
Abstract
By differentially sampling the pheromone gland of females of the moth Heliothis virescens, we explored differences in pheromone on the surface, or outer distal layer(s) of the gland, and that located more proximally. For this, we used two sampling approaches, (i) a solid phase microextraction fiber rub followed by solvent extraction of residual pheromone (SPME rub/extract), and (ii) rapid solvent rinsing followed by solvent extraction of residual pheromone (rinse/extract). The SPME rub showed differences in component ratio between the dorsal and ventral gland surfaces. The rinse sampled a greater amount of pheromone than the SPME rub, sampling the whole gland surface as well as likely deeper into the gland. Compared to the other samplings, pheromone in the rinse was depleted in the minor component; consequently, the corresponding residual extract was highly enriched in the minor component. Further rinses of the gland yielded only small amounts of pheromone, with a similar component ratio as the first rinse, suggesting that the residual pheromone was less accessible and required extraction in solvent to be liberated. Sampling over the photoperiod showed that the more volatile minor component was depleted (relative to the major component) on the surface/outer cuticle over the period when females called. Together, these data suggest that the pheromone is stored, at least in part, on and in the gland cuticle and that distinct pools may be transported to different topographic regions. Females fed with a stable isotope tracer, incorporated label into pheromone in the gland very rapidly, with the labeled pheromone appearing on the gland surface ca. 1 min later.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Most moth species use a volatile sex pheromone for mate recognition and location (Allison and Cardé 2016). Typically, females produce and release pheromone from a specialized gland, a layer of epidermal cells with an overlying cuticle, located between the 8th and 9th abdominal segments (Ma and Ramaswamy 2003). The most common type of pheromone components (called “Type 1”) consists primarily of unsaturated, even-numbered, carbon chains with an oxygenated terminus (Ando et al. 2004). These compounds are produced by de novo biosynthesis of saturated fatty acid from acetyl CoA, followed by chain modification through desaturation, cytosolic β-oxidation (chain shortening) or chain elongation, and finally production of the terminal group by reduction, acetylation or oxidation (Blomquist et al. 2011; Foster 2016). Biosynthesis probably occurs wholly or largely inside gland cells (Fonagy et al. 2000; Hagström et al. 2013), although it has been suggested that the final oxidation step in the production of aldehyde components might occur in the cuticle, where a non-specific oxidase that converts alcohols to aldehydes has been found in two species (Fang et al. 1995; Teal and Tumlinson 1988). Pheromone release usually involves an overt behavior (“calling”), in which a female extends the terminal segments of the abdomen, facilitating the release and evaporation of pheromone components into the air stream (e.g., Conner et al. 1985; Delisle 1992).
In most species of moth, the amount of pheromone in the gland (titer) usually increases after the onset of synthesis, stimulated by release of the pheromone biosynthesis activating neuropeptide (Jurenka 2017). Gland titer typically reaches a peak sometime during the sexually active period (when females are releasing pheromone or mating), before declining back to a low level at the end of the sexually active period (Foster 2016; Groot 2014). This increase in titer results from the rate of synthesis of pheromone being greater than the rate of usage, with the excess pheromone being stored transiently in the gland, until used. In spite of the abundance of studies on pheromone titer changes (Foster 2016), we know relatively little about how or where pheromone is stored in the gland. Is it stored in gland cells or in, or on top of, the cuticle? If a cuticular oxidase is responsible for the conversion of pheromone alcohols to aldehydes (Fang et al. 1995; Teal and Tumlinson 1988), then pheromone must be stored exclusively in the cuticle. However, species that use components with other functional groups (e.g., alcohols or acetates) appear to produce pheromone exclusively within gland cells. For instance, the acetyl transferase that performs the final step in the biosynthesis of (Z)- and (E)-11 tetradecenyl acetates in Argyrotaenia velutinana, has been localized to a microsomal fraction of the cells (Jurenka and Roelofs 1989). In this case, pheromone could be stored in the gland cells or in the cuticle or a combination of both.
One reason we know little about pheromone storage is the lack of sampling methods capable of localizing the very small amounts of pheromone. The most commonly used method for sampling pheromone in moths is dissection and extraction of the entire gland in solvent (Foster 2016). This method has been used to delineate the extent of pheromone producing tissue in the intersegmental membrane (e.g., Ma and Ramaswamy 2003; Raina et al. 2000), but not for localizing pheromone in the distal, outer cuticular layer(s), where it is released, and the more proximal cuticle and/or cellular tissue, where it is produced. However, several methods have been developed for sampling the surface of the gland, primarily for qualitative or non-destructive, repetitive sampling of individuals. These include, a rapid solvent rinse (Cory et al. 1982), rubbing the gland with filter paper soaked in solvent (Zhu et al. 1996), and rubbing the gland with a solid phase micro extraction (SPME) fiber (Frérot et al. 1997; Lievers and Groot 2016).
Given that these methods sample the cuticular surface or (probably for the solvent-based methods) the more distal layers of the gland cuticle, it seemed to us that such methods could be combined with analysis of the residual pheromone from the same gland to elucidate differences in pheromone between gross layers of the gland; i.e., between the surface/outer cuticular layer and deeper into the cuticle and/or intracellular. Furthermore, we have previously demonstrated that stable isotope tracers can be introduced into de novo-biosynthesized pheromone via female moths feeding on labeled glucose (Foster and Anderson 2011). Therefore, we thought that by combining this with the above sampling of the gland, we could also obtain information about the speed of movement of pheromone through the gland; i.e., from the site of production to the cuticular surface for release.
