
Abstract
Aldehydes are components of many moth sex pheromones, and are thought to be produced from analogous alcohols by oxidase(s) in the cell membrane or the gland cuticle. This implies that the two types of components are produced and/or stored in different parts of the gland: alcohols in cells and aldehydes in cuticle. Few studies have investigated the distribution of components in moth pheromone glands. Using rinse/extract sampling, stable isotope tracer/tracee methods, and decapitation/ pheromone biosynthesis activating neuropeptide stimulation, we studied production and distribution of (Z)-11-hexadecenal (Z11–16:Ald) and (Z)-hexadecenol (Z11–16:OH) in the gland of Chloridea virescens (formerly Heliothis virescens). The rinse, which likely sampled the surface and outer cuticle, contained large amounts of aldehyde and small amounts of alcohol. By contrast, the residual extract, which likely sampled cells and less solvent-accessible (inner) cuticle, had large amounts of alcohol and small amounts of aldehyde. When a tracer (U-13C-glucose) was fed to females, the aldehyde had higher isotopic enrichment than the alcohol in the rinse, but not in the residual extract, showing that in the rinse pool, Z11–16:Ald was, on average, synthesized before Z11–16:OH. This is consistent with greater aldehyde than alcohol flux through the cuticle. While our results are consistent with cell/cuticle synthesis sites for alcohol/aldehyde components, we cannot rule out both being synthesized in gland cells. We propose two alternative conceptual models for how site of production, cuticular transport and catabolism/metabolism might explain the relative masses of Z11–16:Ald and Z11–16:OH translocated to the pheromone gland surface in female C. virescens.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Most species of moths use a volatile female-produced sex pheromone during mate location (Allison and Cardé 2016a). The most common types of sex pheromone components produced by female moths are straight chain, unsaturated terminal alcohols, aldehydes or acetate esters (Ando et al. 2004). These so-called “Type 1” compounds are biosynthesized in, and released from, a specific gland, usually part or whole of the intersegmental membrane between the 8th and 9th abdominal segments (Ma and Ramaswamy 2003). Biosynthesis of Type 1 pheromone components involves de novo production of a saturated fatty acid (usually hexadecanoic) from acetyl CoA, itself produced from mitochondrial oxidation of fat or carbohydrate (Foster and Anderson 2015), followed by chain modification through desaturation and/or cytosolic β-oxidation, before the functional group is added at the carboxyl end (Blomquist et al. 2011; Foster 2016). De novo biosynthesis of these compounds is regulated by release of the pheromone biosynthesis activating neuropeptide (PBAN) (Jurenka 2017).
While much work has focused on the biosynthesis of these compounds, particularly the enzymes involved in determining the carbon chain number and unsaturation (Bjostad et al. 1987; Blomquist et al. 2011), relatively little is known about the actual function of the gland, especially in regard to pheromone component storage, translocation of components to the cuticular surface, and release from the surface. These processes are important, not only because they determine the amount of pheromone released by a female, but also because they can determine the relative amounts (i.e., ratio) of different components in the pheromone, a property that is thought to be critical to successful mate recognition (Allison and Cardé 2016b).
We have recently begun to study the function of the pheromone gland in the moth Chloridea virescens (Fabricius) (formerly Heliothis virescens; family Noctuidae] by quantifying and modeling production, storage, catabolism and release of the major pheromone component (Z)-11-hexadecenal (Z11–16:Ald) (Foster et al. 2018), as well as differentially sampling the gland for distribution of Z11–16:Ald and the minor component (Z)-tetradecenal (Z9–14:Ald) (Foster and Anderson 2018). This work has shown that most pheromone produced is catabolized rather than released and that the two components are differentially distributed through the gland.
The analogous alcohol, (Z)-11-hexadecenol (Z11–16:OH), of the major component is found in relatively high abundance in whole gland extracts of C. virescens, but was thought not to be a component of the pheromone (Hillier and Baker 2016). However, a recent study demonstrated that it was released by females at very low rates (relative to Z11–16:Ald) and, when added to a synthetic blend (at 1% of Z11–16:Ald), increased trap catches of males, suggesting it is a pheromone component (Groot et al. 2018). The presence of an analogous alcohol and aldehyde in the pheromone gland of a moth is interesting because the two compounds are very closely related biochemically, with the aldehyde thought to be produced by oxidation of the alcohol (Teal and Tumlinson 1986). In fact, their relationship is even more interesting in that the prevailing thought (Groot et al. 2018; Hillier and Baker 2016; Teal and Tumlinson 1988) is that the two compound classes are produced in distinctly different parts of the gland: alcohols (and also acetate esters; Jurenka and Roelofs 1989) inside gland cells, via reduction of a fatty acid by a fatty acid reductase (Hagström et al. 2013), and aldehydes at the membrane interface or in the cuticle overlaying the cells (Luxová and Svatoš 2006; Teal and Tumlinson 1988). This production of pheromone aldehydes in or just prior to the gland cuticle is based on the finding of non-specific oxidase activity, in the cell membrane or cuticle of several species of moths, able to convert topically applied alcohols to their analogous aldehydes (Fang et al. 1995; Luxová and Svatoš 2006; Teal and Tumlinson 1986; Teal and Tumlinson 1987; Teal and Tumlinson 1988).
