
Abstract
Currently, model-informed precision dosing uses one population pharmacokinetic model that best fits the target population. We aimed to develop a subgroup identification-based model selection approach to improve the predictive performance of individualized dosing, using vancomycin in neonates/infants as a test case. Data from neonates/infants with at least one vancomycin concentration was randomly divided into training and test dataset. Population predictions from published vancomycin population pharmacokinetic models were calculated. The single best-performing model based on various performance metrics, including median absolute percentage error (APE) and percentage of predictions within 20% (P20) or 60% (P60) of measurement, were determined. Clustering based on median APEs or clinical and demographic characteristics and model selection by genetic algorithm was used to group neonates/infants according to their best-performing model. Subsequently, classification trees to predict the best-performing model using clinical and demographic characteristics were developed. A total of 208 vancomycin treatment episodes in training and 88 in test dataset was included. Of 30 identified models from the literature, the single best-performing model for training dataset had P20 26.2–42.6% in test dataset. The best-performing clustering approach based on median APEs or clinical and demographic characteristics and model selection by genetic algorithm had P20 44.1–45.5% in test dataset, whereas P60 was comparable. Our proof-of-concept study shows that the prediction of the best-performing model for each patient according to the proposed model selection approaches has the potential to improve the predictive performance of model-informed precision dosing compared with the single best-performing model approach.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Model-informed precision dosing (MIPD) of antimicrobial agents is increasingly acknowledged. In the case of vancomycin its predictive performance has been evaluated in several studies, including randomized controlled trials [1,2,3], and is used in clinical practice in some hospitals [4]. According to meta-analyses, it significantly improves the target attainment, decreases the time to achieve the target and reduces the risk of adverse effects [5, 6]. However, in many patients, the target is still not attained, e.g., 28.1–56.2% of neonates and infants achieved the vancomycin target in recent trials [4, 7]. Thus, further improvements in MIPD are warranted.
Currently, in MIPD, a model from those available in the used software [1] or the model that best fits the historical group of patients according to the external evaluation of various population pharmacokinetic models is usually chosen [8]. However, in precision medicine, the importance of finding subgroups of patients whose clinical outcome could be improved by providing targeted therapy has been recognized [9]. In line with this, a model selection approach based on the fit of the model to already measured concentrations, i.e., individual predictions, has been described that outperforms the commonly used approach of using the single best model [10]. Still, lower target attainment rates of the first vs. subsequent dosing regimens suggested by MIPD, e.g., 28.1–41.5% vs. 44.3–56.1%, in the case of vancomycin in neonates and infants [4], remains a considerable problem. This is due to poor predictive performance of population predictions compared with individual predictions [10]. However, population predictions are used to optimize the dose when concentrations have not been measured, as may often be the case. Only recently, solutions for improving population predictions used in MIPD have emerged, e.g., estimating individual clearance values from clinical and demographic data [11] or averaging population predictions from different models using weights based on individual features [12]. However, the root mean square error for the area under the vancomycin time-concentration curve prediction was still larger compared with the best-performing population pharmacokinetic models [12]. Thus, further studies are warranted to improve population predictions used in MIPD.
We aimed to develop a population pharmacokinetic model selection approach based on the identification of the subgroups of patients to increase the predictive performance of MIPD by choosing the best-performing model for each patient according to clinical and demographic characteristics. As a test case, we used vancomycin dosing in neonates and infants – one of the most common antimicrobial agents warranting MIPD in a patient group that challenging in terms of pharmacokinetics [4].
Methods
Data
The study included data from neonates and infants from two previous studies: (i) development of software for individualized dosing of vancomycin in neonates and infants [13], and (ii) prospective evaluation of the developed software in a clinical trial (published currently only as an abstract at a conference) [7]. The study of the development of the software included neonates and infants with postnatal age < 90 days who were admitted to the pediatric intensive care unit of Tartu University Hospital between 01.01.2010–31.12.2015, received vancomycin and had at least one vancomycin concentration measurement [13]. The clinical trial for prospective evaluation of the software included the following patients: (i) neonates and infants with postnatal age < 90 days admitted to pediatric intensive care unit of Tartu University Hospital or Tallinn Children’s Hospital between 01.01.2016–16.05.2019, received vancomycin, had at least one vancomycin steady-state concentration measured during the treatment and were not on renal replacement therapy as a control group; (ii) neonates and infants with postnatal age 3 to 90 days expected to survive for at least 72 h who were admitted to PICU of Tartu University Hospital, Tallinn Children’s Hospital or East Tallinn Central Hospital between 29.05.2019–16.06.2021, who were not included in other clinical trials requiring blood samples, had not received vancomycin for more than 24 h before the study intervention (optimization of vancomycin dose with the software) and whose parent(s) had signed informed consent as an intervention group [7]. In both studies, multiple treatment episodes with vancomycin were allowed to be included if the episodes were separated by at least 7 days without receiving vancomycin. Due to rapid changes in physiology during critical illness and thereby in the pharmacokinetics of vancomycin, in this study, treatment episodes from the same neonate or infant were treated as independent. The dataset was randomly divided into training/test (70%/30%) datasets.