In this paper, we investigate differential samplings of the pheromone gland of females of the moth Heliothis virescens. We report on pheromone in the different samplings and demonstrate differences in pheromone component ratio and quantity between outer (surface) and inner samplings of the gland, as well as over the photoperiod for the different samplings. Further, by feeding females U-13C-glucose tracer we found that newly synthesized pheromone was transported to the surface of the gland very rapidly.
Methods and Materials
Insects
Our colony of H. virescens was established from insects at the USDA-ARS, Fargo, ND, and later supplemented with insects from Dr. F. Gould, Department of Entomology, North Carolina State University, Raleigh, NC. Larvae were fed on a wheat germ-casein based diet and maintained at 25 °C, under a 16:8 L:D photoperiod. After they pupated, insects were sexed and the two sexes maintained in separate incubators under the same conditions as the larvae. Newly eclosed adult females were collected each day, and held in small plastic containers without access to any liquid, until used the following day (when nominally 1 d-old).
Pheromone Sampling
Female H. virescens produce and release a sex pheromone consisting of typical “Type 1” moth sex pheromone components (Z)-9-tetradecenal (Z9–14:Ald) and (Z)-11-hexadecenal (Z11–16:Ald), in a ratio of roughly 10/100 (Heath et al. 1991; Klun et al. 1980; Roelofs et al. 1974). We sampled pheromone of individual H. virescens by two different outer/inner gland sampling ‘approaches’. In one approach, we first sampled the outer gland surface by rubbing it with a solid phase microextraction (SPME) fiber, and then extracted the residual (inner) pheromone with n-heptane (SPME rub/extract). In the second, we first sampled the outer surface of the gland by rinsing it with a small amount of n-heptane, before we extracted the inner residual pheromone (rinse/extract). Our rationale for using the two approaches was twofold: (i) the SPME rub sampled the gland surface, but we could not quantify pheromone by this method, because of the inherent difficulties with SPME quantification (Pawliszyn 1997), and the fact that the SPME rubbing was not consistent between samples, possibly due to variation in area rubbed, as well as changes in fiber adsorption properties over time following the physical rubbing, and (ii) the rinse of the outer surface could be readily quantified, but we could not be certain we were sampling only the outer surface of the gland (as solvent might penetrate more deeply into the gland). Thus, we figured using both approaches could give us complementary qualitative and quantitative information about the pools of pheromone on the outside and inside of the gland, as well as test whether the two approaches were sampling the same pools.
SPME Rub/Extract Approach
The gland of a female was forcibly extruded and maintained in position by a vascular clamp (10 × 2.15 mm, Fine Science Tools Inc., Foster City, CA) positioned just anterior to the gland. Then, the exposed fiber of a portable SPME field sampler, fitted with a 100 μm PDMS fiber (Supelco Inc., Bellefonte, PA), was rubbed over both the dorsal and/or ventral portions of the gland for approximately 60 s, before the fiber was retracted. Prior to sampling, the SPME fiber was conditioned in a GC injector at 240 °C for at least 1 h. Immediately following SPME sampling, the gland was excised and placed in a tube containing n-heptane along with 25 ng of (Z)-11-tetradecenyl acetate (Z11–14:Ac) as internal standard. Glands were extracted for at least 1 h at ambient temperature before analysis. Because the handling and sampling took a significant amount of time, we timed the procedures for 10 insects, so as to compensate for differences in actual sampling times. The initial handling of the insect (to clamping) took, on average, 30 s, the SPME sampling 60 s, and gland excision and immersion 30 s. Thus, for example, an insect sampled immediately after feeding (nominal t = 0), was sampled by SPME at real t = 1 min (30 s for initial handling plus the average of 30 s over the 1 min rubbing period), and for gland extract at real t = 2 min (30 s for handling, 60 s for SPME rub, and 30 s for gland excision and immersion).
Rinse/Extract approach
The gland of a female was extruded and maintained in position by a vascular clamp, as above, before the surface was rinsed with ca. 20 μl of n-heptane, applied using a syringe fitted with a 24 gauge needle, and collected in a glass tube. The gland was then excised, dried with a Kimwipe to remove excess solvent and hemolymph, and then placed in a separate glass tube in n-heptane with Z11–14:Ac (25 ng) as internal standard. Again, we timed the procedures and compensated as follows: 30 s for the initial handling (to clamping), 30 s for the rinse, and 30 s for the gland immersion. Thus, for an insect immediately after feeding (nominal t = 0) the gland rinse was conducted at 0.75 min (30 s for handling and 15 s middle of rinsing time), while the gland extraction was at t = 1.25 min (plus 30 s until immersion in solvent). In the case of the latter time, we considered the possibility that that gland rinse might quench biosynthesis; however, since the rinse did not extract all the pheromone (see Results) we added on the extra time before immersion.
Chemical Analysis
All pheromone samples were analyzed by coupled gas chromatography/mass spectrometry (GC/MS) on an Agilent 7890/5978A instrument (Agilent Technologies, Santa Clara, CA) equipped with a splitless injector and a DB-Wax UI (30 m × 0.25 mm × 0.25 mm film thickness) capillary column (Agilent J&W), temperature programmed as follows: 80 °C (delay of 1 min) to 180o at 15 °C.min−1, then to 190o at 5 °C.min−1, and finally to 220o at 20 °C.min−1. The carrier gas was helium at a constant flow of 1.5 ml.min−1. We operated the MS in the selected ion mode with electron ionization (70 eV) and the source and quadrupole temperatures set to 230 °C and 150 °C, respectively. The only difference for the analysis of the different samples was that the injector for the SPME rub samples was fitted with a 0.75 mm i.d. port liner, while a 4 mm i.d., liner was used for the other samples.
