
Abstract
Two luminescent, monoanionic chalcogenide-centered nonanuclear silver clusters stabilized by dichalcogenophosphates were synthesized and fully characterized by various spectroscopies including multinuclear NMR and ESI-mass. Single crystal X-ray diffraction studies on both cluster anions, [Ag9(S){S2P(OEt)2}8]−, 1, and [Ag9(Se){Se2P(OEt)2}8]−, 2, reveal that the nine silver atoms form an extremely distorted tricapped trigonal prism, which has an encapsulating chalcogenide. The coordination geometry of the central chalcogenide appears to be monocapped trigonal prismatic, which was analyzed by DFT calculations. The origin of the yellow emission is assigned by TDDFT calculations to originate from a chalcogen (ligand + encapsulated) → silver charge transfer.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The synthesis of silver chalcogenide or chalcogenolate clusters is of particular interest not only because of their rich structural diversity but also because of their interesting, photocatalytic, semiconducting and photoluminescent properties [1–9]. Very recently the stability of some of these clusters has been proved to have superatom-type characteristics [7–9]. It has previously been reported that a stoichiometric reaction of dichalcogeophosph(in)ate ammonium salts, NH4E2PX2 (E = S, Se; X = R, OR), with Ag(I) in common organic solvents generally will produce either a tetranuclear, [Ag(SSePiPr2)]4 [10], or a hexanuclear molecule, [AgE2P(OR)2]6 [11–13], aside from the polymeric species, [AgE2PX2]n [14]. If the molar ratio of ligands used is greater than that of metal salts, chalcogenide-centered polynuclear species are frequently isolated [15]. The encapsulated chalcogenide results from ligand reduction. It is generated in situ via P–E bond cleavage by the excess of dialkyl diselenophosphate [15]. Intriguingly, upon incorporation of a chalcogenide at the center, the cluster nuclearity can be expanded to eight (an octanuclear silver cage in cubic geometry) [16] and even to ten (a decanuclear silver cluster in a distorted bi-capped tetragonal prism), to afford a hyper-coordinated chalcogen of μ8 and μ10 coordination mode, respectively [13, 16–20]. Since this chemistry of the (spherical) hypercoordinated selenide or sulfide anions can be referred to guest–host supramolecular chemistry, this encouraged us to continue the synthesis of chalcogenide-centered, high nuclearity silver cluster compounds after successful characterizations of [Ag8(E){E2P(OR)2}6] [6, 7], [Ag10(E){E2P(OR)2}8] [13, 19, 20], and [Ag11(S){S2P(OR)2}8](PF6) [21], for the following two reasons. First, the coordination behavior of the anions is similar to the coordination of transition metals such as coordination geometry and coordination number [22]. Therefore it will be of fundamental interest to explore the bonding mode between the central chalcogen and peripherial silver atoms by density functional theory (DFT) calculations. Secondly, nuclearities of silver clusters other than 8, 10, and 11 may be produced to exhibit unusual photophysical properties from which the exact nature of photoluminescence could be deduced. Herein we present a rational synthesis of two new, nonanuclear silver clusters, (Na)[Ag9(S){S2P(OEt)2}8], 1, and (Na)[Ag9(Se){Se2P(OEt)2}8], 2, which display yellow emission at 77 K. The geometry of μ7-S in 1 and μ7-Se in 2 appears to be monocapped trigonal prismatic. Surprisingly both of them are the first monoanionic silver dichalcogenophosph(in)ate clusters, to the best of our knowledge [3].
