
Abstract
Guselkumab, a human IgG1 monoclonal antibody that blocks interleukin-23, has been evaluated in one Phase 2 and two Phase 3 trials in patients with moderate-to-severe psoriasis, in which disease severity was assessed using Psoriasis Area and Severity Index (PASI) and Investigator’s Global Assessment (IGA) scores. Through the application of landmark and longitudinal exposure–response (E–R) modeling analyses, we sought to predict the guselkumab dose–response (D–R) relationship using data from 1459 patients who participated in these trials. A recently developed novel latent-variable Type I Indirect Response joint model was applied to PASI75/90/100 and IGA response thresholds, with placebo effect empirically modeled. An effect of body weight on E–R, independent of pharmacokinetics, was identified. Thorough landmark analyses also were implemented using the same dataset. The E–R models were combined with a population pharmacokinetic model to generate D–R predictions. The relative merits of longitudinal and landmark analysis also are discussed. The results provide a comprehensive and robust evaluation of the D–R relationship.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
During the clinical testing phase of drug development, several dose levels are evaluated to understand the dose–response (D–R) relationship of the agent in the target patient population over a range of treatment durations. This may be facilitated by exposure–response (E–R) modeling, for which drug exposure [including dose and various pharmacokinetic (PK) metrics] is linked to clinical response (including efficacy endpoints and biomarkers). Landmark and longitudinal analyses are commonly used approaches in E–R analyses.
The landmark analysis approach links selected PK parameters [trough drug concentration (Cmin), maximum drug concentration (Cmax), or area under the drug concentration–time curve (AUC)] to the clinical endpoint response at certain time points, typically that employed for the primary analysis or when the clinical response stabilizes. This approach requires the availability of data for both the PK parameter and the clinical endpoint at the selected time points, with the implicit assumption that the PK parameter serves as the “true driver” of response. However, while the landmark analysis has been widely applied and recommended for E–R analysis, in part due to the ease of analysis and interpretation [1], concerns have been raised recently regarding its uncertainty and lack of consistency in such analyses owing to PK parameter and time point selection [2].
A widely used class of longitudinal E–R models includes indirect response (IDR) modeling [3]. These models are most often used to describe pharmacodynamic endpoints with delayed onset. Applications of IDR models to categorical clinical endpoints have emerged in the last decade via the latent-variable approach [4].
In psoriasis, two commonly used efficacy measurements are the Psoriasis Area and Severity Index (PASI), with total scores ranging from 0 to 72 in 0.1-point increments, and the Investigator’s Global Assessment (IGA), a 5-point scale measuring disease severity (0 = cleared, 1 = minimal, 2 = mild, 3 = moderate, 4 = marked or severe) [5, 6]. Clinical trial efficacy is also often measured by the proportions of patients achieving various response thresholds; in the case of psoriasis, patients typically are evaluated for achievement of ≥ 75, 90, or 100% improvement from baseline in the PASI score (i.e., PASI75, PASI90, and PASI100 responses, respectively) and for achievement of an IGA score of 0 or ≤ 1.
In such assessments, mean absolute PASI scores do not accurately predict proportions of patients who achieve given PASI response thresholds (Prt). We recently argued that the main reason for this issue is lack of accuracy in characterizing PASI score distribution [7]. Consequently, the proportions of patients achieving specific Prt, i.e., PASI75, PASI90, and PASI100 responses, may be more effectively analyzed as an ordered categorical endpoint Prt with four possible outcomes: Prt = 0, if achieving PASI100; Prt = 1, if achieving PASI90 but not PASI100; Prt = 2, if achieving PASI75 but not PASI90; and Prt = 3, if not achieving PASI75. IGA scores are most effectively analyzed as an ordered clinical endpoint [8]. Conceivably, because Prt and IGA both assess disease activity, their E–R relationships should share similar characteristics. As such, jointly modeling Prt and IGA allows better integration of information. In previous work [9], we showed that joint modeling of endpoints could be more parsimonious, and yet better describe the individual endpoints, compared with separately modeling the endpoints.
Psoriasis is a chronic immune-mediated skin disorder [10,11,12]. Interleukin (IL)-23 has been implicated as playing a predominant role in the pathogenesis of psoriasis [13,14,15], and agents that block IL-23 have demonstrated efficacy in the treatment of moderate-to-severe plaque psoriasis [14]. Guselkumab is a monoclonal antibody that specifically blocks IL-23. Based on results from a Phase 2b, dose-ranging, clinical trial of guselkumab in psoriasis [16], one dose regimen [guselkumab 100 mg every 8 weeks (q8w) via subcutaneous (SC) administration] was selected and studied in two Phase 3 clinical trials [5, 6]. Prior to regulatory submission, a thorough E–R analysis was conducted to better understand the D–R relationship across different doses of guselkumab, including untested doses. Since the dose-exposure (D–E) relationship, i.e., the PK profile, is typically well characterized, an E–R analysis can help establish the D–R relationship through the dose–exposure–response (D–E–R) relationship. Therefore, these evaluations included the previously established joint E–R model [9], as well as a comprehensive landmark analysis using AUC and trough concentration at Week 16 (primary endpoint time point) and Week 28 (time point at which steady-state PK and clinical responses were expected to be achieved). Predicted D–R relationships and the associated uncertainties under all models were compared, and the potential influence of covariates explored. This manuscript describes the results of these analyses aiming to confirm the optimum dose regimen of guselkumab in treating psoriasis, and provides insights into the utility of landmark and longitudinal analyses.