The Ethics Committee of the University of Tartu approved the studies (256/T-23, 288/T-15). Written informed consent was signed by parents or legal representatives before inclusion in the clinical trial [7].
Pharmacokinetic models
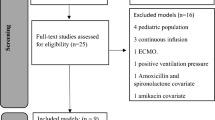
On 20.02.2022 PubMed search „vancomycin AND (population pharmacokinetic* OR nonlinear mixed-effects OR nonlinear-mixed-effects OR NONMEM) AND (neonat* OR newborn* OR infant*)“ was performed to retrieve studies developing population pharmacokinetic model of vancomycin in neonates and infants published since 2010. Models published earlier were excluded due to possible changes in the population of neonates and infants requiring vancomycin and advancements in pharmacokinetic modelling. The studies describing parametric non-linear mixed effects models with all parameter values reported were included in this study. We excluded models with development datasets that did not include infants younger than 3 months and models with covariates missing in our dataset.
Development of model selection approaches
Population predictions from population pharmacokinetic models were calculated using NONMEM version 7.4 (ICON Development Solutions, MD, USA). R was used for all other data analyses. Four approaches for model selection for individualized dosing of vancomycin were evaluated using training dataset – choosing single-best performing model (i) as a reference approach and three proposed subgroup identification-based approaches (ii-iv).
-
(i)
Single best-performing model for the whole training dataset was determined based on median percentage error (PE), median absolute percentage error (APE), percentage of predictions within 20% (P20; shows the percentage of concentrations within 10–15 mg/L if targeting 12.5 mg/L) or 60% (P60; shows the percentage of concentrations within 5–20 mg/L if targeting 12.5 mg/L) of measurement and relative root mean square error (rRMSE) [14]. The models with the smallest PE, APE or rRMSE and largest P20 or P60 were determined as the single best-performing models.
-
(ii)
Clustering based on median APE values was based on the data of median APEs of each model for each neonate and infant in the training dataset (Fig. 1).
Due to a large number of assessed PK models, principal component analysis (PCA) of median APEs of all models was performed to reduce the dimensionality of the data that would result in relatively uniform distances between points, which complicates finding clusters [15]. Subsequently, the first p principal components were used for K-medoid clustering. K-medoid clustering was used as it is relatively robust to outliers [16]. For each cluster, the model with the smallest median APE was determined. Finally, a classification tree using clinical and demographic characteristics (gestational age, postmenstrual age, postnatal age, birth weight, current weight, creatinine, use of inotropes, respiratory support, use of non-steroidal anti-inflammatory agents, sex, small for gestational age) to predict the model for a neonate/infant was developed and its P20 was calculated. The algorithm was performed for p = 2,3,4,5, K = 2,3,4,5 and the maximum depth of classification tree of 2 and 3. The number of principal components p and clusters K and the maximum depth of classification tree was selected using 10-fold cross-validation (CV), i.e., the training dataset was randomly divided into 10 approximately equal parts. For each k = 1,…,10 all except the k-th part was used for analysis up to the development of classification tree and the k-th part was used for the calculation of P20 of the classification tree. Such p, K and the maximum depth of classification tree in which case at least one K-medoid cluster was of size < 20 (due to possibly inaccurate classification tree resulting from such imbalanced data [17]) or principal components explained < 70% of variance [18] were omitted from the ones among which the final tree was determined. The classification tree with the highest mean P20 of all ten CV sets was regarded as the final classification tree. P20, a measure of performance that we eventually want to optimize, rather than the misclassification rate was used to choose the final classification tree because different misclassifications, i.e., predicting a model that is actually not the best model for the patient, may differ in terms of their ‘cost’ [19].
-
(iii)
Clustering based on clinical and demographic characteristics was based on the data on clinical and demographic characteristics of each neonate and infant at the start of the vancomycin treatment. To reduce the dimensionality of the data including continuous and binary variables, factor analysis for mixed data was performed (Fig. 1). Subsequent analysis was similar to clustering based on median APE values.