For analysis of unlabeled pheromone, we monitored m/z 192 (for Z9–14:Ald), 194 (for (Z11–14:Ac; internal standard), and 220 (for Z11–16:Ald). These m/z are moderately intense ions, characteristic of the three compounds, representing the loss of 60 mass units (CH3COOH) for Z9–14:Ac and 18 mass units (H2O) for Z9–14:Ald and Z11–16:Ald.
Mass Isotopomer Distribution Analysis (MIDA)
For the experiment using U-13C-glucose tracer, we only analyzed for the major pheromone component Z11–16:Ald, because of its much greater abundance. In addition to m/z 220 (unlabeled isotopomer), we also monitored m/z 222 and 224, which are the isotopomers with one and two13C2 monomeric units (i.e., from 13C2-acetyl CoA). These three ions allowed us to calculate precursor enrichment of pheromone using MIDA. This technique has been well described (Chinkes et al. 1996; Hellerstein and Neese 1999) and we have previously used MIDA in moth pheromone studies (Foster and Anderson 2011; Foster and Anderson 2015; Foster et al. 2017). Briefly, it is a combinatorial tracer/tracee approach for determining monomeric precursor enrichment (p; i.e., the proportion of stable isotope-labeled monomer tracer) of a polymer. U-13C-glucose ingested is rapidly converted to 13C2- acetyl CoA, which in turn is incorporated into de novo-biosynthesized pheromone. Tracer/tracee ratios (TTRs) of the unlabeled [TRR(M + 0)], singly labeled [TTR(M + 1)] and doubly labeled [TTR(M + 2)] isotopomers (m/z 220, 222 and 224, respectively, for Z11–16:Ald) are determined and the natural contributions of 13C and 2H accounted for, using the following equations:
The ‘pre’ and ‘post’ subscripts, respectively, refer to the intensities of isotopomers before and after tracer (from the U13C-glucose) is introduced. We used theoretically (from known natural abundances) rather than experimentally (analyzing actual unlabeled pheromone) determined values, as previously (Foster and Anderson, 2011; Foster and Anderson, 2012) we found little difference between the two. The term dT1 is the contribution of the M + 1 isotopomer spectrum to the M + 2 isotopomer intensity. In eq. 3, the number (n) of monomeric (acetyl CoA) units in the polymer (Z11–16:Ald) must be accounted for in the binomial expansion; hence the “7” (n-1).
Experiments
-
1)
Comparison of the SPME rub/extract and rinse/extract approaches.
In order to compare pheromone in the two approaches, we collected it from 1 d females and analyzed for ratio of the two components (Z9–14:Ald/ Z11–16:Ald, with the latter normalized to 100) and quantity of pheromone (titer of Z11–16:Ald; but not for the SPME rub). In the rub/extract approach, we sampled the dorsal and ventral (in random order) surfaces of the H. virescens ring gland (Raina et al. 2000) separately, rubbing each surface for 30 s with different SPME fibers.
We analyzed females at various times of the photoperiod, from 2 h before the onset through to 2 h after the end of the scotophase (i.e., 2 h into the subsequent photophase). For subsequent statistical analysis, we separated time of photoperiod into three periods: “pre-calling”, from 2 h before to 2 h into the scotophase, “calling”, from hour 2 to the end of the scotophase, and “post-calling” (only for the rinse/extract approach), 0–2 h of the photophase. These periods correspond approximately to those prior, during and after when most females call (Heath et al. 1991; Ramaswamy 1990). For the rinse/extract approach, we sampled 17–34 individual insects in each of the three periods and for the SPME rub/extract experiment, 11 and 14 insects in the pre-calling and calling periods, respectively. For each time period, we sampled insects at intervals covering the entire span.
For the SPME rub/extract approach, we analyzed mean ratio of Z9–14:Ald/Z11–16:Ald using a two-way ANOVA, with sampling method (extract, ventral rub, dorsal rub) and time period (pre-calling, calling) as categorical variables, with an interaction term in the model, after first confirming the normal distribution and heteroscedasity of the data. As only the extract samples were analyzed for pheromone quantity, we compared quantity between the two periods by one-way ANOVA. For the rinse/extract approach, due to the likely dependence of the extract on the rinse, we analyzed the data separately for rinse and extract, using a one-way ANOVA with time period as the independent variable. Means were separated by Tukey HSD tests with α = 0.05.
-
2)
Fine-scale changes at the beginning of the scotophase
Since the results from the first experiment indicated that the ratio of Z9–14:Ald/Z11–16:Ald on the gland surface changed from the pre-calling to the calling period (see Results), we tested whether this was likely due to the onset of calling. Hence, we analyzed females, both for quantity and ratio of Z9–14:Ald/Z11–16:Ald by the rinse/extract approach over the first 3 h of the scotophase. After the onset of the scotophase, females were kept in the dark until sampled (9–18 females for each time point).
After confirming the normality and heteroscedasity of the data, we analyzed both mean ratio of Z9–14:Ald/Z11–16:Ald and quantity by one-way ANOVA using time as a categorical variable for both the rinse and extract samplings. Means were separated by Tukey HSD tests with α = 0.05.
-
3)
Multiple gland rinses
The first two experiments indicated that pheromone in the rinse was distinct from that of the residual extract (different ratio of Z9–14:Ald/Z11–16:Ald; see Results). However, as the rinse probably sampled pheromone from more than just the surface, likely deeper into the gland, we wished to test whether the rinse sampled a pool of pheromone distinct from that in the residual pheromone or just partially sampled a common pool (albeit yielding a different ratio of Z9–14:Ald/Z11–16:Ald). For this, we sampled a gland by three successive rinses (each 20 μl of heptane), followed by extraction of the residual pheromone (yielding four samples). We reasoned that if the rinse sampled a distinct pool, then the second and third rinses would yield little or no pheromone, each with the same ratio of Z9–14:Ald/Z11–16:Ald. On the other hand, if the rinse partially sampled the extract pool, then the second and third rinses should contain substantial amounts of pheromone, with an increasingly similar ratio of Z9–14:Ald/Z11–16:Ald to that in the residual extract and, consequently, there should be a small amount of residual pheromone. We sampled the gland of 1 d females (N = 6) by this approach at the start of the scotophase.