Experimental
All solvents were purchased from Mallinckrodt Chemicals and purified before use following standard procedures. Other reagent grade chemicals were purchased from Aldrich and used without further purification. All the reactions were carried out under inert atmosphere using Schlenk apparatus. Elemental analyses were done using an Elementar vario EL III analyzer. Multinuclear (1H, 31P, 77Se) NMR spectra were recorded with a Bruker Advance DPX300 FT-NMR spectrometer. Both 31P{1H} and 77Se{1H} NMR spectra were referenced externally against 85 % H3PO4 (δ = 0 ppm) and PhSeSePh (δ = 463 ppm), respectively. While NH4S2P(OEt)2 and NaSH were obtained from Aldrich, NH4Se2P(OEt)2 [13], NaSeH [23], and [Ag5{S2P(OEt)2}4(PF6)]∞ were prepared by following the literature method [24]. UV-visible absorption spectra were measured on a Perkin Elmer Lambda 750 spectrophotometer using quartz cells with path length of 1 cm recording in the 250-750 nm region. Emission spectra were recorded on a Cary Eclipse B10 fluorescence spectrophotometer. ESI-mass spectra were recorded on a Fison Quattro Bio-Q (Fisons Instruments, VG Biotech, UK). Melting points were measured by using a Fargo MP-2D melting point apparatus. The lifetime was recorded on Fluorescence Spectrometers Edinburgh FLSP920 at 77 K. TGA was measured with a PerkinElmer TGA 4000 thermogravimetric analyzer.
Synthesis of (Na)[Ag9(S){S2P(OEt)2}8] (1)
Method (a): NaSH (3.76 mg, 0.067 mmol) were added into 20 mL of acetone solution of [Ag5{S2P(OEt)2}4(PF6)]∞ (191.0 mg, 0.134 mmol) in a Schlenk flask. The resulting mixture was stirred for 15 h at −20 °C under a nitrogen atmosphere, and the color changed from light-yellow to deep-green. After filtration, the black powder was removed and the filtrate was evaporated to dryness under vacuum. It was then extracted with CHCl3 (50 mL) to afford a yellow solution which upon evaporation formed a yellow solid. Yield: 0.140 g (83 %). Anal. Calcd for C32H80Ag9NaO16P8S17·Me2CO: C, 16.38; H, 3.38; S, 21.25 %. Found: C, 16.00; H, 3.25; S, 21.34 %. 1H NMR (300 MHz, acetone-d6, δ, ppm): 4.20 [m; 32H, OCH 2], 1.34 [t, 3 J HH = 7.0 Hz, 48H, CH 3]. 31P{1H} NMR (121.49 MHz, acetone-d6, δ, ppm): 109.0. ESI-MS (m/z) (Cal.): 2483.9 (2484.0). m.p. 122 °C(dec.).
Method (b): Ag(CH3CN)4PF6 (46.2 mg, 1.108 mmol) and NH4S2P(OEt)2 (200 mg, 0.985 mmol) were mixed in a 100 mL Schlenk flask containing 30 mL of acetone. The reaction was stirred for one hour at −20 °C under a nitrogen atmosphere to yield colorless solution from which one equiv. of NaSH (6.90 mg, 0.123 mmol) was added. The resulting mixture was stirred for 15 h at −20 °C, and then the solution color changed to deep-green. After filtration, the filtrate was extracted with CHCl3 (75 mL) to afford a yellow solution which upon evaporation formed a yellow solid. Yield: 0.235 g (76 %).
Synthesis of (Na)[Ag9(Se){Se2P(OEt)2}8] (2)
Ag(CH3CN)4PF6 (0.316 g, 0.76 mmol) and NH4Se2P(OEt)2 (0.200 g, 0.67 mmol) were dissolved in acetone (30 mL) and the mixture was stirred at 0 °C for 30 min to form a light-yellow solution. NaSeH (0.17 mmol) was added into the yellow solution and continued to stir at 0 °C for 1 h until the solution color turned black. After filtration affords yellow-green solution from which yellow-green powders were yielded under vacuum. The residue was washed with hexane, and the resulting solid was dried under vacuum to give 2 as a yellow powder. Yield: 0.102 g (36 %) Anal. Calcd for C32H80Ag9NaO16P8Se17 ·C6H14: C, 13.46; H, 2.79 %. Found: C, 13.89; H, 3.19 %. 1H NMR (300 MHz, acetone-d6, δ, ppm): 4.20 [m; 32H, OCH 2], 1.33 [t, 3 J HH = 7.0 Hz, 48H, CH 3]. 31P{1H} NMR (121.49 MHz, acetone-d6, δ, ppm): 83.2 (J P-Se = 681 Hz, 8P). 77Se{1H} NMR (57.24 MHz, acetone-d6, δ, ppm): 85.9 (d, J SeP = 683 Hz, 16Se), −1702.7 (1Se). ESI-MS (m/z) (Cal.): 3286.4 (3285.1). mp: 115 °C(dec.).