-
(iv)
Model selection by genetic algorithm was used to find subset of models into which patients can be grouped according to their best-performing models. A reduced number of models for grouping patients was required due to the large number of models to avoid small groups of patients and, thereby, difficulties in building classification tree. Genetic algorithm is an optimization algorithm mimicking the mechanisms of evolution [20]. This iterative method uses information in previous iterations (called generations) to guide the search for the best solution, avoiding assessment of all possible solutions [20]. Genetic algorithm used in this study was adapted from a genetic algorithm used in population pharmacokinetic-pharmacodynamic model selection by Ismail et al. [20]. . The algorithm started with a random selection, without replacement, of 100 subsets of M = 2,3,4,5 models, followed by five steps repeated in 100 generations (Fig. 1). Ten-fold cross-validation was used to choose M and the maximum depth of classification tree of 2 or 3. First, fitness (defined as the mean of P20 of classification tree of ten cross-validation sets) was calculated for each subset of models. For that purpose, the best model was determined for each neonate and infant based on median APE values and classification tree was built to predict the best model for each neonate and infant. The mean of P20 of 10 CV sets was used as the fitness metric. The fitness metric was set to 0 for subsets of models in which case at least one model was the best-performing for < 20 patients to reduce the inaccuracy of such classification tree resulting from imbalanced data [17]. Second, tournament selection was performed, where for each subset of models, a random opponent was chosen and the one with better fitness was retained. Third, in the crossover step, two subsets of models were randomly chosen and randomly chosen models were exchanged. Fourth, in the mutation step a random replacement of models with probability of 0.2 within each subset of models was performed. Fifth, in the elitism step, one subset of models was randomly chosen and replaced with the subset of models with the highest fitness. These five steps were repeated 100 times, called generations. The resulting modified set of the initial 100 subset of models was carried on to the next generation, where the described five steps were repeated. The final classification tree was developed using the subset of models with the highest P20 of 10 CV sets in the last (100-th) generation. Genetic algorithm was repeated twice for each M and the maximum depth of classification tree with mutation rates 0.1, 0.2 and 0.5 to ensure convergence to the best subset of models.

Subgroup identification-based approaches with subsequent development of classification trees to predict the best-performing model for each neonate and infant
* Classification trees and clustering based on clinical and demographic characteristics included the following: gestational age (GA), postmenstrual age, postnatal age, birth weight (BW), current weight, creatinine, use of inotropes, respiratory support, use of non-steroidal anti-inflammatory agents, sex, small for gestational age
APE – absolute percentage error; CV – cross-validation; P20 – the percentage of predictions within 20% of measurement
Evaluation of the approaches
The single best-performing models based on all five performance metrics and the final classification trees from approaches (ii) to (iv) were evaluated on the test dataset by calculating P20 and P60.
Results
A total of 245 patients were included, with a total of 296 vancomycin treatment episodes divided into training (n = 208) and test (n = 88) dataset. The characteristics of the study population is shown in Table 1. A total of 699 vancomycin concentrations were obtained (497 in the training, 202 in the test dataset), median (range) of 2 (1–3) per patient. Altogether, 35 (7.0%) and 16 (7.9%) were trough concentrations (< 0.5 h before the next dose), and 7 (1.4%) and none were peak concentrations (up to 2 h after the dose) in the training and test set, respectively. All doses were administered as a 1-hour infusion. A total of 30 vancomycin population pharmacokinetic models identified from the literature were included in the analysis (Fig. 2).

Flowchart of literature search and selection of pharmacokinetic (PK) models included in this study
The performance metrics varied largely between the 30 models (Fig. 3). All performance metrics resulted in different single best-performing model: Zhao et al. (based on median APE) [21], Mehrotra et al. (P20) [22], Jacqz-Aigrain et al. (P60) [23], De Cock et al. (median PE) [24], Oudin et al. (rRMSE) [25]. The single best-performing models according to P20, P60 and median APE had similar performance in terms of P20, P60 (Table 2) and median APE (27.8%, 27.1% and 26.5%, respectively) (Fig. 3). Compared with these, the single best-performing models according to median PE and rRMSE had considerably smaller P20 (Table 2) and larger median APE (36.7% and 34.2%, respectively) (Fig. 3). The single best-performing models had P20 in the training dataset 27.0-39.4% (Table 2).

Performance of all 30 models in the training dataset in terms of median percentage error (PE), median absolute percentage error (APE), relative root mean square error (rRMSE), percentage of predictions within 20% (P20) or 60% (P60) of measurement
The single best-performing models according to the performance metrics – Zhao et al. (based on median APE), Mehrotra et al. (P20), Jacqz-Aigrain et al. (P60), De Cock et al. (median PE), Oudin et al. (rRMSE) – are shown with colored bars (red, green, blue, yellow, pink, respectively). Grey bars show models that were not the best-performing according to any performance metric. Asterisks above the bars indicate models that were predicted by the classification trees.