We surmised that using our techniques for solvent sampling the inner and outer parts of the pheromone gland (Foster and Anderson 2018), combined with stable isotope tracer-tracee techniques for tracking newly synthesized pheromone (Foster and Anderson 2011), we could learn more about the distribution and conversion of alcohols to aldehydes in the pheromone gland of female C. virescens. Specifically, in this paper we set out to address, (i) the variation of whole gland titers of the two components over time and with decapitation/PBAN stimulation, (ii) the distribution of the two components through the inner and outer parts of the gland, and (iii) the translocation of newly synthesized (labeled) components in the gland.
Methods and Materials
Insects
We used moths from a laboratory colony that was initiated with insects from USDA-ARS (Fargo, ND) and later supplemented with insects supplied by Dr. Fred Gould (Department of Entomology, North Carolina State University). Larvae were fed on a wheatgerm-casein diet. After pupation, the insects were sexed and females and males placed in separate containers at 25 °C and under a 16:8 L:D photoperiod. Newly emerged females were collected each day and stored without access to liquid, under the same conditions, until used. Typically, starvation for 1 d has little effect on pheromone titer of young virgin females (Foster and Johnson 2010).
Chemical Sampling/Extraction
For analysis of whole glands, pressure was applied to the abdomen of a female to extrude the gland, which was excised with fine forceps. The gland was placed in 10 μl of heptane, along with 50 ng of (E)-11-tetradecenal internal standard, and allowed to extract for at least 1 h at ambient temperature before analysis by gas chromatography/mass spectrometry (GC/MS).
The gland was also sampled for pheromone by a rinse/extract method (Foster and Anderson 2018). Briefly, the surface of a female gland was rinsed with 20 μl of heptane using a 100 μl gas tight syringe. The rinse was collected in a small tube along with 50 ng of internal standard. Thereafter, the gland was excised, dried on a Kimwipe and extracted in 10 μl of heptane along with internal standard, as above. Thus, we obtained two samples from an individual female: the initial rinse, which effectively samples the outer cuticle and surface of the gland, and the extract, which samples the residual pheromone located more inside (e.g., in cells) the gland (Foster and Anderson 2018). In one experiment, we collected three successive individual rinses of a gland before obtaining the residual extract.
Chemical Analysis
GC/MS was carried out using an Agilent 7890/5978A (Agilent Technologies, Santa Clara, CA) fitted with a DB-Wax UI (30 m × 0.25 mm × 0.25 mm film thickness) capillary column (Agilent J&W). The column oven was temperature programmed from 80 °C (delay of 1 min) to 180 °C at 15 °C.min−1, then to 190 °C at 5 °C.min−1, and finally to 220 °C at 20 °C.min−1. Injection was splitless, with helium as carrier gas at a constant flow of 1.5 ml.min−1. The MS was operated in the selected ion mode with electron ionization (70 eV).
For pheromone quantitation, we monitored m/z 192 (for Z9–14:Ald and E11–14:Ald), 220 (for Z11–16:Ald), and 222 (for Z11–16:OH), all moderately intense ions characteristic of the compounds, representing the loss of 18 mass units (H2O).
Mass Isotopomer Distribution Analysis (MIDA)
To determine isotopic enrichment of pheromone components following introduction of a stable isotope tracer, we used the technique of MIDA (Chinkes et al. 1996; Hellerstein and Neese 1992). We have described this technique applied to pheromone biosynthesis in detail previously (e.g., Foster and Anderson 2011; Foster and Anderson 2018). Briefly, MIDA is a combinatorial approach for determining monomeric precursor enrichment in a polymer. For this, a stable isotope tracer is introduced and the resultant distribution of isotopomers experimentally determined by GC/MS. The intensities of only two labeled isotopomers plus the unlabeled isotopomer are needed. Usually, the intensities of the isotopomers including one and two labeled monomers [(M + 1) and (M + 2), respectively] are used to calculate tracer: tracee ratios (TTRs). Precursor enrichment (PE; the proportion of labeled tracer monomer in the polymer) is determined thus:
The post and pre subscripts denote isotopomer intensities after and before label has been introduced into the system; this accounts for the contribution of naturally occurring isotopes to the (M + 1) isotopomer intensity. We used theoretically calculated (based on natural isotopic abundances) pre values since we have found these differ little from experimentally determined ones (Foster and Anderson 2011). The dT1 term is the contribution of the (M + 1) isotopomer spectrum to the (M + 2) isotopomer. Finally, n is the number of monomeric units in the polymer. For Z11–16:Ald (and Z11–16:OH), this is 8 units of acetyl CoA, and for Z9–14:Ald, 7 units.