X-ray Crystallography
Crystallographic data of (Na)[Ag9(S){S2P(OEt)2}8], 1, and [Ag9(Se){Se2P(OEt)2}8]− 2, which has a solvated hexane molecule, are given in Table 1, and selected bond distances and angles are listed in Tables S1, S2. Yellow crystals of 1 and 2 were obtained by diffusion a layer of hexanes into an acetone solution. Intensity data of 1 were collected at 223(2) K and 2 at 296(2) K on a Bruker APEX-II CCD diffractometer (Mo-Kα radiation, λ = 0.71073 Å). The unit cell parameters were calculated and refined from the full data set. The SMART software was used for data acquisition [25], and the SAINT-Plus software was used for data reduction [26]. The absorption corrections were performed with the help of the SADABS program [27]. The structure was solved by direct methods and refined by full-matrix least-squares on F 2 by using the SHELXTL-2014/7 software package incorporated in SHELXTL/PC version 6.14 [28]. Ag5 and Ag5A of 1, which are related to the crystallographic two-fold axis, are each in 50 % occupancy. Two ethoxy groups, O7 and O8, connecting to P4 were disordered over two positions and the occupancy ratio is 60:40. All non-hydrogen atoms were refined anisotropically. Both the solvated hexane molecule and the Ag2 atom of 2 were disordered in two positions and the latter was refined in a 60:40 occupancy ratio. However the sodium counter cation could not be located precisely from the final Fourier difference map due to a severely disordered issue [29].
Computational Details
DFT calculations were carried out using the Gaussian 09 package [30], employing PBE1PBE (PBE0) functional [31]. Geometry optimizations were performed with a standard double-ξ polarized basis set, namely the LANL2DZ set [32–36], augmented with Ahlrichs polarization functions [36] on all the atoms. Analytical calculations of the vibrational frequencies were performed on all the optimized geometries to verify that these structures are local minima on the potential energy surface. The geometries obtained from DFT calculations were used to perform natural atomic orbital (NAO) population analysis and calculate Wiberg indices with the NBO 5.0 program [37]. The compositions of the molecular orbitals were calculated using the AOMix program [38].
The gauge including atomic orbital [39–43] method has been used to compute the 1H and 77Se NMR chemical shifts, with tetramethylsilane and diphenyldiselenide as references, respectively. The all-electron triple-ξ polarized Def2-TZVP basis set from EMSL basis set exchange library [44], was used for these calculations. The UV-visible singlet–singlet transitions were calculated by means of time-dependent DFT (TDDFT) calculations, at the PBE0/LANL2DZ level.
Results and Discussion
Synthesis
We have previously demonstrated that one-dimensional polymeric chains, [Ag5{S2P(OR)2}4(PF6)]∞ (R = Et [24], iPr[18]), were exclusively synthesized from the reaction of silver salts with dtp ligands (dtp = dithiophosphate [S2P(OR)2]−) in a 4:3 molar ratio. More importantly, this pentanuclear 1D chain reorganizes into discrete, octanuclear silver clusters in the presence of spherical anions such as halides or hydride [24]. When tetrahedrally shaped anions (sulfate, selenate, chromate, and molybdate) are used in the cluster build-up, EO4 2−@Ag16 clusters, which dimerize via intermolecular Ag–S bonds to form Ag32 clusters in the solid state [45], were characterized. Trigonal pyramidal oxyanions, EO3 2− (E = S, Se, Te), were also utilized as a template in the synthesis of Ag16 clusters, which eventually dimerize to form Ag32 clusters stabilized by 1,1-dithiolate ligands [46]. Stereochemically active lone-pair electrons on the latter appear to play structure-directing effects. Thus, these examples nicely demonstrate the template effect played by the anions in the cluster assembly from the 1D chain.