Methods
Study designs
One Phase 2 dose-ranging study [16] and two pivotal Phase 3 studies [5, 6] of guselkumab conducted in patients with moderate-to-severe plaque psoriasis were included in the current modeling analyses. All studies were randomized, double-blinded, multicenter, placebo- and active-controlled trials. In the Phase 2 dose-ranging study, approximately 240 patients were treated with SC injections of guselkumab 5, 50, or 200 mg at Week 0, Week 4, and then every 12 weeks (q12w); guselkumab 15 or 100 mg q8w; or placebo. Patients randomized to the placebo group crossed over to receive guselkumab 100 mg q8w at Week 16. The last SC injection of guselkumab was administered at Week 40. The last PK sample and efficacy data were collected at Week 52. Further details of the trial design and participants [16], as well as its population PK and E–R modeling results [7], have been reported.
In one pivotal Phase 3 trial, approximately 450 patients, in a 2:1 ratio, were treated with guselkumab SC 100 mg at Weeks 0, 4, 12, and q8w through Week 44; or placebo at Weeks 0, 4, and 12 followed by guselkumab 100 mg at Weeks 16 and 20, and q8w through Week 44. The second pivotal Phase 3 trial was similarly designed, enrolled approximately 750 patients to receive guselkumab or placebo (2:1 randomization), and included randomized withdrawal beginning at Week 28. In both Phase 3 studies, guselkumab concentrations and clinical efficacy were evaluated through Week 48. Further details of the trials’ designs and participants have been reported [5, 6].
PK, PASI, and IGA assessments
In each study, approximately 10 trough serum samples were collected from each patient. An additional random guselkumab PK sample was collected from each of the 450 patients treated with guselkumab in the first Phase 3 trial. The PASI and IGA evaluations were performed every 4 weeks (q4w) from Weeks 0–40 across trials and were conducted at Week 2 in the Phase 3 trials [5, 6, 16]. The final dataset contained 1459 patients with 13,031 PK measurements, 17,580 Prt observations, and 18,986 IGA scores. The numbers of patients and observations in the longitudinal E–R modeling dataset by treatment group are provided in Table 1.
Population PK model
A confirmatory population PK analysis based on the previously developed model [17, 18] was implemented to describe guselkumab PK in patients with psoriasis. The model is one-compartment with first-order absorption and elimination [apparent clearance (CL/F), apparent volume of distribution (V/F), and absorption rate constant (ka)]. Between-subject random effects on CL/F, V/F, and ka were included using log-normal distributions. Correlation between the between-subject variability (BSV) on CL/F and V/F also were included, as were baseline body weight (BWT) effects on CL/F and V/F using a power model standardized to the median BWT of 87.1 kg. Diabetes and race (Caucasian vs. Non-Caucasian) both demonstrated a marginal effect on CL/F and were included in the final PopPK model. Details of the population PK modeling results have been reported [19].
Longitudinal E–R model
The E–R model is given by the Prt and IGA response components. Based on previously established latent-variable modeling [4], Prt was modeled by the following mixed-effect probit regression:
where Φ is the cumulative normal distribution, prob() is the probability of Prt reaching categories k = 0,1,2; αk,Prt are intercepts; LPrt(t) = fp(t) + fd(t) represents placebo and drug effect over time; and η is BSV modeled with normal distribution η ~N(0, ω2).
To stabilize parameter estimation, αk,Prt, k = 0,1,2, are re-parameterized, namely as (α1,Prt,d0,Prt, d2,Prt) with d0, Prt, d2,Prt > 0 such that α0,Prt= α1,Prt − d0,Prt, and α2,Prt = α1,Prt + d2,Prt.
The placebo effect was modeled empirically as:
where Fp is the maximum placebo effect and r is the rate of onset. The drug effect was modeled with:
where DE represents the maximal drug effect and, following a previous approach [8, 20], the drug effect was assumed to be driven by a latent variable R(t) governed by the Type I IDR model below:
where Cp is the PK model predicted drug concentration at time t, and kin (disease formation rate), IC50 (half-maximal inhibitory concentration), and kout (disease amelioration rate) are model parameters. It was further assumed that R = 1 at baseline, i.e., R(0) = 1, yielding kin= kout.