Experiments
Whole Gland Z11–16:Ald and Z11–16:OH Titers
We ran two experiments quantifying the variation in titers of the two compounds in the whole gland. In the first, at the start of the scotophase, 1 d females were either decapitated or left intact and sampled for Z11–16:Ald and Z11–16:OH over the next 7 h. Components were quantified relative to the internal standard. Six to ten decapitated or intact females were sampled for each time point.
In the second experiment, females (1 d) were decapitated, left for ca. 24 h, and then injected with 5 pmole of synthetic Helicoverpa zea PBAN (Bachem. Torrance, CA) in physiological saline (2 μl) and the amounts of Z11–16:Ald and Z11–16:OH in the gland quantified at various times. Five to eleven females were sampled for each time point.
Gland Distribution of Z11–16:Ald and Z11–16:OH
Two hours into the scotophase, the gland of a 1 d female was subjected to three successive solvent rinses before it was excised and extracted in heptane. Internal standard was added to each of the four samples (rinses 1, 2, 3 and residual extract) and the samples quantified for both Z11–16:Ald and Z11–16:OH. Six females were analyzed.
Next, intact (1 d), and decapitated (1 d, decapitated 16 h prior) and PBAN-injected (5 pmol) females were subjected to the (single) rinse/extract sampling and quantified for Z11–16:Ald and Z11–16:OH. To ensure that the quantities of pheromone were similar in both types of female, intact females were sampled 3 h into the scotophase, while PBAN-injected females were sampled 2 h after injection. Ten females were analyzed for each of the four treatments.
Enrichment Dynamics
Females were allowed to feed on a 12.5 μl drop of 10% (w/v) U-13C-glucose (99% enrichment; Cambridge Isotope Laboratories, Cambridge, MA) after which individual whole glands were sampled and analyzed for Z11–16:Ald, Z11–16:OH and Z9–14:Ald at various times. Only females (7–15 for each time point) that consumed the entire drop were sampled. The appropriate m/z of Z9–14:Ald (192, 194, 196), Z11–16:Ald (220, 222, 224) and Z11–16:OH (222, 224, 226) were quantified and used for MIDA. These m/z correspond to the loss of water from (M + 0), (M + 1) and (M + 2) isotopomers of the respective intact carbon skeletons of the compounds.
Finally, females were fed on U-13C-glucose and sampled by the rinse/extract method at four times after feeding. Precursor enrichments for Z11–16:Ald and Z11–16:OH in both the rinse and extract were calculated as above. We did not analyze for Z9–14:Ald enrichment because preliminary samples showed the amount in rinse samples was too low for reliable detection. Note that, because we were not strictly making comparisons between the rinse and the extract, only between compounds within the rinse or extract, we did not compensate for the time difference between the two types of sampling (the rinse is taken a little before the gland is excised and extracted; see Foster and Anderson 2018). Therefore, we give only the nominal time (when the rinse commenced after females had finished the glucose) for both types of sampling. Eight or nine females were sampled at each time.
Statistical Analysis
Titers and ratios of Z11–16:Ald and Z11–16:OH were analyzed by ANOVA, after first checking heteroscedasity and normality of data. For the multiple rinse experiment, we analyzed amounts of each of the compounds and their ratio in the different samplings (three rinses and residual extract) by mixed regression models, using insect identity as a random effect. Differences among means of ratio and titer of each compound across the different samplings were compared by post-hoc Tukey HSD tests (α set at 0.05). The PBAN/intact female rinse/extract experiment was analyzed similarly but included a female treatment (PBAN-injected or intact) effect.
The final precursor enrichment of the each of the three components was determined from the asymptote of a 3P exponential model fit (JMP 2012) to the precursor enrichment/time data. In both the rinse and extract samplings of precursor enrichment, we used paired t tests to assess the differences between Z11–16:Ald and Z11–16:OH precursor enrichments (matched pair analysis, JMP 2012) at each of the four times.