We analyzed both amount and ratio of Z9–14:Ald/Z11–16:Ald in the different samplings (three rinses and extract) by mixed regression models using insect identity as a random effect. Tukey HSD tests (α set at 0.05) were used to separate means.
-
4)
Latency between de novo-synthesized pheromone inside and on surface of gland
To determine how quickly de novo-biosynthesized pheromone moves from inside the gland, at the point of synthesis, to the cuticular surface, we fed females U-13C-glucose and then sampled them by the SPME rub/extract approach at various times following glucose ingestion. Briefly, one hour into the scotophase, a female fed on a 12.5 μl drop of a 10% (w/v) solution of U-13C-glucose (99% isotopically enriched; Cambridge Isotope Laboratories, Cambridge, MA). When females had entirely consumed the drop, they were either sampled immediately (nominal time = 0) by a SPME rub, after which the gland was excised and extracted, or placed back into the dark and sampled at an appropriate time. We sampled 6–11 insects for each time point. Samples were analyzed by GC/MS and MIDA to derive precursor enrichments for both samples with respect to time after feeding.
For a further comparison of the two sampling approaches, we carried out a similar experiment, but sampled females by the rinse/extract method. Samples were analyzed by GC/MS and MIDA to derive precursor enrichments, as for the first part of the experiment. We sampled 5–13 individual insects for each time point.
We used a non-linear three-parameter exponential model (JMP Version 12.2.0) to fit a curve to the precursor enrichment data for each of the four sampling methods. We then calculated the x-axis intercept (i.e., when p = 0) as an indicator of when enrichment of pheromone was first observed (i.e., when label was first incorporated into de novo-biosynthesized pheromone). We obtained the latency between a pair of sampling methods by subtracting the respective x-axis intercepts.
-
5)
Pheromone released by females
To compare ratio of Z9–14:Ald/Z11–16:Ald released by females with that obtained in the other sampling methods, as well as to detect any changes over time, we collected pheromone released by females over the first (0–4 h) and second (4–8 h) halves of the scotophase. Just prior to the start of the scotophase, 2 × 1 d-old females were placed in a 400 ml glass vessel with charcoal-filtered air pumped (pump model NMS010S, KNF Neuberger, Trenton, NJ) through. Air exiting the glass vessel at 300 ml.min−1 passed through a Pasteur pipette loaded with 400 mg Tenax TA (Supelco Inc., Bellefonte, PA) and with glass wool at either end to retain the adsorbent. Before collection, adsorbent tubes were conditioned at 220 °C for at least 1 h with nitrogen flow. The adsorbent tube was changed 4 h into the scotophase and pheromone collected for the second half of the scotophase. After collection, 25 ng of Z11–14:Ac was placed on a glass wool plug and the chemicals desorbed with 1.5 ml of n-hexane. The eluent was concentrated to ca. 10 μl using a gentle stream of nitrogen before analysis by GC/MS. We sampled from the same nine pairs of insects for both periods.
Component ratios of Z9–14:Ald/Z11–16:Ald between the two periods were compared by ANOVA (α set at 0.05), while pheromone amount was compared by a Wilcoxon rank sum test, since the data were not normally distributed.
Results
-
1)
Comparison of the SPME rub/extract and rinse/extract approaches.
For the SPME rub/extract approach, there were significant effects of time of photoperiod (F1,69 = 11.9, P < 0.001) and sampling method (F2,69 = 15.0, P < 0.001) on ratio of Z9–14:Ald/Z11–16:Ald, but no significant interaction between the two (F2,69 = 1.4, P = 0.26). In particular, the ratio of Z9–14:Ald/Z11–16:Ald was higher in the pre-calling, than in the calling period, while the ratios of Z9–14:Ald/Z11–16:Ald in the different samples followed the order, dorsal rub>extract>ventral rub (Fig. 1a). Extract had a higher quantity of pheromone (F1,23 = 4.1, P < 0.05) during the calling period than during the pre-calling period (Fig. 1b).

Ratios of (Z)-9-tetradecenal/(Z)-11-hexadecenal (Z9–14:Ald/Z11–16:Ald) and titers of Z11–16:Ald in paired samplings of the pheromone gland of female Heliothis virescens at different periods of the day. (a) Solid phase microextraction fiber rubs of the dorsal and ventral surfaces of the gland, and extract of residual gland pheromone after the rubs. (b) Extract of residual gland pheromone after the rubs. (c, d) Solvent rinse and subsequent solvent extract of residual pheromone. Pre-calling = 2 h before to hour 2 of the scotophase, calling = hours 2–8 of the scotophase and post-calling = hours 0–2 h of the subsequent photophase. Numbers of individual insects analyzed are in parentheses in the legends. SEMs are given atop the bars. In (c) and (d), different letters of the same case atop bars indicate means that are different (P < 0.05; Tukey HSD test)
For the rinse/extract approach, the ratio of Z9–14:Ald/Z11–16:Ald showed an effect of sampling period for the rinse (F2,79 = 3.54, P = 0.034) but not for the residual extract (F2,79 = 2.25, P = 0.11) (Fig. 1c). For the rinse, the ratio showed a decline over the three successive periods, with ratio in the pre-calling period being greater (P < 0.05; Tukey-Kramer HSD) than that in the post-calling period. It was noticeable that the ratio in the extract was much higher (i.e., enriched in Z9–14:Ald) than that in the rinse. In terms of amount of pheromone (Fig. 1d), there was a significant effect of period for both rinse (F2,79 = 3.54, P = 0.034) and extract (F2,79 = 12.2, P < 0.001). For the rinse, the amount of pheromone was greater (P < 0.05; Tukey-Kramer HSD) during the calling period than for the other two periods, while for the extract, the amount in the pre-calling and calling periods was greater (P < 0.05; Tukey-Kramer HSD) than that in the post-calling period.