The template effect of thiol, SH−, was subsequently applied in our continued foray into this silver cluster chemistry. A new compound, (Na)[Ag9(S){S2P(OEt)2}8], was prepared in 83 % yield from the reaction of [Ag5{S2P(OEt)2}4(PF6)]∞ and NaSH in a molar ratio of 2:1 in acetone at −20°. It can also be synthesized in 76 % yield from the reaction of Ag(CH3CN)4PF6, NH4S2P(OEt)2, and NaSH in 9:8:1 molar ratios at −20° (Scheme 1). Compound 1 was fully characterized by elemental analysis, multinuclear NMR (1H, 31P), and single crystal X-ray diffraction techniques. 31P NMR spectrum displayed a singlet at δ 109.0 ppm at ambient temperature, whereas the precursor [Ag5{S2P(OEt)2}4(PF6)]∞ showed a singlet at δ 103.9 ppm. Thus the ~5 ppm shift suggested a new species was formed. This, coupled with a set of resonances of ethyl protons detected from 1H NMR spectrum, indicated that all eight dtp ligands are equivalent in solution (vide infra). In addition, its composition is primarily determined by negative ion ElectroSpray ionization-mass spectrometry, which displayed a band centered at 2483.9 corresponding to the molecular ion peak of [Ag9(S){S2P(OEt)2}8]− (m/z = 2484.0). Its simulated isotopic pattern matches well with the experimental one (Fig. S1). Two fragment bands centered at 2190.6 and 1897.3, which corresponded to [Ag8(S){S2P(OEt)2}7]− (m/z = 2191.6) and [Ag7(S){S2P(OEt)2}6]− (m/z = 1898.2), respectively, were also detected. Finally the compound purity was further confirmed by the satisfactory C, H, and S elemental analyses. However the exact structure of this cluster anion could not be realized until single crystal X-ray diffraction studies were carried out (vide infra).

Synthesis of (Na)[Ag9(S){S2P(OEt)2}8] and (Na)[Ag9(Se){Se2P(OEt)2}8]
On the other hand, a selenium analogue of the nonanuclear silver cluster anion, [Ag9(Se){Se2P(OEt)2}8]−, 2, could only be prepared by mixing Ag(CH3CN)4PF6, NH4Se2P(OEt)2, and NaSeH in 9:8:2 molar ratios (Scheme 1). If one equiv. of NaSeH was used in the synthesis, the isolated product is [Ag10(Se){Se2P(OEt)2}8] instead [13]. Cluster 2 was fully characterized by various spectroscopic methods including negative ESI-mass spectrometry (Fig. S2). The equivalency of eight diselenophosphato (dsep) ligands in solution is revealed from both 31P NMR and 77Se NMR spectra: a singlet peak flanked with a set of selenium satellite (J P-Se = 681 Hz) and a doublet peak (J Se–P = 683 Hz) arising from the coupling to the phosphorus nuclei. The chemical shift of the central selenide is at −1702.7 ppm, which appears to be the most up-field shift among the reported selenium-containing species [47]. This large up-field value is confirmed by DFT calculations (see below).