Results
Whole Gland Z11–16:Ald and Z11–16:OH Titers
In intact females, Z11–16:Ald titer increased initially, rising to a peak some 3 h into the scotophase before declining (ANOVA, F4,32 = 3.40, p = 0.020). Z11–16:OH titer was fairly constant through the scotophase but dropped by the end (ANOVA, F4,32 = 3.86, p = 0.011) (Fig. 1a). There was a strong positive correlation (correlation coefficient = 0.78, F1,29 = 45.8; p < 0.001) between titers of Z11–16:Ald and Z11–16:OH; consequently, the ratios of Z11–16:OH: Z11–16:Ald were fairly constant (ca. <1; ANOVA, F4,32 = 1.43, p = 0.25) throughout much of the scotophase (Fig. 1b). Decapitation resulted in a decline in titer of both components (ANOVA, F4,32 = 0.91, p = 0.011 for Z11–16:Ald and F4,32 = 8.08, p < 0.001 for Z11–16:OH) (Fig. 1a,). There was a strong correlation (correlation coefficient = 0.61, F1,30 = 17.6; p < 0.001) between titers of Z11–16:Ald and Z11–16:OH. The ratio of Z11–16:OH: Z11–16:Ald was relatively constant following decapitation (Fig. 1b; ANOVA, F4,32 = 0.79, p = 0.54).

(a) Mean titers, and (b) ratios of (Z)-11-hexadecenal (Ald) and (Z)-11-hexadecenol (OH) in whole glands of intact and decapitated Chloridea virescens females at various times. The time scale represents hours of the scotophase for intact females and hours after decapitation for decapitated females. Lines were included in (a) to distinguish the different series. SEMs are given
Z11–16:Ald titer varied with time after PBAN injection, similar to that for the dynamic changes in intact females, with titer rising to a peak some 2 h following injection and then declining (F8,47 = 11.2, p < 0.001) (Fig. 2a). Z11–16:OH titer also increased to a peak at ca. 2 h following injection before declining (F8,47 = 7.35, p < 0.001). Generally, titer of Z11–16:OH was less than that of Z11–16:Ald (Fig. 2a). Again, there was a very strong correlation (correlation coefficient = 0.82, F1,53 = 104.9; p < 0.001) between titers of Z11–16:Ald and Z11–16:OH. Ratio of Z11–16:OH: Z11–16:Ald showed a significant change (F8,47 = 5.70, p < 0.001) over the course of the experiment (Fig. 2b), but this was mainly due to relatively high ratios at the beginning of the experiment; after this, ratios were relatively constant. It should be noted that in contrast to the intact females, both Z11–16:Ald and Z11–16:OH titers at the beginning of this experiment were initially very low and rose rapidly.

Mean (a) titers, and (b) ratios of (Z)-11-hexadecenal (Ald) and (Z)-11-hexadecenol (OH) in whole glands of decapitated Chloridea virescens females injected with 5 pmol pheromone biosynthesis activating neuropeptide. The time scale represents hours after injection. SEMs are given
Gland Distribution of Z11–16:Ald and Z11–16:OH
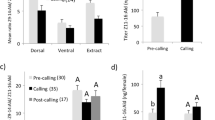
There was a significant effect (F = 7.71, p = 0.002, DF = 3, Kenward Roger F test, mixed regression model) of treatment on Z11–16:Ald titer over the three successive rinses. The first rinse contained the greatest amount, with the succeeding rinses containing progressively smaller amounts. There was still a substantial amount of Z11–16:Ald left in the residual extract (Fig. 3). Z11–16:OH titer also showed an effect of treatment (F = 40.8, p < 0.001, DF = 3, Kenward Roger F test, mixed regression model). In this case, all three rinses contained small amounts, with the residual extract having by far the greatest amount. Ratio of Z11–16:OH: Z11–16:Ald showed a steady increase through the three rinses but was highest in the residual extract and lowest in the first rinse (F = 18.3, p < 0.001, DF = 3, Kenward Roger F test, mixed regression model; Tukey HSD test).

Mean titers (ng/female) of (Z)-11-hexadecenal (Ald) and (Z)-11-hexadecenol (OH) and mean ratios (OH: Ald) in the pheromone gland of Chloridea virescens females sampled by three successive solvent rinses (Rinses 1–3) and then placed in solvent to extract the residual (Extract). Ratios are given on a scale 10 x their actual value. SEMs are given and different letters of the same case and subscript indicate means of a given compound or ratio that are different among treatments (mixed regression model, Tukey HSD test, p < 0.05)
The distribution of amounts of both Z11–16:Ald and Z11–16:OH in the respective rinse and residual extract were remarkably similar for both intact and PBAN-injected females (Fig. 4). The mixed regression model showed no effect of female treatment (PBAN/intact) but a strong effect of sampling (rinse/extract) for both Z11–16:Ald (DF 1, F = 0.002, P = 0.98 for treatment, F = 8.36, p = 0.006 for sampling) and Z11–16:OH (DF 1, F = 0.04, P = 0.94 for treatment, F = 61. 6, p < 0.001).