Clustering based on median APE values had mean P20 32.8–39.9% in CV-sets, clustering based on clinical and demographic characteristics 32.5–39.3% and model selection by genetic algorithm 43.4–47.6%. The best-performing clustering based on APE values had p = 5 and K = 3, clustering based on clinical and demographic values p = 5 and K = 3, genetic algorithm M = 4. All had maximum depth of classification tree of 2. The best-performing subgroup identification-based methods had P20 41.6–48.7% in the training set (Table 2).
In the test dataset, P20 of the single best-performing model approach was 26.2 to 42.6% and P20 of subgroup identification-based methods 44.1–45.5% (Table 2), an improvement by 3.5–73.7% from the single best-performing model approaches.
Subgroup identification-based methods did not have consistently larger P60 compared with single best-performing models, having smaller or equal P60 than single best-performing models based on P60 in the training dataset and based on median APE, P20 or P60 in test dataset (Table 2).
Classification trees had the depth of one or two and predicted two to four models with probabilities ranging between 0.59 and 0.81 (Fig. 4). Among the seven models used in at least one tree were three models that were the single best-performing models (Mehrotra et al. [22], Jacqz-Aigrain et al. [23] and Oudin et al. [25]), two models that had relatively good performance (Colin et al. [26]. and Tang et al. [27]. with median APE 27.0% and 29.1%, P20 36.4% and 36.8%, median PE -5.1% and − 4.0%, respectively) and two models with considerably poorer performance (Song et al. [28]. and Hughes et al. [29]. with median APE 43.4% and 119.3%, P20 22.3% and 10.9%, median PE 37.9% and − 119.3%, respectively) (Fig. 3).

The final classification trees of the clustering based on median absolute percentage error (APE) values (uppermost tree) or clinical and demographic characteristics (middle tree) and model selection by genetic algorithm (lowermost tree) used
The leaf nodes show the best-performing model for this group and the probability of this model
Discussion
This study described approaches to derive a model selection algorithm based on clinical and demographic characteristics to improve population predictions for model-informed precision dosing. The improvement of P20 by 3.5–73.7% from the single best-performing model approach shows the potential of such a model selection, although P20 in the test dataset remained low (44.1–45.5%).
Our proposed approach aims to improve the predictive performance of population predictions by selecting an appropriate model based on clinical and demographic characteristics. A simpler solution could be choosing a model for each group of patients that was developed on a similar population, e.g., a model based on critically ill patients preferred in critically ill patients [30]. In large part, the predicted models by classification trees agree with this principle. For example, the model by Colin et al. [26]. , predicted by two classification trees for neonates and infants with postmenstrual age ≥ 33 weeks or gestational age ≥ 35 weeks, was built on a population varying from premature neonates to elderly in contrast to studies by Tang et al. [27]. and Jacqz-Aigrain et al. [23]. where median postmenstrual age was 33 and 32 weeks, respectively, and Mehrotra et al. [22]. with median gestational age 32.7 weeks. The only exceptions to this rule were the terminal nodes that were preceded by splits at two different levels, e.g., the model by Jacqz-Aigrain et al. [23]. was predicted if creatinine is < 55 µmol/L and Tang et al. [27]. if ≥ 55 µmol/L, although median creatinine levels in the study populations were reversed, 54 and 28 µmol/L, respectively. Such disagreement could be explained by differing covariate-covariate [31] or covariate-parameter interactions [32] between populations for model development and our target population that lead to biased predictions. However, this discrepancy may only be apparent as the distributions of the variables within the subgroups formed by the classification trees were not described in the original papers. Such classification of patients exemplifies why the choice of the best model for a particular subgroup of patients warrants more detailed analysis than constructing subgroups of patients based on some broad criteria, e.g., receiving renal replacement therapy or not. Subgroup identification allows revealing obscured associations that can be hard to detect by describing pre-defined subgroups of patients, particularly in complex situations [33], such as pharmacokinetics that changes considerably during critical illness [34].