Theoretically, the representation of drug effect in Eqs. 1–4 has been shown to be equivalent to a change-from-baseline latent-variable IDR model [21], under which kout can be interpreted as the rate of drug effect onset and offset, and DE can be interpreted as the baseline of the latent variable prior to normalization [4].
While IDR models have been widely used, directly applying them to model-ordered categorical variables would cause over-parametrization, because the latent variable is determined only up to a constant and therefore needs to be normalized [4, 22]. For this reason, change-from-baseline IDR models [21, 23] are especially suitable for the modeling of categorical endpoints. Further details on the theoretical characteristics of latent-variable IDR models have been reported [4].
For the IGA score component, as previously implemented [7], IGA scores > 3 were merged with IGA score = 3, i.e., only the levels 0 to ≥3 were modeled. The probability of achieving IGA scores of k ≤ 0, 1, 2, or 3 was modeled in a manner similar to Prt with:
where αk, IGA are intercepts, re-parameterized as (α1, IGA, d0, IGA, d2, IGA, d3, IGA) with d0, IGA, d2, IGA and d3, IGA > 0 such that α0, IGA = α1, IGA − d0, IGA, α2, IGA= α1, IGA+ d2, IGA, and α3, IGA= α2, IGA+ d3, IGA. LIGA(t) represents placebo and drug effect and was modeled similarly as in Eqs. 2–4 above in a separate modeling approach.
Joint E–R modeling of Prt and IGA scores
Equations 1–5 were first fitted to Prt and IGA data separately to serve as a starting point, and then simultaneously. As noted previously [7], maximum sharing can occur if the underlying latent variables for the two endpoints differ by only a scale factor, in which case only one parameter, Sc, could be used to jointly model Prt and IGA, by using LIGA(t) = SC LPrt(t) in Eq. 5. This implies that the placebo and drug effects are the same for Prt and IGA. This maximum sharing was previously shown to be parsimonious by testing intermediary models with less sharing [7]. This led to the following joint model specification:
with fp(t) and fd(t) given by Eqs. 2–4. As noted previously [5], fitting Eqs. 6 and 7 simultaneously to the Prt and IGA data reduces the total number of fixed and random effect parameters by four and one, respectively, compared with separately modeling the endpoints.
Landmark analyses at Week 16 and Week 28
Landmark analyses were performed for Prt and IGA measures separately at Week 16, which is the primary analysis time point, and Week 28, when PK, Prt, and IGA measures were expected to have reached steady state. The exposure measure used for Week 16 was the individual cumulative AUC from time 0 to Week 16 (AUC0−W16) predicted using the population PK model. The observed concentration at Week 16 was not considered appropriate for this analysis, because Week 16 was not a pre-dose visit for patients from the two Phase 3 studies. The exposure measures used for Week-28 analyses were the predicted individual average weekly steady-state AUC (AUCss) and the steady-state trough concentrations (Css). This led to a total of six landmark analyses, including those for Prt and IGA scores.
The landmark analyses required the availability of Prt/IGA measurements at the selected timepoints. For Week 16, 1423 and 1424 patients, respectively, including patients who received placebo treatment, were included in the analysis of Prt and IGA responses. For analyses at Week 28, patients randomized to receive placebo were excluded from the analyses due to crossover to guselkumab at Week 16. Thus, 912 and 913 patients, respectively, were included in the analysis of Prt and IGA using AUCss, and 933 and 934 patients, respectively, were included in the analysis of Prt and IGA using Css. Mixed-effect logistic regression with maximum drug effect in logit scale (Emax) models were used for all landmark analyses, as follows:
where logit(x) = log[x/(1−x)]; Y is either Prt or IGA; βk, k = 0,1, 2 for Prt analysis and k = 0,1 for IGA analysis are intercepts; EM is the exposure metric; and Emax and guselkumab exposure metrics to reach 50% maximum drug effect (EC50) are model parameters.
Covariate modeling
To avoid spurious covariate findings [17, 18], baseline covariates investigated in the E–R analyses were prespecified based on previous experience and physiological/pharmacological relevance. The covariates included BWT and the following disease characteristics: C-reactive protein, baseline PASI score, disease duration, baseline IGA score, and presence or absence of psoriatic arthritis. For categorical covariates, i.e., psoriatic arthritis status and baseline IGA score, those categories appearing in fewer than 20 patients, representing just 5% of patients evaluated, were either dropped from consideration or combined with other categories. Correlations between covariates were examined to ensure that no highly correlated covariates remained simultaneously in the analysis.
In longitudinal E–R modeling, exploratory covariate modeling was conducted by an initial screening of plotting the random effect η against the covariates, and subsequently testing the effects on structural model parameters deemed appropriate.