Crystal Structure
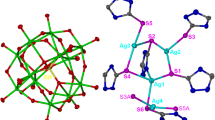
Compound 1 crystallizes in the monoclinic space group C2/c with four molecules per unit cell. The geometry of the Ag9S core can be best described as an extremely distorted, tricapped trigonal prism of C 2 symmetry centered by a sulfur atom lying on the C 2 axis and in an unusual μ7 bridging mode (Fig. 1a). Three silver atoms, Ag1, Ag2, Ag4, and their partners generated by the C 2 axis, Ag1A, Ag2A, and Ag4A form six vertices of a distorted trigonal prism. While the lengths of a triangular base range from 3.189(1) to 3.533(1) Å, one of the stick lengths, which link two triangles, is very short, 3.126(2) Å, comparing to the other two sticks which are as long as 3.812(1) Å. Thus instead of three parallelograms typically observed in a trigonal prism, two trapezoidal faces, each consisting of four vertices (Ag1, Ag1a, Ag2, and Ag4A) and capped by Ag3, are produced in compound 1. The only identified parallelogram is capped by the Ag5 atom. Thus, the encapsulated sulfide, S9, appears to adopt the unusual μ7 coordination mode of a monocapped trigonal prism, being bonded to all the Ag atoms, except to the capping Ag3 and Ag3A ones which are 3.338 Å away from S9. The distances to these seven silver atoms are in the range 2.496(3)~2.8166(14) Å, which are similar to the Ag–μ 7 S and Ag–μ 6 S separations observed in [Ag70S16(SPh)34(PhCO2)4(triphos)4] and [Ag14(μ6–S)(tab)12(PPh3)8]12+ (2.936 Å), respectively [48, 49].

a The drawing of [Ag9(μ7-S)] core. b Thermal ellipsoid drawing (30 %) of [Ag9(S){S2P(OEt)2}8]− with ethoxy groups omitted for clarity. (Symmetry code: A = −x + 1, y, −z + 3/2)
In a tricapped trigonal prism, the three capping vertices delineate twelve (3 × 4) triangular faces with the prism vertices. Among these twelve faces of the Ag9S core, six of them are bridged by one dtp ligand, whereas two couples of two adjacent faces constituting the Ag2/Ag3/Ag4/Ag5 and Ag2A/Ag3A/Ag4A/Ag5A diamonds are each capped by one dtp ligand. Among the six dtp ligands which cap a triangular face, four are in trimetallic triconnectivity (μ3: η2, η1) and the two diamond-capping ligands corresponding to P2 and P2A are in a tetrametallic tetraconnectivity (μ4: η2, η2). Finally, the two ligands corresponding to P4 and P4A which are bonded to the 50 % disordered Ag5 and Ag5A positions (Ag5–Ag5A = 0.985 Å) are in trimetallic tetraconnectivity (μ3: η2, η2). Removing one of this positions regenerates a fully ordered structure in which one of these two ligands is now (μ3: η2, η1) triconnected, the other one remaining in the (μ3: η2, η2) tetraconnectivity. As a result, all silver atoms are each coordinated to three sulfur atoms, not considering the bonding with the encapsulated sulfide. It is noteworthy that if the ordered structure was of perfect C 2 symmetry (Ag5 lying on the C 2 axis), then both dtc ligands related to P4 and P4A would be really (μ3: η2, η2) tetraconnected and Ag5 would be tetracoordinated (not considering the encapsulated atom). This point will be discussed in the theoretical part below. The Ag–S(dtp) distances (2.333(4)–2.890(3) Å) are comparable to those observed in [Ag9(i-MNT)6(PPh3)6]3− (2.492–2.707 Å) and [Ag9(Tab)8(MeCN)8]2(PF6)18·4MeCN 2.397(5)–2.654(5) Å [50, 51]. The average intraligand, S…S bite distance is 3.412 Å and the S–Ag–S bond angles range from 73.44(7)° to 146.99(14)°.
The differences in the ligand bridging patterns coupled with the extremely distorted silver skeleton observed in 1 strongly suggest that the number of 31P chemical shifts should be greater than one. Thus, VT 31P{1H} spectra of 1 were recorded at acetone-d6 and significant peak broadening was observed as soon as the temperature was lowered to 0 °C (Fig. S3). Surprisingly the broad peak did not change its line shape all the way down to −80 °C. Clearly the ligand exchanging rate is so fast on the NMR timescale that not much structure information can be retrieved from the VT 31P{1H} NMR experiment. On the other hand in contrast to 1 the VT 31P{1H} NMR spectra of 2, exhibit more complex splitting patterns which could be due to its distorted asymmetric geometry (Fig. S7).