We believe that the improvement in P20 by subgroup identification-based approaches compared with the single best-performing model approach could be higher than in our study if relevant aspects of dataset are considered. First, a larger training dataset could allow grouping of patients into a larger number of subgroups, as there could be a higher risk of possible inaccuracy of the prediction model if built using highly unbalanced data [17]. Second, a comprehensive list of factors that influence pharmacokinetics, e.g., surgery, renal replacement therapy, plasma protein levels [35], could improve predictions of the classification trees, even if these have been tested as covariates in the models. As the datasets of model and algorithm development may differ in size and characteristics of the population, previously undetected relationships, including interactions, may be detected. This is shown by our study, where postmenstrual age and creatinine values, which are commonly used as covariates in the models, were also used as predictors in two trees. Such interaction between creatinine and postmenstrual age is possibly due to different reflection of renal function by creatinine in various age groups [36]. Third, a larger number of concentrations per treatment episode could allow splitting episodes into shorter periods and fitting models separately to the obtained periods due to changes in pharmacokinetics during critical illness [34]. This prevents selecting a model that has ‘on average’ the best fit to the data from one treatment episode but is outperformed by other models during some time periods. This is exemplified by findings that the predictive performance of individual predictions from pharmacokinetic models deteriorates as the time from measured concentrations increases [37]. Finally, the inclusion of concentrations measured at various times during dosing interval, e.g., peak concentrations, may allow testing timing of concentration prediction as a predictor of model selection. Models differ in their predictions of peak or trough concentrations [37], likely due to the differences in the model development dataset. Thus, different models may be selected for prediction of peak or trough concentrations or peak concentration-based metrics, like area under the curve (AUC).
The subgroup identification-based approaches performed quite comparably, although model selection by genetic algorithm slightly outperformed other methods in CV sets and the training dataset, but only slightly in the test dataset. The reason for the smaller improvement in the test dataset could be the grouping of patients by the model that is the best for the patient, even if the model is only slightly better than for others. Thus, grouping is more pronouncedly affected by randomness leading to the risk of overfitting. Although the differences in P20 were small, possible overfitting is exemplified by its lower P20 in the test compared with the training dataset, in contrast to other subgroup identification-based approaches. Still, the other subgroup identification-based methods also have some disadvantages. Due to the inclusion of all models, clustering based on median APE values is in part driven by models that fit poorly to the data. Thus, this may not result in finding clusters that each have a well-defined best-performing model. However, using methods to improve clustering, like weighting [38] or variable selection [39], to downweigh or omit overall poorly performing models may not lead to optimal solution. This is exemplified by the inclusion of overall relatively poorly performing models by Hughes et al. [29]. and Song et al. [28]. into the prediction algorithm by genetic algorithm. Our results suggest that for some patients models performing poorly for the entire population may be the best-fitting ones. Indeed, these models were the best for eight and nine patients in the training and test dataset with median of median APEs of 8.9% and 15.4%, respectively (data not shown). The result of clustering based on clinical and demographic characteristics depends on the variables considered. If some variables affecting pharmacokinetics are omitted and some with no influence are included, the resulting clusters are less homogeneous in terms of pharmacokinetics and thus the best-performing models for the clusters are not optimal. This will remain a challenge for this approach, as the knowledge of what affects pharmacokinetics warrants further research [35].
Our study has some limitations. First, the small dataset with sparse sampling and few variables did not allow the grouping of patients into a larger number of subgroups and, thereby, better predictive performance. Second, the neonates and infants in the test dataset were from the same population as those in the training dataset. Thus, we do not know how well the algorithms may perform in external evaluation – a critical step in the assessment of models that can describe overfitting or underspecification [40]. Third, the results of the developed subgroup identification-based approaches are most likely influenced by performance metrics, as in the case of selecting the single-best performing models. Several metrics were used for the latter to present the variability of performance evaluation between various studies [41] and compare the proposed approach with already existing approaches. While P20 that we used in selecting the final classification trees is not a commonly used metric, 20% as the cut-off value for the prediction error has been claimed to be in line with clinical practice when concentrations 15–25 mg/L are targeted during continuous infusion [42]. Similarly, it can also be appropriate in the case of targeting 10–15 mg/L in neonates and infants. Median APE value was used in the initial grouping of patients due to its continuity (in contrast to few different values of P20 in one patient) and its alignment with P20 (both use absolute prediction errors, and median APE 20% means P20 equals 50%). In assessing the performance, we additionally used P60 as this can reflect the percentage of concentrations in the range of 5 to 20 mg/L, a target also used in neonates and infants [4]. Of note, the metrics used in the proposed approaches can be changed to tailor the evaluation to the ultimate goals [41]. Fourth, we did not assess how well the algorithms predict AUC. Further research with richer datasets is warranted to assess the predictions of AUC and possibly develop model selection algorithms for the prediction of AUC [2]. . Fifth, the assumption of finding discrete groups of patients may not hold. Although some patients could be clearly grouped separately from others based on some variables, e.g., those receiving renal replacement therapy, pathophysiological processes follow more of a continuum rather than form discrete groups [43]. Thus, there will be inherently some heterogeneity within each subgroup, although less than in the whole population, resulting in a variable performance of the best-performing model in different patients in the subgroup. However, similar to our approach, sepsis phenotypes, for example, have been described [44] to inform on personalized care, supporting our concept of identification of subgroups of patients for improved MIPD. Sixth, we did not assess to what extent missing or erroneous covariate data may undermine the performance of such algorithms as this would need a dataset with more often recorded covariate values. However, our results should be applicable to the clinical setting, as we used data that is also available at the point of care when the need for vancomycin is decided and when acquiring more accurate data, e.g., creatinine values at that time point, would delay the start of treatment and thereby hinder the success of treatment. Finally, the best-performing pharmacokinetic model chosen according to developed approaches improves predictive performance for optimizing a priori dose in MIPD. However, it should be highlighted that improved predictive performance may not hold for dose optimization after concentrations have been measured.