A rough comparison of the two approaches showed that the residual extract in the SPME rub/sampling approach had greater amounts of pheromone than the residual extract in the rinse/extract approach; in fact, the former was roughly similar to the total amount from the combined rinse/extract samplings. Further, the ratio of Z9–14:Ald/Z11–16:Ald in the rinse samples was much higher than in all the other sampling methods.
-
2)
Fine-scale changes at the beginning of the scotophase
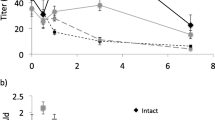
We again saw a large difference in ratio of Z9–14:Ald/Z11–16:Ald between the rinse and extract samplings, with the latter being more highly enriched in Z9–14:Ald (Fig. 2a). During the sampling period (first 3 h of the scotophase), there were differences in the ratio of Z9–14:Ald/Z11–16:Ald in the rinse (ANOVA, F6,79 = 3.96, P = 0.002). In particular, there was a decrease in the ratio around the 1 h mark, such that by 1.5 h into the scotophase, the ratio was lower than 0.5 h into the scotophase (Tukey HSD test, P < 0.05). By contrast, there was no change in component ratio in the residual extract over the experiment (ANOVA, F6,79 = 1.66, P = 0.14).

(a) Ratios of (Z)-9-tetradecenal/ (Z)-11-hexadecenal (Z11–16:Ald/Z9–14:Ald) and (b) titers of Z11–16:Ald in paired rinse/extract samplings of the pheromone gland of female Heliothis virescens during the first three hours of the scotophase. Different letters atop points indicate ratios or titers that are different (P < 0.05; Tukey’s HSD test). SEMs are given
In general, both the rinse and extract samplings showed a tendency for quantity to increase over the first 3 h of the scotophase (Fig. 2b). Only in the case of the rinse sampling was there a significant increase (from 0.5 to 3 h; Tukey HSD test, P < 0.05).
-
3)
Multiple rinses of the gland
There was an effect of sampling method on ratio of Z9–14:Ald/Z11–16:Ald (F = 58.9, P < 0.001, DF = 3; Kenward-Roger F test, mixed regression model). Notably, the three rinses all had similar ratios; all lower than that of the residual extract (Fig. 3a; Tukey HSD test, P < 0.05). Similarly, there was an effect (F = 19.1, DF = 3; P < 0.001) of sampling method on amount of pheromone. Most notable was that the amounts in the second and third rinses were very low, lower than that of the first rinse and of the residual extract (Fig. 4b, DF = 3; Tukey HSD test, P < 0.05).
-
4)
Latency between de novo-synthesized pheromone inside and outside of gland

(a) Ratios of (Z)-9-tetradecenal/ (Z)-11-hexadecenal (Z9–14:Ald/Z11–16:Ald) and (b) titers of Z11–16:Ald in rinse/extract samplings. Three successive solvent rinses were collected (in order 1–3) before the gland was excised and extracted. Different letters atop bars indicate ratios or titers that are different (P < 0.05; Tukey’s HSD test). SEMs are given

Change in precursor enrichment (p) of (Z)-11-hexadecenal in the pheromone gland of female Heliothis virescens after females had fed on U-13C-glucose, from (a) solid phase microextraction rub and a solvent extract of the residual pheromone, and (b) heptane rinse and solvent extract of the residual pheromone. SEM is given for each time point. Time was adjusted for handling and sampling procedures as detailed in the methods. The 3-parameter equations of the fitted curves are given
After feeding on U-13C-glucose, label incorporated rapidly into pheromone in the extract, as indicated by the rapid increase in precursor enrichment (Fig. 4a). Shortly thereafter, labeled pheromone appeared in the SPME rub (Fig. 4a). Over the first 10 min, precursor enrichment in the extract was always slightly greater than that in the corresponding SPME rub. However, after 10–12 min, precursor enrichment between the two samples was very similar, with both curves starting to plateau. Our 3 parameter exponential models showed that the equlibrated precursor enrichments for the extracts and rub were, respectively, 0.20 ± 0.01 and 0.22 ± 0.02, confirming that the labeled pheromone in both probably derived from the same pool of precursor (acetyl CoA). Interpolating our models back to p = 0 gave adjusted times of 0.086 and 1.015 min, respectively, for the extract and rub, a difference of 0.93 min. That is, label started appearing in the pheromone on the surface of the gland 56 s after labeled pheromone was first observed in the gland.
We observed a similar pattern for precursor enrichment for the gland rinse and residual extract, with both showing a rapid increase following feeding, before starting to plateau after 10 min (Fig. 4b). However, in this case, the differential between the two during the first 10 min was less pronounced, although precursor enrichment was consistently higher in the extract than the rinse. Our 3 parameter exponential model indicated isotopically equilibrated precursor enrichments of 0.19 ± 0.01 and 0.18 ± 0.01 for the extract and rinse, respectively, again demonstrating that both probably derived from the same precursor pool. Interpolating the curves back to p = 0 gave adjusted times of 1.28 and 1.62 min, for the extract and rinse, respectively, a difference of 0.34 min. That is, label appeared in the pheromone in the rinse some 20 s after it appeared in the gland.