Mean titers (ng/female) of (Z)-11-hexadecenal (Ald) and (Z)-11-hexadecenol (OH) in the pheromone gland of Chloridea virescens females sampled by a solvent rinse followed by residual extraction of the gland in solvent. Two types of females were sampled this way: intact females 3 h into the scotophase, and decapitated females injected with 5 pmol of pheromone biosynthesis-activating neuropeptide (PBAN). SEMs are given
Enrichment Dynamics
In the whole gland extract, the three components had similar precursor enrichment increases (Fig. 5), rising rapidly and plateauing around 10–15 min as the precursor pool reached equilibrium (i.e., when the labeled acetyl CoA from ingested glucose equilibrated with the other sources of acetyl CoA used for de novo pheromone production). Using an exponential 3P model, we calculated the equilibrated precursor enrichments of Z11–16:Ald, Z11–16:OH and Z9–4:Ald to be 0.20 ± 0.02, 0.22 ± 0.2 and 0.19 ± 0.02, respectively, indicating that the three components were probably made from the same precursor pool.

Mean precursor enrichment (molar percent excess) of (Z)-11-hexadecenal (Z11–16:Ald), (Z)-11-hexadecenol (Z11–16:OH) and (Z)-9-tetradecenal (Z9–16:Ald) in whole pheromone gland extracts after female Chloridea virescens were fed U-13C-glucose (at time = 0). SEMs and a 3P exponential model fit of the Z11–16Ald data are given
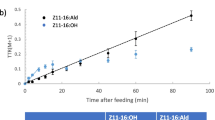
At each of the four times in the rinse sampling (Fig. 6a), there was a difference between the precursor enrichment of Z11–16:Ald and that of Z11–16:OH (matched pair analysis, P < 0.05; see Table 1). Notably, the precursor enrichment of Z11–16:Ald was always greater than that of Z11–16:OH, and substantially so at 20 min. By contrast, in the residual gland extract sampling (Fig. 6b), there was no difference between the precursor enrichments of Z11–16:Ald and Z11–16:OH (matched pair analysis; Table 1).

Mean precursor enrichment (molar percent excess) of (Z)-11-hexadecenal (Ald) and (Z)-11-hexadecenol (16:OH) in the pheromone gland after female Chloridea virescens were fed U-13C-glucose (at time = 0). (a) Solvent gland rinse and (b) solvent extract of gland residual. SEMs are given
Discussion
Whole Gland Z11–16:Ald and Z11–16:OH Titers
During pheromone biosynthesis, in both intact and PBAN-injected females, whole gland titers of Z11–16:Ald and Z11–16:OH were highly correlated. This, combined with the fact that decapitation resulted in rapid and correlated declines in both compounds, suggests that their biosynthesis is closely linked and controlled by the same factor (i.e., PBAN). While ratios of the two compounds were fairly constant through the different experiments, there was a consistent trend of relatively high ratios of Z11–16:OH: Z11–16:Ald when titers of the compounds were low or dropping rapidly (after decapitation), perhaps suggesting the alcohol as a precursor to the aldehyde.
Gland Distribution of Z11–16:Ald and Z11–16:OH
The solvent rinse/residual extract samplings of the gland showed differences in composition between the two samplings, with most Z11–16:Ald in the rinse and most Z11–16:OH in the residual extract. It is unlikely that this difference was due solely (or largely) to different solubility of the two non-polar compounds in heptane, because successive rinses of the gland failed to sample much more Z11–16:OH and Z11–16:Ald, even though the final residual extract contained substantial amounts of both (especially Z11–16:OH). Thus, this likely shows that the two compounds are distributed unevenly through the gland, with relatively more Z11–16:Ald in the outer part, and relatively more Z11–16:OH in the inner part. We also note that the ratio of components in the rinse is much closer, than that in the whole gland extract, to that released into the air by females (Groot et al. 2018). This highlights again the unreliability of whole gland extracts for determining actual pheromone component ratios released by females (Allison and Cardé 2016c). Further, it may show how sampling closer to the gland surface is likely to yield the final blend of components available for release, with the actual final ratio released being determined by the respective surface abundances and vapor pressures of the compounds.
This distribution of the two compounds occurred regardless of whether a female was intact and producing pheromone or decapitated and injected with PBAN. Since decapitated females do not release pheromone (Foster et al. 2018), this shows that the broad distribution of Z11–16:OH and Z11–16:Ald through the gland is independent of actual release; i.e., pheromone is translocated toward the gland surface so long as it is produced. Muscular pumping of the gland region during pheromone release has been observed in numerous species of moths (e.g., Conner et al. 1985), including C. virescens (Hillier and Vickers 2004), and has been suggested to be involved directly with movement of pheromone to the gland surface (Raina et al. 2000). We have not observed this behavior in decapitated C. virescens females (Foster and Anderson, unpublished). It should be noted that our solvent rinse sampling is somewhat crude and likely samples more broadly than just pheromone on the gland surface (e.g., pheromone located in pores through the cuticle). Thus, in a normal female, if more pheromone were located in the outer part, rather than on the surface, of the cuticle, our sampling might not show surface content differences. While pheromone still translocates through the gland in the absence of release, abdominal/gland pumping could aid the final movement of pheromone to the surface for release.