The structural characteristics of cluster 1, suggested us the possibility in the isolation of the known sulfide-centered undecanuclear silver cluster, [Ag11(S){S2P(OEt)2}8]+ (hexacapped trigonal prism) [21], by simply adding two supplementary Ag+ ions to cluster 1 as sketched in Scheme 2. Unfortunately, the proposed reaction did not succeed, no matter which silver salts were added into a solution containing clusters 1.

Synthesis of [Ag11(S){S2P(OEt)2}8]+ by adding two equivalent of Ag+ ions to [Ag9(S){S2P(OEt)2}8]− was unsuccessful
Slow diffusion of hexane into an acetone solution of 2 afforded crystals suitable for X-ray diffraction. Compound 2 crystallizes in the P21 space group and has a solvated hexane molecule, which is disordered, in the asymmetric unit. Similarly as in 1, the Ag9 skeleton of 2 (Fig. 2a) displays a distorted tricapped trigonal prism made of the two Ag2/Ag8/Ag9 and Ag3/Ag5/Ag6 triangles and with Ag1, Ag4 and Ag7 as the three capping atoms. Only eleven Ag–Ag edge lengths among a total of twenty-one edges of this elongated tricapped trigonal prism are shorter than 3.44 Å, which is twice the van der Waals radius of a silver atom. As in 1, the central selenium atom (Se17) links to seven silver atoms in the range of 2.554(4)–3.066(2) Å to yield a μ7-Se monocapped trigonal prismatic arrangement. This particular coordination mode has been reported by Fenske et al. in the ligand-stabilized silver chalcogenide cluster, [Ag154Se77(PPh2CH2C6H4CH2PPh2)18] [52]. In addition, Ag–Ag stick lengths, 3.428(5), 3.543(3), and 4.378(2) Å, within the trigonal prism are significant longer than those in compound 1. These values correspond well to the larger ionic size of selenide, as opposed to sulfide. From this size point of view, it is noteworthy that, contrarily to what is observed in 1, the two capping Ag atoms which are not considered as bonded to the encapsulated chalcogenide in 2 (Ag1 and Ag4) are lying at a rather short non-bonding separation from Se17 (3.193(3) and 3.123(2) Å, respectively). The question of a possible interaction associated with these contacts will be discussed below.

a The drawing of [Ag9(μ7-Se)] core. b Thermal ellipsoid drawing (30 %) of [Ag9(Se){Se2P(OEt)2}8]− with ethoxy groups omitted for clarity
The topology of the capping pattern of the eight dsep ligands in 2 is similar to that of the eight dtp ligands in 1, except that one of the two tetracoordinated (μ3: η2, η2) ligands in 1 is in a (μ3: η2, η1) coordination mode in 2. Thus, 2 has one Ag–E bond less than 1. It results that, whereas in 1, all the Ag atoms are triconnected (not considering the bonding with the encapsulated sulfide), in 2, all but one (Ag8, digonal, but with an additional long contact with Se1 (3.293(2) Å) are triconnected (still not considering the Ag–(μ7-Se) bonding).
The geometry of a nine-atom cluster is usually described as deriving from either ideal distorted tricapped trigonal prismatic or ideal capped square antiprismatic arrangements as those typically observed in the Zintl ions [53]. In addition, an elongated tricapped trigonal prismatic metallic skeleton is also known in the ligand-stabilized soluble Ge9 cluster compound, [Ge9{Si(SiMe3)3}]− [54], reported by Schnepf. Although the Ag–Ag bonding in cluster compounds 1 and 2 appears not very significant (weak argentophillic (d10–d10 bonding) interactions) [55], the structure characteristics present here are a nice addition to the well-known nine-atom cage having a tricapped trigonal prismatic geometry.