-
5)
Pheromone released by females
There were no differences in ratio of Z9–14:Ald/Z11–16:Ald [13.2 ± 4.1 Vs 8.3 ± 5.8; F1,16 = 1.31, P = 0.27, ANOVA] or amount (14.3 ± 4.1 Vs 12.1 ± 2.8 ng Z11–16:Ald/h; χ2 = 0.049, P = 0.83, Wilcoxon rank sum test) of pheromone released between the two time periods (1st Vs 2nd half of scotophase, respectively). The ratio of Z9–14:Ald/Z11–16:Ald was generally higher in Z9–14:Ald than that found in surface samplings in other experiments.
Discussion
We used combinations of sampling methods to analyze pheromone on or near the gland surface, and compared these with methods sampling pheromone stored deeper within the gland to address a number of questions related to sampling method and pheromone storage.
What Do The Different Approaches Sample?
Of the sampling methods, only the SPME rub probably sampled only the cuticular surface of the gland; i.e., the pheromone about to be released. While we could not quantify the amount in this sampling, the relatively large amount in the associated residual extract, similar to the total in the combined rinse/extract approach or in whole gland extracts (e.g., Heath et al. 1991; Lievers and Groot 2016), suggest that the amount was relatively small. The rub, however, was not a complete sampling of the gland surface; hence we cannot be certain that this means that only a small quantity of pheromone is present on the gland surface. Although there were differences in ratio of Z9–14:Ald/Z11–16:Ald among the rubs and extract, these differences were not great. The other difference observed between pheromone on the surface, in the rubs, and in the residual gland extract was a temporal one: the U13C-glucose experiment indicated that de novo-synthesized pheromone on the outside of the gland lagged that inside the gland by approximately one minute.
As we could not quantify the amount of pheromone on the surface using the SPME rub, we used a rapid solvent rinse of the surface as an alternative. However, it was clear that this sampled pheromone differently from the SPME rub. In particular, the rinse sampled a much larger quantity of pheromone, indicated by the reduced amount in the residual extract, and with a lower enrichment of Z9–14:Ald compared to all other sampling methods. This low enrichment in the rinse was matched by a high enrichment in the residual extract. These indicate that the rinse probably sampled pheromone from the cuticular surface and from below the surface, possibly elsewhere in the cuticle. There are two possible explanations for the low enrichment of Z9–14:Ald in the rinse: the rinse might selectively dissolve Z11–16:Ald over Z9–14:Ald, or Z11–16:Ald in the gland might be more accessible to solvent rinsing than Z9–14:Ald (see later). The latter seems more plausible given the very similar chemical structures (and properties) of the two components, with both highly soluble in non-polar solvents. This is further supported by the successive rinses, with all three having a similar ratio of Z9–14:Ald/Z11–16:Ald, and with the last two rinses containing very small quantities of pheromone, while the amount in the residual extract was still quite large. Thus, the second and third rinses seem to have sampled the same part of the gland (pool of pheromone) as the first rinse, and not the part (pool) sampled by the residual extract. The consistent latency in precursor enrichment between the rinse and extract samplings in the U-13C-glucose experiment further supports a difference between Z11–16:Ald in the rinse and extract.
In summary, the SPME rub sampled a portion of the cuticular surface of the gland, and its residual extract the entire gland (minus the portion sampled from the surface), while the rinse likely sampled the entire surface of the gland plus some pheromone (predominantly Z11–16:Ald) below the surface, with its residual extract sampling the remainder (enriched in Z9–14:Ald) from below the surface.
How Is Pheromone Distributed In The Gland?
Our results indicate that the two components are not distributed evenly on the gland surface. In particular, the dorsal surface was more enriched in Z9–14:Ald than the ventral surface. That this was observed during the pre-calling period shows that this was not due to differential release of the two components from the dorsal and ventral surfaces. This suggests that the two components may exist, at least to some degree, in spatially distinct pheromone “pools” in the gland, which are transported through topographically distinct pore canals/filaments (Raina et al. 2000) to the surface. For instance, greater activity of the cytosolic β-oxidation system that produces (Z)-9-tetradecenoate from (Z)-11-hexadecenoate (Choi et al. 2005) in dorsal, compared to ventral, gland cells, could result in a higher ratio of Z9–14:Ald/Z11–16:Ald on the dorsal cuticle than on the ventral cuticle. Such differences in distribution of ratios of Z9–14:Ald/Z11–16:Ald through the gland, especially if associated with different pore densities, lengths or diameters, might also explain why solvent rinses were relatively depleted in Z9–14:Ald. Regardless, this emphasizes the need for more detailed ultrastructural characterization, combined with precise localization of pheromone molecules, precursors and enzymes throughout the gland, in order to understand storage and transport of pheromone.
Our gland rinses indicated that a large proportion (ca. 50%) of pheromone was stored in a part relatively accessible to solvent, while extended immersion of the gland in solvent was needed to extract the remaining pheromone. Immersion likely ruptured gland cell membranes, but whether the rinse did or not is less certain, especially since repeated rinsing did not extract substantial quantities of pheromone. For moths producing Type I pheromones, pheromone is largely or entirely produced in gland cells (Hagström et al. 2013), suggesting that some, if not most, pheromone in the gland could be stored intracellularly. However, the suggestion that a cuticular oxidase is responsible for the final step in the production of Type 1 aldehydes (Fang et al. 1995; Teal and Tumlinson 1988), as in H. virescens, implies that glandular storage of pheromone aldehyde components should be in the cuticle. While our rinse data support that a considerable portion of transiently stored pheromone is indeed on or in the cuticle, the necessity for the immersion to extract the remainder suggests that some pheromone is stored deeper in the gland and perhaps intracellularly. While this demonstrates that pheromone is distributed on and through the gland, it again shows the need for fine-scale localization studies to pinpoint exactly where pheromone is stored.