In conclusion, our proof-of-concept study shows that one size does not fit all also in terms of choosing the best-performing pharmacokinetic model for MIPD. Our proposed subgroup identification-based approaches to predict the best-performing model for each patient has the potential to improve the precision of individualized dosing.
References
Ewoldt TMJ, Abdulla A, Rietdijk WJR et al (2022) Model-informed precision dosing of beta-lactam antibiotics and ciprofloxacin in critically ill patients: a multicentre randomised clinical trial. Intensive Care Med 48:1760–1771. https://doi.org/10.1007/S00134-022-06921-9
Neely MN, Kato L, Youn G et al (2018) Prospective trial on the Use of Trough Concentration versus Area under the curve to determine therapeutic vancomycin dosing. Antimicrob Agents Chemother 62. https://doi.org/10.1128/AAC.02042-17
Roggeveen LF, Guo T, Fleuren LM et al (2022) Right dose, right now: bedside, real-time, data-driven, and personalised antibiotic dosing in critically ill patients with sepsis or septic shock—a two-centre randomised clinical trial. Crit Care 26:1–11. https://doi.org/10.1186/S13054-022-04098-7/TABLES/4
Frymoyer A, Schwenk HT, Zorn Y et al (2020) Model-informed Precision Dosing of Vancomycin in Hospitalized Children: implementation and adoption at an academic children’s hospital. Front Pharmacol 11:551. https://doi.org/10.3389/FPHAR.2020.00551/BIBTEX
He N, Su S, Yan Y et al (2020) The benefit of Individualized Vancomycin Dosing Via Pharmacokinetic Tools: a systematic review and Meta-analysis. Ann Pharmacother 54:331–343. https://doi.org/10.1177/1060028019887363/ASSET/IMAGES/LARGE/10.1177_1060028019887363-FIG4.JPEG
Sanz-Codina M, Bozkir HÖ, Jorda A, Zeitlinger M (2023) Individualized antimicrobial dose optimization: a systematic review and meta-analysis of randomized controlled trials. Clin Microbiol Infect. https://doi.org/10.1016/J.CMI.2023.03.018
Kalamees R, Soeorg H, Metsvaht T, Lutsar I (2023) Prospective validation of model-informed precision dosing of vancomycin in neonates and infants. In: The 41st Annual Meeting of the European Society for Paediatric Infectious Diseases
Roggeveen LF, Fleuren LM, Guo T et al (2019) Right dose right now: bedside data-driven personalized antibiotic dosing in severe sepsis and septic shock - rationale and design of a multicenter randomized controlled superiority trial. Trials 20. https://doi.org/10.1186/S13063-019-3911-5
Stanski NL, Wong HR (2019) Prognostic and predictive enrichment in sepsis. Nature Reviews Nephrology 2019 16:1 16:20–31. https://doi.org/10.1038/s41581-019-0199-3
Uster DW, Stocker SL, Carland JE et al (2021) A Model Averaging/Selection Approach improves the predictive performance of Model-Informed Precision Dosing: Vancomycin as a case study. Clin Pharmacol Ther 109:175–183. https://doi.org/10.1002/CPT.2065
Tang BH, Guan Z, Allegaert K et al (2021) Drug Clearance in neonates: a combination of Population Pharmacokinetic Modelling and Machine Learning approaches to improve individual prediction. Clin Pharmacokinet 60:1435–1448. https://doi.org/10.1007/S40262-021-01033-X/TABLES/5
Chan A, Peck R, Gibbs M, van der Schaar M (2023) Synthetic model combination: a new machine-learning method for pharmacometric model ensembling. CPT Pharmacometrics Syst Pharmacol 12:953–962. https://doi.org/10.1002/PSP4.12965
Tasa T, Metsvaht T, Kalamees R et al (2017) DosOpt: a tool for personalized bayesian dose adjustment of Vancomycin in neonates. Ther Drug Monit 39:604–613. https://doi.org/10.1097/FTD.0000000000000456
Gastmans H, Dreesen E, Wicha SG et al (2022) Systematic comparison of hospital-wide Standard and Model-based therapeutic drug monitoring of vancomycin in adults. Pharmaceutics 14:1459. https://doi.org/10.3390/PHARMACEUTICS14071459/S1
Steinbach M, Ertöz L, Kumar V (2004) The challenges of Clustering High Dimensional Data. New Dir Stat Phys 273–309. https://doi.org/10.1007/978-3-662-08968-2_16