In the landmark analysis, the covariates were searched for all structural parameters, i.e., the intercept, Emax, and EC50. An additive model was assumed for covariate effects on intercept, and a power model was applied for covariate effects on Emax and EC50. A stepwise forward-addition-backward-deletion procedure was used with the criteria of nonlinear mixed effects modeling (NONMEM) objective function changes of 6.63 and 10.83 in the forward-addition and backward-deletion steps, respectively, when the degree of freedom is 1.
Model estimation and evaluation
In longitudinal E–R modeling, the “PPP&D” (Population PK Parameters and Data) approach described by Zhang and colleagues [24] was used for estimation by fixing the population PK model parameter estimates and retaining the individual concentrations in the dataset to allow individual PK profiles to be determined. As in previous implementations [7], the Importance Sampling estimation option in the software NONMEM (v. 7.3, http://www.iconplc.com/innovation/nonmem/) was used [25]. The first-order conditional estimation method with interaction was used for PopPK analysis. In landmark analyses, the estimation option does not affect the analysis results. The empirical Bayesian estimates of the PK parameters based on the population PK model were used to predict PK exposure (AUC and Css) at Weeks 16 and 28.
A decrease in the NONMEM minimum objective function value (OFV) of 10.83, corresponding to a nominal p value of 0.001, was considered the threshold criterion for determining whether including an additional model parameter improved the model fit. Visual predictive check (VPC) was used for model evaluation by simulating 200 replicates in longitudinal E–R modeling and in landmark analyses [26].
Simulations
The longitudinal and landmark E–R modeling results were each combined with the population PK model to generate the predicted D–R relationships at Week 16 and Week 28 for the treatment regimen as administered in the Phase 3 trials, accounting for parameter estimation uncertainties. In the longitudinal model predictions, 10,000 patients were simulated in each replicate to approximate the true responses for the treatment regimens, and 400 replicates from the parameter estimation uncertainty distribution were simulated to generate 90% confidence intervals (CIs) of the predictions. The landmark analysis model predictions employed 200 replicates, and each replicate contained all patients (> 1300) in the original datasets. The numbers of replicates were sufficient due to the relatively large number of patients in the Phase 3 studies, which was confirmed by initial assessments using larger numbers of replicates.
Results
Demographics characteristics
BWT, the only influential PK covariate, ranged from 45 to 198 kg (mean ± SD: 89 ± 21 kg). Details of baseline patient characteristics and covariates have been reported [5, 6, 16] and are summarized in abbreviated form in the Supplementary Materials (Tables S1 and S2).
Longitudinal modeling
Compared with separately estimating the placebo and drug effect parameters using the same IDR model structure, joint modeling of Prt and IGA achieved an improvement in NONMEM objective function by approximately 1500 despite having fewer parameters, indicating significant improvement in the fit. As shown previously [7], this improvement is due to the sharing of BSV and the improvement of its estimation.
Subsequent exploratory analyses suggested a BWT influence on kout and IC50, in that patients with higher BWT would have slower onset of effect and be less sensitive to guselkumab. These effects were modeled as:
where subscript i indicates the parameter value for the ith patient, and BWT is baseline body weight in kg. Including these two parameters resulted in an improvement in NONMEM objective function of 67, indicating improvement of the fit. This was considered as the final model, and the parameter estimates are shown in Table 2. Estimation was considered precise, with associated relative standard errors (RSEs) within 7% for structural model parameters. ETA-shrinkage of the BSV was low (2.8%). The magnitude of BSV was in line with our previous experiences with latent variable IDR models [7,8,9, 21, 27, 28], after adjusting by a factor of π2/3 when logistic regression was used instead of probit regression [9].
Figures 1 and 2 show the VPC results of the joint model for the Prt and IGA responses for all treatment groups. In general, the model adequately described the data. Some discrepancies occurred, most notably for the low-dose regimens of 5 and 15 mg, potentially due to the relatively small sample sizes of the Phase 2 trial’s treatment groups, which had relatively wide prediction intervals (PIs). The VPC results of the joint model for Prt and IGA responses for all treatment groups in the Phase 3 trials, stratified by BWT categories of < 90 and ≥ 90 kg, are shown in Figs. S1 and S2 in Supplementary Materials. Treatment groups from the Phase 2 trial were not included due to their small sample sizes. Because Eqs. 8 and 9 are nonlinear, the BWT effect on kout and IC50 cannot be expressed on a ‘per kg’ basis. Based on the 10th (64.5 kg) and 90th (115 kg) percentiles of the observed BWT distribution, however, the 90% CIs for BWT effects for a reference 90-kg patient were calculated to be (+ 15%, − 10%) for kout and (− 37.5%, + 42%) for IC50. The cut-off of 90 kg was employed based on the mean BWT of 89 kg. This stratified VPC was considered instead of a prediction-corrected VPC to allow a clear visualization of the BWT effect. The model reasonably described the data.