How Does Pheromone Storage Change Across the Photoperiod?
Both pheromone quantity and ratio of Z9–14:Ald/Z11–16:Ald changed in the various sampling methods across the photoperiod. With respect to quantity, we observed changes for both the rinse and the residual extract consistent with changes observed previously for whole gland titer (Foster 2005; Heath et al. 1991); namely, high amounts during the calling period and lower amounts outside this period. When we sampled the first three hours of the scotophase in greater resolution, we found that quantity rose in the rinse but not in the residual extract, consistent with results in the experiment sampling a greater span of the photoperiod. This may indicate a more limited capacity for storing pheromone in the inner part of the gland (i.e., that not sampled by the rinse) or that the flux of pheromone release is less than the flux of pheromone transport from the site of synthesis to the cuticular surface, resulting in increased accumulation over time in the outer part of the gland.
Ratios of Z9–14:Ald/Z11–16:Ald also changed throughout the photoperiod, with both sampling approaches showing lower ratios during, compared to outside, the calling period. The higher resolution sampling over the first 3 h of the scotophase showed that the ratio of Z9–14:Ald/Z11–16:Ald decreased around 0.5–1.5 h into the scotophase, a time corresponding to when most females start calling in this species (Heath et al. 1991; Ramaswamy 1990; Foster, Anderson and Casas, unpublished results). This effect was likely due to the higher evaporation rate of the more volatile Z9–14:Ald at the cuticular surface when females called, and further confirms that the rinse primarily sampled pheromone on or close to the surface. The enrichment of Z9–14:Ald in the released pheromone was confirmed by our volatile collections (ca. 8–13% Z9–14:Ald relative to Z11–16:Ald, compared to <5% in both of our surface samplings during the calling period). This enrichment of more volatile components in released pheromone versus gland extract has been noted previously (Allison and Cardé 2016).
How Quickly Is Newly Synthesized Pheromone Transported For Release?
In the SPME rub/extract, the pheromone precursor enrichment curves suggested that newly synthesized pheromone was present in the gland within roughly 5 s after feeding ceased. Since feeding on the glucose drop took approximately 1 min, glucose was absorbed, circulated to the gland, and used in de novo pheromone biosynthesis roughly within a minute after females started to feed. Furthermore, the SPME rub showed that labeled pheromone appeared on the cuticular surface of the gland approximately a minute after it first appeared in the gland. This latency between the two fitted curves was largely maintained over the first 8–10 min before the enrichment of the two samplings converged. We obtained a similar result with the rinse/extract approach, although in this case there was a slightly shorter latency between the two samplings, perhaps reflecting the greater similarity of the pheromone pools sampled. While we did not determine the flux of pheromone from the extract to the surface, both approaches demonstrate that pheromone is made very rapidly from ingested sugar and that this newly synthesized pheromone can be transported very rapidly to the gland surface for release.
In summary, different sampling methods of a moth pheromone gland sample different “pools” of pheromone. Using these different methods enabled us to detect differences in both pheromone quality (ratio of Z9–14:Ald/Z11–16:Ald) and quantity in different parts of the gland, as well as determine that pheromone can be biosynthesized and transported to the surface of the gland very rapidly. While this showed that pheromone storage differs in relation to both position in the gland and time of photoperiod, more precise pheromone sampling and localization methods are needed to track the intra-gland flux of pheromone from point of synthesis to release.
References
Allison JD, Cardé RT (eds) (2016) Pheromone communication in moths: evolution, behavior and application. University of Caifornia Press, Oakland
Ando T, Inomata S, Yamamoto M (2004) Lepidopteran sex pheromones. In: Schulz S (ed) The Chemistry of Pheromones and Other Semiochemicals I, vol 239. Topics in Current Chemistry. Springer Berlin / Heidelberg, pp 51–96. doi:https://doi.org/10.1007/b95449
Blomquist GJ, Jurenka R, Schal C, Tittiger C (2011) Pheromone production: biochemistry and molecular biology. In: Gilbert LI (ed) Insect Endocrinology. Academic Press, San Diego, pp 523–567
Chinkes DL, Aarsland A, Rosenblatt J, Wolfe RR (1996) Comparison of mass isotopomer dilution methods used to compute VLDL production in vivo. Am J Physiol Endocrinol Metab 271:E373–E383
Choi MY, Groot A, Jurenka RA (2005) Pheromone biosynthetic pathways in the moths Heliothis subflexa and Heliothis virescens. Arch Insect Biochem Physiol 59:53–58
Conner WE, Webster RP, Itagaki H (1985) Calling behaviour in arctiid moths: The effects of temperature and wind speed on the rhythmic exposure of the sex attractant gland. J Insect Physiol 31:815–820. https://doi.org/10.1016/0022-1910(85)90074-5
Cory HT, Daterman GE, Daves GD, Sower LL, Shepherd RF, Sanders CJ (1982) Chemistry and field evaluation of the sex pheromone of western spruce budworm. Choristoneura occidentalis, Freeman J Chem Ecol 8:339–350. https://doi.org/10.1007/bf00987782