Component Translocation
Both compounds, as well as Z9–14:Ald, are produced very rapidly and from the same precursor pool, as shown by the same precursor enrichments in whole gland extracts. However, we saw differences in translocation through the gland by enrichment differences between Z11–16:Ald and Z11–16:OH in the rinse but not in the residual gland extract. The consistently greater enrichment of Z11–16:Ald over that of Z11–16:OH in the rinse indicates that, on average, Z11–16:Ald (incorporating tracer) in this pool was produced later than Z11–16:OH (incorporating tracer). There are two possible explanations for this: (i) Z11–16Ald is produced in the pathway before Z11–16:OH, or (ii) Z11–16:Ald flux is greater than that of Z11–16:OH through the rinse pool.
If the former is correct, then it implies that Z11–16:OH is made from Z11–16:Ald, the reverse of how the two components are thought to be biosynthesized (Choi et al. 2005; Hagström et al. 2012). Thus, Z11–16:OH, the product of fatty acid reduction (Hagström et al. 2013), which is rapidly oxidized to Z11–16:Ald, would have to be subsequently reduced back to Z11–16:OH. Since the greater enrichment of alcohol over aldehyde was only apparent in the rinse, this would mean that the alcohol in the residual extract and rinse were, effectively, biosynthetically distinct. While this is feasible, as Z11–16:Ald is catabolized in the gland to unknown product(s) (Foster et al. 2018), the second explanation is the more parsimonious: the enrichment difference between the two compounds in the rinse is due to more rapid turnover of Z11–16:Ald than that of Z11–16:OH, effectively yielding increasingly higher average enrichment as it is replaced by higher isotopically enriched compound. To turn over the much larger Z11–16:Ald pool more rapidly than the smaller Z11–16:OH pool (in the rinse) requires a greater flux of the former compound into and out of the pool. The two paths for compounds out of this pool are release to the environment and catabolism/metabolism (Foster et al. 2018). A recent study on pheromone release by female C. virescens found that females released <0.1% the mass of Z11–16:OH than of Z11–16Ald (Groot et al. 2018), much lower than the roughly 7% Z11–16:OH: Z11–16Ald ratio found in the rinse fraction in our study, creating a large flux differential between the two compounds.
Catabolism/metabolism also likely plays a major role in this flux differential. Most Z11–16:Ald produced is catabolized rather than released (Foster et al. 2018). As most Z11–16:Ald is stored in the rinse pool and decapitated females do not release pheromone (so only catabolize), it seems safe to assume that most catabolism of Z11–16:Ald occurs while it is in this pool. However, the whole gland extracts indicated that decapitated females also lost Z11–16:OH mass at a comparable rate to that of Z11–16:Ald mass. In this case, though, Z11–16:OH is metabolized to Z11–16:Ald, rather than catabolized to other product(s). Hence, this produces a high flux of Z11–16:Ald and a relatively low flux of Z11–16:OH into the rinse pool. With little or no catabolism of the alcohol, there is a slow turnover of this component as it passes through to the surface.
While the above is consistent with the prevailing view of the alcohol being produced in gland cells and the aldehyde in the cuticle (Groot et al. 2018; Hagström et al. 2013; Hillier and Baker 2016; Morse and Meighen 1987; Teal and Tumlinson 1988), we cannot rule out that the aldehyde is also produced in gland cells, especially as repeated gland rinses left a small, but substantial, amount in the residual extract. Alternatively, this result could be due to inefficient solvent extraction of the cuticle by the rinses, especially if the conversion took place in the inner (less accessible) part of the cuticle, perhaps at the membrane interface (Luxová and Svatoš 2006). If aldehyde were produced inside gland cells, then the distribution of components in the residual extract and rinse would require a slower mass translocation of Z11–16:OH out of the cells and through the cuticle than that of Z11–16:Ald. While this might seem unlikely, given the similar chemical properties of the two compounds, we know little about the processes driving the translocation of pheromone components through the gland. Because of this, we propose two alternative models, incorporating site of synthesis, catabolism/metabolism and translocation rate through the gland (Fig. 7).