Longitudinal model of predicted median and 90% PIs, at planned observation times, in overlay with observed PASI responses

Longitudinal model of predicted median and 90% PIs at planned observation times, in overlay with observed IGA responses
Landmark analyses
The Emax models were successfully developed in all six analyses linking Prt and IGA to the PK metrics at Week 16 and Week 28. All six prespecified covariates yielded Spearman correlation coefficients < 0.3, and thus were retained in the stepwise search. All analyses identified a BWT influence on the intercept. Furthermore, in the Week-28 analyses, EC50 was found to be influenced by baseline PASI scores in the IGA analyses, and influenced by baseline disease duration in the Prt analyses. It is noted that greater BWT or longer disease duration may indicate more severe disease, perhaps in different ways. Therefore, collectively, all analyses suggest that patients with more severe disease may be less sensitive to treatment (Table 3).
In the landmark analyses for the endpoints at Week 16, estimation was reasonable, with all RSEs within 30%. However, the landmark analyses for the endpoints at Week 28 yielded relatively high RSEs for some key parameters, including intercept and EC50. The lack of precision for these estimations primarily resulted from lack of placebo data in these analyses. Figure 3 shows the VPC results for Prt at Week 16 using AUC0−W16; additional landmark analysis VPC results are provided in Figs. S3–S5 in Supplementary Materials. These demonstrated that the models reasonably described the data.

Visual predictive check of landmark analysis of PASI at Week 16. The observed PASI 75/90/100 response rates (red circles) were determined according to 10 bins of the model-predicted guselkumab exposure metrics and were plotted at the median exposure within each bin. The red circles are the observed response rates in each bin. The blue solid lines are the simulated median responses. The blue dotted lines and the shaded areas both represent the simulated 90% PIs from 1000 replicates (Color figure online)
Simulations
The longitudinal E–R model-predicted medians (90% CIs) of the population D–R relationship for Prt at Week 16 and Week 28 are shown in Fig. 4. At the 100-mg q8w dose regimen evaluated in the Phase 3 trials, the predicted D–R relationship was flat at Week 16 but moderately increased at Week 28. This is consistent with the estimate of kout = 0.0212/day in Table 2, which corresponds to a half-life of 4.67 weeks, indicating that Week 28 may be closer to pharmacodynamic steady state, at which point patients’ sensitivity to treatment would be expected to show larger influences. The narrow CI ranges are direct consequences of the small RSEs of model parameter estimates shown in Table 2. The results for IGA were similar and shown in Fig. S6 in Supplementary Materials. Simulation of the D–R in a range of 15–200 mg q8w indicated that 100 mg q8w guselkumab treatment aligns with the region of imminent response plateau. This supported the appropriateness of the 100-mg q8w regimen, given the favorable safety profile of guselkumab [5, 6].

Longitudinal model of predicted dose–response for PASI at Week 16 and Week 28 for the guselkumab 50-, 100-, 150-, and 200-mg q8w dose regimen in the Phase 3 psoriasis trials
A landmark analysis E–R relationship plot for Prt with the median and 90% PIs of exposure achieved by the dosing regimens [1] is shown in Fig. 5. Similar to results of longitudinal simulations, guselkumab 100 mg q8w was considered to result in systemic exposures that provide high efficacy approaching the plateau of the E–R curve at Weeks 16 and 28. However, when compared with the Week-16 simulation, a wide CI was predicted for Week-28 responses, resulting in less informative conclusions. The analysis results for IGA were similar (shown in Fig. S7 in Supplementary Materials). The landmark analysis-predicted D–R relationships for Prt and IGA at Week 16 also are shown in Fig. 6. Overall, in line with results of the longitudinal analysis, these analyses were considered to consistently indicate that the guselkumab 100-mg q8w treatment regimen was approaching the plateau of the D–R relationships of Prt and IGA.