Mannor S, Jin X, Han J et al (2011) K-Medoids clustering. Encyclopedia Mach Learn 564–565. https://doi.org/10.1007/978-0-387-30164-8_426
Zhu Y, Li C, Dunson DB (2021) Classification trees for Imbalanced Data: surface-to-volume regularization. J Am Stat Assoc. https://doi.org/10.1080/01621459.2021.2005609/SUPPL_FILE/UASA_A_2005609_SM9089.PDF
Jollife IT, Cadima J (2016) Principal component analysis: a review and recent developments. Philosophical Trans Royal Soc A: Math Phys Eng Sci 374. https://doi.org/10.1098/RSTA.2015.0202
Hand DJ (2006) Classifier technology and the illusion of progress. Stat Sci 21:1–14. https://doi.org/10.1214/088342306000000060
Ismail M, Sale M, Yu Y et al (2022) Development of a genetic algorithm and NONMEM workbench for automating and improving population pharmacokinetic/pharmacodynamic model selection. J Pharmacokinet Pharmacodyn 49:243–256. https://doi.org/10.1007/S10928-021-09782-9/TABLES/1
Zhao W, Lopez E, Biran V et al (2013) Vancomycin continuous infusion in neonates: dosing optimisation and therapeutic drug monitoring. Arch Dis Child 98:449–453. https://doi.org/10.1136/ARCHDISCHILD-2012-302765
Mehrotra N, Tang L, Phelps SJ, Meibohm B (2012) Evaluation of Vancomycin Dosing regimens in Preterm and Term neonates using Monte Carlo simulations. Pharmacotherapy: J Hum Pharmacol Drug Therapy 32:408–419. https://doi.org/10.1002/J.1875-9114.2012.01029.X
Jacqz-Aigrain E, Leroux S, Thomson AH et al (2019) Population pharmacokinetic meta-analysis of individual data to design the first randomized efficacy trial of Vancomycin in neonates and young infants. J Antimicrob Chemother 74:2128–2138. https://doi.org/10.1093/JAC/DKZ158
De Cock RFW, Allegaert K, Sherwin CMT et al (2014) A neonatal amikacin covariate model can be used to predict ontogeny of other drugs eliminated through glomerular filtration in neonates. Pharm Res 31:754–767. https://doi.org/10.1007/S11095-013-1197-Y
Oudin C, Vialet R, Boulamery A et al (2011) Vancomycin prescription in neonates and young infants: toward a simplified dosage. Arch Dis Child Fetal Neonatal Ed 96:F365–F370. https://doi.org/10.1136/ADC.2010.196402
Colin PJ, Allegaert K, Thomson AH et al (2019) Vancomycin Pharmacokinetics throughout Life: results from a Pooled Population analysis and evaluation of current dosing recommendations. Clin Pharmacokinet 58:767–780. https://doi.org/10.1007/S40262-018-0727-5/TABLES/5
Tang Z, Guan J, Li J et al (2021) Determination of Vancomycin exposure target and individualised dosing recommendations for neonates: model-informed precision dosing. Int J Antimicrob Agents 57:106300. https://doi.org/10.1016/J.IJANTIMICAG.2021.106300
Song L, He C-Y, Yin N-G et al (2017) A population pharmacokinetic model for individualised dosage regimens of Vancomycin in Chinese neonates and young infants. Oncotarget 8:105211–105221. https://doi.org/10.18632/ONCOTARGET.22114
Hughes JH, Tong DMH, Lucas SS et al (2021) Continuous learning in Model-Informed Precision Dosing: a Case Study in Pediatric Dosing of Vancomycin. Clin Pharmacol Ther 109:233–242. https://doi.org/10.1002/CPT.2088
Cunio CB, Uster DW, Carland JE et al (2021) Towards precision dosing of Vancomycin in critically ill patients: an evaluation of the predictive performance of pharmacometric models in ICU patients. Clin Microbiol Infect 27. https://doi.org/10.1016/J.CMI.2020.07.005. :783.e7-783.e14
Dhaese SAM, Farkas A, Colin P et al (2019) Population pharmacokinetics and evaluation of the predictive performance of pharmacokinetic models in critically ill patients receiving continuous infusion meropenem: a comparison of eight pharmacokinetic models. J Antimicrob Chemother 74:432–441. https://doi.org/10.1093/JAC/DKY434
Cella M, Knibbe C, de Wildt SN et al (2012) Scaling of pharmacokinetics across paediatric populations: the lack of interpolative power of allometric models. Br J Clin Pharmacol 74:525–535. https://doi.org/10.1111/J.1365-2125.2012.04206.X