Proposed models utilizing site of production, translocation and catabolism/metabolism to explain relative quantities of (Z)-11-hexadecenal (Z11–16:Ald) and (Z)-11-hexadecenol (Z11–16:OH) translocated to the cuticular surface of the pheromone gland of female Chloridea virescens. In (i) Z11–16:OH is produced in gland cells and (mostly) converted (shown by arrows) to Z11–16:Ald in the cuticle or at the membrane-cuticle interface (as originally proposed by Teal and Tumlinson 1986), while in (ii) both components are produced in gland cells but Z11–16:Ald is transported much more rapidly to the cuticular surface than is Z11–16:OH. Note the difference between the two models in where catabolism of Z11–16:Ald can occur
The first model (i) essentially follows the prevailing view, with Z11–16:OH produced in gland cells that, when translocated to the cuticle, is rapidly oxidized to aldehyde. A small amount of alcohol makes it through the part of the cuticle where oxidation occurs and eventually to the surface. Catabolism is specific to Z11–16:Ald and occurs exclusively in the cuticle. Compounds reaching the cuticular surface evaporate according to their vapor pressure, surface area on the gland, and ambient temperature and windspeed (Nielsen et al. 1995). In the second model (ii), alcohol is produced and rapidly oxidized to aldehyde all within gland cells; however mass translocation out of the cell to the surface is much greater for Z11–16:Ald than for Z11–16:OH. As for the first model, it is not necessary to invoke catabolism of Z11–16:OH (although it could still occur) due to rapid metabolic oxidation to Z11–16:Ald within cells. Catabolism of Z11–16:Ald could occur in both gland cells and cuticle although, due to its distribution (especially in decapitated females), most Z11–16:Ald is likely catabolized in the cuticle.
These models illustrate how multiple factors, including site and rate of synthesis, rate of catabolism/metabolism, and rate of translocation through the gland can determine the blend of compounds on the gland surface available for release. It should be possible to distinguish between these two models, as well as learn more about moth pheromone gland function, through development of higher resolution gland sampling techniques, as well as through characterization and localization of oxidases and catabolic enzymes.
References
Allison JD, Cardé RT (eds) (2016a) Pheromone communication in moths: evolution, behavior and application. University of Caifornia Press, Oakland
Allison JD, Cardé RT (2016b) Pheromone: reproductive isolation and evolution in moths. In: Allison JD, Cardé RT (eds) Pheromone communication in moths: evolution, behavior and application. University of California Press, Oakland, pp 11–23
Allison JD, Cardé RT (2016c) Variation in moth pheromone: causes and consequences. In: Allison JD, Cardé RT (eds) Pheromone communication in moths: evolution, behavior and application. University of California Press, Oakland, pp 25–41
Ando T, Inomata S, Yamamoto M (2004) Lepidopteran sex pheromones. In: Schulz S (ed) The chemistry of pheromones and other Semiochemicals I, vol 239. Topics in current chemistry. Springer, Berlin / Heidelberg, pp 51–96. https://doi.org/10.1007/b95449
Bjostad LB, Wolf WA, Roelofs WL (1987) Pheromone biosynthesis in lepidopterans: desaturation and chain shortening. In: Prestwich GD, Blomquist GJ (eds) Pheromone biochemistry. Academic Press, New York, pp 77–120
Blomquist GJ, Jurenka R, Schal C, Tittiger C (2011) Pheromone production: biochemistry and molecular biology. In: Gilbert LI (ed) Insect endocrinology. Academic Press, San Diego, pp 523–567
Chinkes DL, Aarsland A, Rosenblatt J, Wolfe RR (1996) Comparison of mass isotopomer dilution methods used to compute VLDL production in vivo. Am J Physiol Endocrinol Metab 271:E373–E383
Choi MY, Groot A, Jurenka RA (2005) Pheromone biosynthetic pathways in the moths Heliothis subflexa and Heliothis virescens. Arch Insect Biochem Physiol 59:53–58
Conner WE, Webster RP, Itagaki H (1985) Calling behaviour in arctiid moths: the effects of temperature and wind speed on the rhythmic exposure of the sex attractant gland. J Insect Physiol 31:815–820. https://doi.org/10.1016/0022-1910(85)90074-5
Fang NB, Teal PEA, Tumlinson JH (1995) Characterization of oxidase(s) associated with the sex pheromone gland in Manduca sexta (L) females. Arch Insect Biochem Physiol 29:243–257
Foster SP (2016) Toward a quantitative paradigm for sex pheromone production in moths. In: Allison JD, Cardé RT (eds) Pheromone communication in moths: evolution, behavior and application. University of California Press, Oakland, pp 113–126
Foster S, Anderson K (2011) The use of mass isotopomer distribution analysis to quantify synthetic rates of sex pheromone in the moth Heliothis virescens. J Chem Ecol 37:1208–1210