Landmark analysis of predicted exposure–response for PASI at Week 16 and Week 28. Solid red lines and shaded area represent the model-predicted median response and 90% PIs, respectively, from 200 replicates. The open circles and blue horizontal segments show the median and 5th/95th percentiles, respectively, of the predicted guselkumab AUC0−W16 and AUCss for the 50-, 100-, and 200-mg q8w dose regimens. The black dotted lines indicate the 5th/95th percentile range of AUC0−W16 and AUCss from the 100 mg q8w treatment (Color figure online)

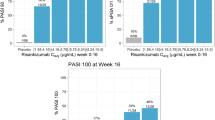
Landmark analysis of predicted responses for PASI and IGA ≤ 1/IGA0 at Week 16 for the guselkumab 50-, 100- and 200-mg q8w dose regimens. Simulations from 200 replicates with parameters obtained from variance–covariance matrices of the estimates from final population pharmacokinetics and landmark exposure–response models. The boxes represent the median (black dot) limited by the 25th and 75th percentiles
Discussion
A previously established population E–R joint model [7] for two ordered categorical endpoints, i.e., Prt (PASI75, PASI90, and PASI100) and IGA scores, was applied to a larger dataset composed of one Phase 2 and two Phase 3 studies. The joint model, with a shared latent variable between the two ordered categorical endpoints, again achieved significant improvement in model fit in terms of NONMEM OFV, while using fewer parameters than required when separately modeling the endpoints. As described previously [7], the improvement was due to the similarity between Prt and IGA, which accounted for the correlation between BSVs for Prt and IGA in the joint model. This confirms that Prt and IGA intrinsically measure overlapping disease characteristics, as well as the advantages of the novel joint modeling approach. Furthermore, as described previously [7], the joint modeling improved parameter estimation precision, as evidenced by small RSEs (Table 2), achieved by using all relevant information. This translated to the narrow range of CIs and thus increased confidence in the predicted D–R relationships. In drug development, improving precision generally allows the trial objectives to be achieved with fewer patients and reduced costs [2].
Landmark analysis is commonly used in clinical development [1], primarily due to its ease of conduct and ability to draw conclusions that are easily explainable to clinicians. Landmark analyses provide a straightforward approach for characterizing D–R relationships and support drug development when sufficient data are available. In general, a wide exposure range achieved from well-planned dose-ranging studies is needed. The relatively high uncertainty in model parameter estimation for the endpoints at Week 28 compared with those at Week 16 suggest that inclusion of placebo or low-dose level data can be critical for landmark analysis. It could be difficult to determine the E–R relationship by using the PK and efficacy data from only relatively high dose levels, which are typically evaluated in Phase 3 studies. Extrapolation using such an E–R relationship with large uncertainty sometimes can be misleading. In addition, selection of appropriate PK metrics is important, as is the accuracy of the exposure metrics used. The landmark analyses for Prt and IGA at both Week 16 and Week 28 support the guselkumab 100-mg q8w regimen as an efficient dose for treating patients with moderate-to-severe psoriasis, similar to the longitudinal models. Nevertheless, with placebo data, Week-16 landmark models were generally more reliable and yielded parameters with better precision. Simulations using the established models also suggested that Week-16 models generated predictions with less uncertainty and are thus more informative in supporting dose selection.
The analyses also suggested an influence of BWT on the E–R relationship. While this influence could potentially relate to more severe disease in heavier patients, the exact mechanism is unclear. Therefore, it is difficult to determine exactly which specific E–R model parameters are influenced by BWT. Additional stratified simulations (not shown) evaluating dose regimens suggested the lack of influence of BWT, likely due to the 100-mg q8w regimen being at or near the plateau of the D–R relationship. In this context, BWT was not considered to warrant dose adjustment for lighter patients, given the overall favorable safety profile of guselkumab, and the BWT influence on the E–R relationship for guselkumab found here should be treated as an exploratory finding pending future confirmation. In the landmark analysis, in addition to BWT, baseline PASI score and disease duration were found to influence EC50 in models for endpoints at Week 28 but not Week 16. The lack of influential covariates at Week 16 could be due to the requirement of reaching steady state to detect such relationships.
Often, uncertainties in E–R analysis conclusions are typically ignored [2], especially for landmark analyses. Our analyses aimed to resolve these potential shortcomings via a comprehensive plan, with inclusion of Phase 2 dose-ranging data, investigation of multiple reasonable exposure metrics for all endpoints, and the evaluation of uncertainties for the predicted D–R relationship. In conjunction with the longitudinal analysis, this helped to provide a thorough and robust understanding of study conclusions [2].