Delisle J (1992) Age related changes in the calling behaviour and the attractiveness of obliquebanded leafroller virgin females, Choristoneura rosaceana, under different constant and fluctuating conditions. Entomologia experimentalis et applicata 63:55–62
Fang NB, Teal PEA, Tumlinson JH (1995) Characterization of oxidase(s) associated with the sex pheromone gland in Manduca sexta (L) females. Arch Insect Biochem Physiol 29:243–257
Fonagy A, Yokoyama N, Okano K, Tatsuki S, Maeda S, Matsumoto S (2000) Pheromone-producing cells in the silkmoth, Bombyx mori: identification and their morphological changes in response to pheromonotropic stimuli. J Insect Physiol 46:735–744
Foster S, Anderson K (2011) The use of mass isotopomer distribution analysis to quantify synthetic rates of sex pheromone in the moth Heliothis virescens. J Chem Ecol 37:1208–1210
Foster SP (2005) Lipid analysis of the sex pheromone gland of the moth Heliothis virescens. Arch Insect Biochem Physiol 59:80–90. https://doi.org/10.1002/arch.20058
Foster SP (2016) Toward a quantitative paradigm for sex pheromone production in moths. In: Allison JD, Cardé RT (eds) Pheromone communication in moths: evolution, behavior and application. University of California Press, Oakland, pp 113–126
Foster SP, Anderson KG (2015) Sex pheromones in mate assessment: analysis of nutrient cost of sex pheromone production by females of the moth Heliothis virescens. J Exp Biol 218:1252–1258. https://doi.org/10.1242/jeb.119883
Foster SP, Anderson KG, Casas J (2017) Sex pheromone in the moth Heliothis virescens is produced as a mixture of two pools: de novo and via precursor storage in glycerolipids. Insect Biochem Mol Biol 87:26–34. https://doi.org/10.1016/j.ibmb.2017.06.004
Frérot B, Malosse C, Cain A-H (1997) Solid-phase microextraction (SPME): A new tool in pheromone identification in lepidoptera. J High Resolut Chromatogr 20:340–342. https://doi.org/10.1002/jhrc.1240200609
Groot AT (2014) Circadian rhythms of sexual activities in moths: a review. Front Ecol Evol 2. https://doi.org/10.3389/fevo.2014.00043
Hagström ÅK, Walther A, Wendland J, Löfstedt C (2013) Subcellular localization of the fatty acyl reductase involved in pheromone biosynthesis in the tobacco budworm, Heliothis virescens (Noctuidae: Lepidoptera). Insect Biochem Mol Biol 43:510–521. https://doi.org/10.1016/j.ibmb.2013.03.006
Heath RR, McLaughlin JR, Proshold F, Teal PEA (1991) Periodicity of female sex pheromone titer and release in Heliothis subflexa and H. virescens (Lepidoptera: Noctuidae). Ann Entomol Soc Am 84:182–189
Hellerstein MK, Neese RA (1999) Mass isotopomer distribution analysis at eight years: theoretical, analytic, and experimental considerations. Am J Physiol Endocrinol Metab 276:E1146–E1170
Jurenka R (2017) Regulation of pheromone biosynthesis in moths. Current Opinion in Insect Science 24:29–35. https://doi.org/10.1016/j.cois.2017.09.002
Jurenka RA, Roelofs WL (1989) Characterization of the acetyltransferase used in pheromone biosynthesis in moths: specificity for the Z isomer in Tortricidae. Insect Biochem 19:639–644
Klun JA et al (1980) Sex pheromone chemistry of the female tobacco budworm moth, Heliothis virescens. J Chem Ecol 6:177–184
Lievers R, Groot AT (2016) Disposable Polydimethylsiloxane (PDMS)-Coated Fused Silica Optical Fibers for Sampling Pheromones of Moths. PLoS One 11:e0161138. https://doi.org/10.1371/journal.pone.0161138
Ma PWK, Ramaswamy SB (2003) Biology and ultrastructure of sex pheromone-producing tissue. In: Blomquist GJ, Vogt RC (eds) Insect pheromone biochemsitry and molecular biology. Elsevier Academic Press, London, pp 19–51
Pawliszyn J (1997) Solid phase microextraction: theory and practice. Wiley-VCH, New York
Raina AK, Wergin WP, Murphy CA, Erbe EF (2000) Structural organization of the sex pheromone gland in Helicoverpa zea in relation to pheromone production and release. Arthropod Structure and Development 29:343–353
Ramaswamy SB (1990) Periodicity of oviposition, feeding, and calling by mated female Heliothis virescens in a field cage. J Insect Behav 3:417–427. https://doi.org/10.1007/BF01052118
Roelofs WL, Hill AS, Cardé RT, Baker TC (1974) Two sex pheromone components of the tobacco budworm moth, Heliothis virescens. Life Sci 14:1555–1562. https://doi.org/10.1016/0024-3205(74)90166-0
Teal PEA, Tumlinson JH (1988) Properties of cuticular oxidases used for sex pheromone biosynthesis by Heliothis zea. J Chem Ecol 14:2131–2145
Zhu J, Löfstedt C, Bengtsson BO (1996) Genetic variation in the strongly canalized sex pheromone communication system of the European corn borer, Ostrinia nubilalis Hübner (Lepidoptera, Pyralidae). Genetics 144:757–766
Acknowledgments
This work was funded in part by a United States Department of Agriculture Hatch Project ND02388 (to SPF). We thank the United States Department of Agriculture–National Institute of Food and Agriculture for an Instrument Grant, 2015-07238 (to SPF), contributing to the purchase of the GC/MS system. We are grateful to Professor Jérôme Casas for comments on an earlier version of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Foster, S.P., Anderson, K.G. Differential Pheromone Sampling of the Gland of Female Heliothis Virescens Moths Reveals Glandular Differences in Composition and Quantity. J Chem Ecol 44, 452–462 (2018). https://doi.org/10.1007/s10886-018-0954-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10886-018-0954-0