Foster SP, Anderson KG (2015) Sex pheromones in mate assessment: analysis of nutrient cost of sex pheromone production by females of the moth Heliothis virescens. J Exp Biol 218:1252–1258. https://doi.org/10.1242/jeb.119883
Foster SP, Anderson KG (2018) Differential pheromone sampling of the gland of female Heliothis virescens moths reveals glandular differences in composition and quantity. J Chem Ecol 44:452–462. https://doi.org/10.1007/s10886-018-0954-0
Foster SP, Johnson CP (2010) Feeding and hemolymph trehalose concentration influence sex pheromone production in virgin Heliothis virescens moths. J Insect Physiol 56:1617–1623
Foster SP, Anderson KG, Casas J (2018) The dynamics of pheromone gland synthesis and release: a paradigm shift for understanding sex pheromone quantity in female moths. J Chem Ecol 44:525–533. https://doi.org/10.1007/s10886-018-0963-z
Groot AT et al (2018) Alcohol contributes to attraction of Heliothis (= Chloridea) virescens males to females. J Chem Ecol 44:621–630. https://doi.org/10.1007/s10886-018-0995-4
Hagström ÅK, Liénard MA, Groot AT, Hedenström E, Löfstedt C (2012) Semi–selective fatty acyl reductases from four Heliothine moths influence the specific pheromone composition. PLoS One 7:e37230. https://doi.org/10.1371/journal.pone.0037230
Hagström ÅK, Walther A, Wendland J, Löfstedt C (2013) Subcellular localization of the fatty acyl reductase involved in pheromone biosynthesis in the tobacco budworm, Heliothis virescens (Noctuidae: Lepidoptera). Insect Biochem Mol Biol 43:510–521. https://doi.org/10.1016/j.ibmb.2013.03.006
Hellerstein MK, Neese RA (1992) Mass isotopomer distribution analysis: a technique for measuring biosynthesis and turnover of polymers. Am J Physiol Endocrinol Metab 263:E988–1001
Hillier NK, Baker TC (2016) Pheromones of heliothine moths. In: Allison JD, Cardé RT (eds) Pheromone communication in moths: evolution, behavior and application. University of California Press, Oakland, pp 301–333
Hillier NK, Vickers NJ (2004) The role of heliothine hairpencil compounds in female Heliothis virescens (Lepidoptera: Noctuidae) behavior and mate acceptance. Chem Senses 29:499–511. https://doi.org/10.1093/chemse/bjh052
JMP (2012) JMP version 10. SAS Institute Inc., Cary
Jurenka R (2017) Regulation of pheromone biosynthesis in moths. Current Opinion in Insect Science 24:29–35. https://doi.org/10.1016/j.cois.2017.09.002
Jurenka RA, Roelofs WL (1989) Characterization of the acetyltransferase used in pheromone biosynthesis in moths: specificity for the Z isomer in Tortricidae. Insect Biochem 19:639–644
Luxová A, Svatoš A (2006) Substrate specificity of membrane-bound alcohol oxidase from the tobacco hornworm moth (Manduca sexta) female pheromone glands. J Mol Catal B: Enzymatic 38:37–42
Ma PWK, Ramaswamy SB (2003) Biology and ultrastructure of sex pheromone-producing tissue. In: Blomquist GJ, Vogt RC (eds) Insect pheromone biochemsitry and molecular biology. Elsevier Academic Press, London, pp 19–51
Morse D, Meighen E (1987) Pheromone biosynthesis: enzymatic studies in Lepidoptera. In: Prestwich GD, Blomquist GJ (eds) Pheromone biochemistry. Academic Press, New York, pp 121–158
Nielsen F, Olsen E, Fredenslund A (1995) Prediction of isothermal evaporation rates of pure volatile organic compounds in occupational environments—a theoretical approach based on laminar boundary layer theory. The Annals of Occupational Hygiene 39:497–511. https://doi.org/10.1016/0003-4878(95)00032-A
Raina AK, Wergin WP, Murphy CA, Erbe EF (2000) Structural organization of the sex pheromone gland in Helicoverpa zea in relation to pheromone production and release. Arthropod Structure and Development 29:343–353
Teal PEA, Tumlinson JH (1986) Terminal steps in pheromone biosynthesis by Heliothis virescens and H. zea. J Chem Ecol 12:353–366
Teal PEA, Tumlinson JH (1987) The role of alcohols in pheromone biosynthesis by two noctuid moths that use acetate pheromone components. Arch Insect Biochem Physiol 4:261–269. https://doi.org/10.1002/arch.940040404
Teal PEA, Tumlinson JH (1988) Properties of cuticular oxidases used for sex pheromone biosynthesis by Heliothis zea. J Chem Ecol 14:2131–2145
Acknowledgments
We are grateful for funding of this work by a United States Department of Agriculture Hatch Project ND02388. We also thank the United States Department of Agriculture–National Institute of Food and Agriculture for an Instrument Grant, 2015-07238 contributing, in part, to the purchase of the GC/MS system.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Foster, S.P., Anderson, K.G. Production and Distribution of Aldehyde and Alcohol Sex Pheromone Components in the Pheromone Gland of Females of the Moth Chloridea virescens. J Chem Ecol 45, 9–17 (2019). https://doi.org/10.1007/s10886-018-1041-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10886-018-1041-2