References
Overgaard RV, Ingwersen SH, Tornoe CW (2015) Establishing good practices for exposure-response analysis of clinical endpoints in drug development. CPT Pharmacomet Syst Pharmacol 4(10):565–575. https://doi.org/10.1002/psp4.12015
Hu C, Zhou H, Sharma A (2017) Landmark and longitudinal exposure-response analyses in drug development. J Pharmacokinet Pharmacodyn 44(5):503–507. https://doi.org/10.1007/s10928-017-9534-0
Sharma A, Jusko WJ (1996) Characterization of four basic models of indirect pharmacodynamic responses. J Pharmacokinet Biopharm 24(6):611–635
Hu C (2014) Exposure-response modeling of clinical end points using latent variable indirect response models. CPT Pharmacomet Syst Pharmacol 3:e117. https://doi.org/10.1038/psp.2014.15
Blauvelt A, Papp KA, Griffiths CEM, Randazzo B, Wasfi Y, Shen YK, Li S, Kimball AB (2017) Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the continuous treatment of patients with moderate to severe psoriasis: results from the phase III, double-blinded, placebo- and active comparator-controlled VOYAGE 1 trial. J Am Acad Dermatol 76(3):405–417. https://doi.org/10.1016/j.jaad.2016.11.041
Reich K, Armstrong AW, Foley P, Song M, Wasfi Y, Randazzo B, Li S, Shen YK, Gordon KB (2017) Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the treatment of patients with moderate to severe psoriasis with randomized withdrawal and retreatment: results from the phase III, double-blind, placebo- and active comparator-controlled VOYAGE 2 trial. J Am Acad Dermatol 76(3):418–431. https://doi.org/10.1016/j.jaad.2016.11.042
Hu C, Randazzo B, Sharma A, Zhou H (2017) Improvement in latent variable indirect response modeling of multiple categorical clinical endpoints: application to modeling of guselkumab treatment effects in psoriatic patients. J Pharmacokinet Pharmacodyn 44(5):437–448. https://doi.org/10.1007/s10928-017-9531-3
Hu C, Szapary PO, Yeilding N, Zhou H (2011) Informative dropout modeling of longitudinal ordered categorical data and model validation: application to exposure-response modeling of physician’s global assessment score for ustekinumab in patients with psoriasis. J Pharmacokinet Pharmacodyn 38(2):237–260. https://doi.org/10.1007/s10928-011-9191-7
Hu C, Zhou H (2016) Improvement in latent variable indirect response joint modeling of a continuous and a categorical clinical endpoint in rheumatoid arthritis. J Pharmacokinet Pharmacodyn 43(1):45–54. https://doi.org/10.1007/s10928-015-9453-x
Koo J (1996) Population-based epidemiologic study of psoriasis with emphasis on quality of life assessment. Dermatol Clin 14(3):485–496
Krueger GG, Duvic M (1994) Epidemiology of psoriasis: clinical issues. J Invest Dermatol 102(6):14S–18S
Schon MP, Boehncke WH (2005) Psoriasis. N Engl J Med 352(18):1899–1912
Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ (2005) IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med 201(2):233–240
Rozenblit M, Lebwohl M (2009) New biologics for psoriasis and psoriatic arthritis. Dermatol Ther 22(1):56–60. https://doi.org/10.1111/j.1529-8019.2008.01216.x
Yawalkar N, Karlen S, Hunger R, Brand CU, Braathen LR (1998) Expression of interleukin-12 is increased in psoriatic skin. J Invest Dermatol 111(6):1053–1057. https://doi.org/10.1046/j.1523-1747.1998.00446.x
Gordon KB, Duffin KC, Bissonnette R, Prinz JC, Wasfi Y, Li S, Shen YK, Szapary P, Randazzo B, Reich K (2015) A phase 2 trial of guselkumab versus adalimumab for plaque psoriasis. N Engl J Med 373(2):136–144. https://doi.org/10.1056/NEJMoa1501646
Hu C, Zhang J, Zhou H (2011) Confirmatory analysis for phase III population pharmacokinetics. Pharmaceut Stat 10(1):14–26. https://doi.org/10.1002/pst.403
Hu C, Zhou H (2008) An improved approach for confirmatory phase III population pharmacokinetic analysis. J Clin Pharmacol 48(7):812–822. https://doi.org/10.1177/0091270008318670
Yao Z, Hu C, Zhu Y, Xu Z, Randazzo B, Wasfi Y, Chen Y, Sharma A, Zhou H (2018) Population pharmacokinetic modeling of guselkumab, a human IgG1λ monoclonal antibody targeting IL-23, in patients with moderate-to-severe plaque psoriasis. J Clin Pharmacol. https://doi.org/10.1002/jcph.1063
Hu C, Yeilding N, Davis HM, Zhou H (2011) Bounded outcome score modeling: application to treating psoriasis with ustekinumab. J Pharmacokinet Pharmacodyn 38(4):497–517. https://doi.org/10.1007/s10928-011-9205-5
Hu C, Xu Z, Mendelsohn A, Zhou H (2013) Latent variable indirect response modeling of categorical endpoints representing change from baseline. J Pharmacokinet Pharmacodyn 40(1):81–91. https://doi.org/10.1007/s10928-012-9288-7
Hutmacher MM, Krishnaswami S, Kowalski KG (2008) Exposure-response modeling using latent variables for the efficacy of a JAK3 inhibitor administered to rheumatoid arthritis patients. J Pharmacokinet Pharmacodyn 35(2):139–157