Loftus TJ, Shickel B, Balch JA et al (2022) Phenotype clustering in health care: a narrative review for clinicians. Front Artif Intell 5:184. https://doi.org/10.3389/FRAI.2022.842306/BIBTEX
Gous AGS, Dance MD, Lipman J et al (1995) Changes in Vancomycin Pharmacokinetics in critically ill infants. 23:678–682. https://doi.org/101177/0310057X9502300603
Mørk ML, Andersen JT, Lausten-Thomsen U, Gade C (2022) The blind spot of Pharmacology: a scoping review of drug metabolism in prematurely born children. Front Pharmacol 13:351. https://doi.org/10.3389/FPHAR.2022.828010/BIBTEX
Rios DR, Rhee CJ, Elizondo L et al (2020) Creatinine filtration kinetics in critically ill neonates. Pediatr Res 2020 89(4):952–957. https://doi.org/10.1038/s41390-020-0977-4
Broeker A, Nardecchia M, Klinker KP et al (2019) Towards precision dosing of Vancomycin: a systematic evaluation of pharmacometric models for bayesian forecasting. Clin Microbiol Infect 25. https://doi.org/10.1016/J.CMI.2019.02.029. :1286.e1-1286.e7
Ackerman M, Ben-David S, Brânzei S, Loker D (2021) Weighted clustering: towards solving the user’s dilemma. Pattern Recognit 120:108152. https://doi.org/10.1016/J.PATCOG.2021.108152
Pierre-Jean M, Deleuze JF, Le Floch E, Mauger F (2020) Clustering and variable selection evaluation of 13 unsupervised methods for multi-omics data integration. Brief Bioinform 21:2011–2030. https://doi.org/10.1093/BIB/BBZ138
Cabitza F, Campagner A, Soares F et al (2021) The importance of being external. Methodological insights for the external validation of machine learning models in medicine. Comput Methods Programs Biomed 208:106288. https://doi.org/10.1016/J.CMPB.2021.106288
El Hassani M, Marsot A (2023) External evaluation of Population Pharmacokinetic models for Precision Dosing: current state and knowledge gaps. Clin Pharmacokinet 62:533–540. https://doi.org/10.1007/S40262-023-01233-7
Guo T, van Hest RM, Roggeveen LF et al (2019) External evaluation of population pharmacokinetic models of Vancomycin in large cohorts of intensive care unit patients. Antimicrob Agents Chemother 63. https://doi.org/10.1128/AAC.02543-18/ASSET/5C8EC4AB-AB87-488C-8ED0-97E560B2A643/ASSETS/GRAPHIC/AAC.02543-18-F0003.JPEG
Ruiz-Rodriguez JC, Plata-Menchaca EP, Chiscano-Camón L et al (2022) Precision medicine in sepsis and septic shock: from omics to clinical tools. 11:1–21. https://doi.org/10.5492/WJCCM.V11.I1.1. http://www.wjgnet.com/
Koutroulis I, Velez T, Wang T et al (2022) Pediatric sepsis phenotypes for enhanced therapeutics: an application of clustering to electronic health records. J Am Coll Emerg Physicians Open 3:e12660. https://doi.org/10.1002/EMP2.12660
Funding
This study was funded by Tartu University Hospital development fund 8090 through project “Digital solutions to improve the effectiveness and safety of antibiotic treatment in Tartu University Hospital” (562/2021).
Author information
Authors and Affiliations
Contributions
Author Contributions. Acquired and formatted clinical data: RK, HS. Designed the original studies from which datasets are included: TM, IL, RK. Designed the current analysis: HS. Performed the data analysis and interpreted the results: HS. Contributed to the interpretation of the data analysis: IL, TM. Wrote the manuscript: HS.
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflicts of interest.
Ethics approval
The Ethics Committee of the University of Tartu approved the studies (256/T-23, 288/T-15).
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Soeorg, H., Kalamees, R., Lutsar, I. et al. Subgroup identification-based model selection to improve the predictive performance of individualized dosing. J Pharmacokinet Pharmacodyn 51, 253–263 (2024). https://doi.org/10.1007/s10928-024-09909-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10928-024-09909-8