N, Raveh B, Movshovitz-Attias D, Schueler-Furman O (2010) Can self-inhibitory peptides be derived from the interfaces of globular protein-protein interactions? Proteins 78:3140–3149
Agrawal P, Singh H, Srivastava HK, Singh S, Kishore G, Raghava GPS (2019) Benchmarking of different molecular docking methods for protein-peptide docking. BMC Bioinform 19:S426
Acknowledgements
This work was supported by the YCH funds.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All the authors declared that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Wu, Y., Jia, G., Chi, H. et al. Integrated In Silico-In Vitro Identification and Optimization of Bone Morphogenic Protein-2 Armpit Epitope as Its Antagonist Binding Site. Protein J 39, 703–710 (2020). https://doi.org/10.1007/s10930-020-09937-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10930-020-09937-6