Woo S, Pawaskar D, Jusko WJ (2009) Methods of utilizing baseline values for indirect response models. J Pharmacokinet Pharmacodyn 36(5):381–405. https://doi.org/10.1007/s10928-009-9128-6
Zhang L, Beal SL, Sheiner LB (2003) Simultaneous vs. sequential analysis for population PK/PD data I: best-case performance. J Pharmacokinet Pharmacodyn 30(6):387–404
Beal SL, Sheiner LB, Boeckmann A, Bauer RJ (2009) NONMEM user’s guides (1989–2009). Icon Development Solutions, Ellicott City
Karlsson MO, Holford NHG (2008) A tutorial on visual predictive checks. http://www.page-meeting.org/?abstract=1434. Accessed Jan 3 2018
Hu C, Szapary P, Mendelsohn A, Zhou H (2014) Latent variable indirect response modeling of a continuous and a categorical clinical endpoint. J Pharmacokinet Pharmacodynam 41(4):335–349. https://doi.org/10.1007/s10928-014-9366-0
Hu C, Szapary P, Yeilding N, Zhou H (2011) Informative dropout modeling of longitudinal ordered categorical data, model validation: application to exposure-response modeling of physician’s global assessment score for ustekinumab in patients with psoriasis. J Pharmacokinet Pharmacodynam 38(2):237–260. https://doi.org/10.1007/s10928-011-9191-7
Acknowledgements
The authors thank Michelle L. Perate MS, a professional medical writer funded by Janssen Scientific Affairs., LLC, for editorial support.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
10928_2018_9581_MOESM1_ESM.eps
Supplementary material 1 (EPS 730 kb). Fig. S1. Longitudinal model of predicted median and 90% PIs by BWT categories, at planned observation times, in overlay with observed PASI responses
10928_2018_9581_MOESM2_ESM.eps
Supplementary material 2 (EPS 748 kb). Fig. S2. Longitudinal model of predicted median and 90% PIs by BWT categories, at planned observation times, in overlay with observed IGA responses
10928_2018_9581_MOESM3_ESM.eps
Supplementary material 3 (EPS 585 kb). Fig. S3. Visual predictive check of landmark analysis of IGA ≤ 1/IGA0 at Week 16. The observed IGA ≤ 1/IGA0 response rates (red circle) were determined according to bins of the model-predicted guselkumab exposure metrics and were plotted at the median exposure within each bin. The n’s are the numbers of patients in each bin. The blue solid lines are the simulated median responses. The blue dotted lines and the shaded areas both represent the simulated 90% PIs from 1000 replicates
10928_2018_9581_MOESM4_ESM.eps
Supplementary material 4 (EPS 802 kb). Fig. S4. Visual predictive check of landmark analysis of PASI responses at Week 28. The observed PASI 75/90/100 response rates (red circle) were determined according to bins of the model-predicted guselkumab exposure metrics and were plotted at the median exposure within each bin. The n’s are the numbers of patients in each bin. The blue solid lines are the simulated median responses. The blue dotted lines and the shaded areas both represent the simulated 90% PIs from 1000 replicates
10928_2018_9581_MOESM5_ESM.eps
Supplementary material 5 (EPS 701 kb). Fig. S5. Visual predictive check of landmark analysis of IGA ≤ 1/IGA0 at Week 28. The observed IGA ≤ 1/IGA0 responses (red circle) were determined according to bins of the model-predicted guselkumab exposure metrics and were plotted at the median exposure within each bin. The n’s are the numbers of patients in each bin. The blue solid lines are the simulated median responses. The blue dotted lines and the shaded areas both represent the simulated 90% PIs from 1000 replicates
10928_2018_9581_MOESM6_ESM.eps
Supplementary material 6 (EPS 349 kb). Fig. S6. Longitudinal model of predicted dose–response for IGA ≤ 1/IGA0 at Week 16 and Week 28, for the guselkumab 50-, 100-, 150-, and 200-mg q8w dose regimens in the Phase 3 psoriasis trials
10928_2018_9581_MOESM7_ESM.eps
Supplementary material 7 (EPS 693 kb). Fig. S7. Landmark analysis of predicted exposure–response curve for IGA ≤ 1/IGA0 at Week 16 and Week 28. Solid red lines and shaded area represent the model-predicted median response and 90% CIs, respectively, from 200 replicates. The open circles and blue horizontal segments show the median and 5th/95th percentiles, respectively, of the predicted guselkumab AUC0-W16 and AUCss for the guselkumab 50-, 100-, and 200-mg q8w dose regimens. The black dotted lines indicate the 5th/95th percentile range of AUC0-W16 and AUCss for the guselkumab 100–mg q8w dose regimen
Rights and permissions
About this article

Cite this article
Hu, C., Yao, Z., Chen, Y. et al. A comprehensive evaluation of exposure–response relationships in clinical trials: application to support guselkumab dose selection for patients with psoriasis. J Pharmacokinet Pharmacodyn 45, 523–535 (2018). https://doi.org/10.1007/s10928-018-9581-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10928-018-9581-1