Absorption Spectrum and Luminescence Properties
The cluster compound, [Ag11(S){S2P(OEt)2}8]+, was reported to display a broad, near-IR emission centered at 720 nm in both solid state and solution at ambient temperature [21]. Surprisingly, the sulfide-centered Ag9 cluster displays a yellow emission only at 77 K. A UV-vis absorption spectrum of compound 1 was recorded in dichloromethane solution (Fig. 3). It exhibited an intense absorption band centered at 369 nm and the spectrum can be reproduced satisfyingly by TDDFT calculations (vide infra). The large extinction coefficient of ~104 indicates a fully allowed transition. Compound 1 displays strong yellow emission at 77 K under UV-irradiation in both solid state and solution. Accordingly, excitation at 370 nm resulted in a broad yellow emission centered at 587 nm at 77 K in CH2Cl2 glass. The life-time data recorded at 77 K for powder samples reveals a double-exponential behavior with life time of 41.4 and 76.6 μs. The lifetime on the microsecond scale coupled with a large Stokes shift (~10,000 cm−1) suggests a spin-forbidden triplet excited state. Since the dtp ligands are not emissive in both DCM and solid state, the emission origin of (Na)[Ag9(S){S2P(OEt)2}8] can be reasonably inferred as a 3LMCT charge transfer.

Absorption (green, ε = 8760 (L mol−1cm−1)) and photoluminescence spectra (excitation, red; emission, orange) of 1 at 77 K (Color figure online)
The absorption maximum red-shifts to ~400 nm in compound 2 in comparison with its sulfur homologue, 1. While a similar emission spectrum centered at ~580 nm was observed at 77 K, a low energy excitation peak at ~ 450 nm besides the one centered at 395 nm was also revealed (Table 2). Both lower energy absorption and excitation bands identified in 2 are in line with the easier oxidation of selenium as compared to sulfur, an indicator of chalcogen-to-silver charge transfer [56]. A triple-exponential behavior with life time of 6.0, 10.6, and 120.0 μs recorded at 77 K for 2 in powder forms suggests a triplet excited state, hence phosphorescence in nature.
Theoretical Investigation
Full geometry optimizations (see computational details) were performed by DFT calculations on compounds 1 and 1′, the later defined as the model [Ag9(S){S2P(OH)2}8]−. We have shown previously that similar substituent simplifications on the dtp and dsep ligands do not modify significantly the computed results in terms of cluster structure and properties, but allow considerable sparing of computer time and space [21, 56, 57]. The experimental crystal structure of 1 is of C 2 symmetry, but only if the 50 % disorder on the Ag5 position is considered (see above). Localizing Ag5 on one of its two disordered positions lowers the symmetry to C 1 (Ag5 coordinated to S9, S8, S8A and S4 or, in its 180°-rotated image, to S9, S8, S8A and S4A). The optimized geometry of 1 was found to be of C 1 symmetry and similar in topology to its X-ray structure, i.e., not that far from C 2 symmetry if not considering the Ag5 position and environment. Relevant energetic and metrical data are given in Table 3.
As usually found at the considered level of calculations which do not take properly into account the metallophilic interactions, the optimized cluster cage is somehow expanded (by ~5 %) when compared to the experimental structure of 1. Interestingly, a geometry optimization of 1 under the C 2 symmetry constraint yielded a structure (Table 3) which is only 0.6 kcal/mol higher in total energy (isoenergetic in free energy) and is associated with a unique (and weak) vibrational imaginary frequency of 17i cm−1. Such small values lie within the range of computational accuracy and cannot ascertain the proper symmetry group of 1 in vacuum but suggest that there is a very flat energy potential surface associated with the displacement of Ag5 near the “C 2 axis”. This displacement can be described as an oscillation between two structures in which Ag5 is alternatively bonded to S4 and S4A and going through an intermediate geometry in which it is bonded to both S4 and S4A. This description is fully consistent with the disordered Ag5 position in the crystal structure. To ascertain this view, similar calculations were performed on the simplified model 1′. It turns out that, in the case of 1′, both C 1 and C 2 were found nearly isoenergetic in total and free energy (see Table 3), the “computationally true” minimum being of C 2 symmetry. The Ag5 atom in this C 2 “minimum” is tetracoordinated (not considering bonding with the encapsulated S9 sulfur), lying in an approximate square-planar configuration and with two short and two long Ag–S distances of 2.648 Å (Ag5–S8) and 2.851 Å (Ag5–S4), respectively. An interesting structural feature of this C 2 minimum is that the Ag3 and Ag3A atoms which are not bonded to the encapsulated S9 sulfide in the X-ray structure of 1 (Table 3) lie now much closer to it (2.996 Å). Even, the computed Wiberg bond indices suggest stronger Ag1–S9 and Ag1A–S9 bonds than the Ag5–S9 bond, for example. Actually, the same tendency for nonacoordination is conserved in the C 1 geometry of 1′ (Table 3). These results are consistent with the view of a rather mobile encapsulated S9 sulfide, rattling around the center of the large and unsymmetrical Ag9 cage. The covalent interaction between S9 and the Ag9 cage occurs between its occupied 3s and 3p AOs and proper combinations of the accepting 5s/5p hybrids on the nine AgI centers. It is associated with significant electron transfer from the sulfide to the Ag+ centers. Consistently, the NAO configuration of S9 in 1 is 3s1.84 3p5.59, corresponding to an atomic charge significantly lower than −2 (−1.43). Similar values were found for 1′. Full geometry optimizations were also carried out on 2 and its simplified model [Ag9(Se){Se2P(OH)2}8]− (2′). Contrarily to that of 1, in its crystal structure cluster 2 has no symmetry element involving a disordered metal position, despite of the fact that one of the silver positions (Ag2) is also disordered. The optimized geometry of 2 (C 1 symmetry) was found to be rather similar to its X-ray structure, with some differences however (Table 4). The tricoordination of all the silver atoms except Ag8 is confirmed, but in the case of Ag8, the additional long experimental Ag8–Se1 contact (3.293 Å (X-ray, see above)) is shorter (3.125 Å (DFT)) and its computed Wiberg index (0.094) indicates a weak, but real, bond. Another difference comes from the ordering of the nine Ag–Se17 distances. In the optimized structure, the larger separations concern Ag4 and Ag9, the latter metal belonging to the distorted trigonal prism. Anyway, all the Ag–Se17 Wiberg indices are consistent with some bonding interactions, although weaker in the case of the longer contacts (Table 4). A geometry optimization of 2 was also carried out under the C 2 symmetry constraint in the same way as it was done for 1 (see above). This optimized structure was found to lie only 0.9 kcal/mol in total energy (4.3 kcal/mol in free energy) higher than the C 1 one described just above. Calculations on the simplified model 2′ reproduce most of the results obtained with 2 (Table 4). As in the case of 1 and 1′, the longer distances between the encapsulated chalcogenide and the metal atoms are not the same as in the X-ray structure, suggesting easy chalcogenide motion inside its cavity.
The computed proton NMR chemical shifts of 2 (averaged values: 4.3 and 1.3) are in an excellent agreement with their experimental counterparts (4.20 and 1.33 ppm, respectively). Furthermore, the computed 77Se NMR chemical shifts of the encapsulated selenide (−1769.4 ppm) is also in agreement with the experimental value (−1702.7 ppm).
As a whole, the computational results described above indicate that 1 and 2 adopt the same molecular structure, which is the result of a compromise between various geometrical constraints (mainly related to metal–ligand bonding), offering to the encapsulated chalcogenide a rather large cavity size in which it can freely rattle. As a result the whole molecular shape is rather flexible.
The UV–vis transitions of 1 and 2 were calculated by TDDFT. Their simulated absorption spectra [58] are shown in Fig. S11. The transitions of lowest energy computed for 1, one at 389 nm (HOMO → LUMO) and one at 367 nm (HOMO-1 → LUMO) are in a fairly good agreement with the experimental values (Table 2). These two transitions are associated with a sulphur (ligand + encapsulated) → silver charge transfer. The lowest triplet state corresponds to the single-occupation of the LUMO which is mainly of metal character. The computed emission associated with this triplet state (542 nm) is in good agreement with the experimental one (587 nm). A similar computed behaviour for 2 yielded an emission transition at 611 nm (Table 2).
Conclusions
In this contribution, the synthesis, structure, characterization and bonding analysis of the first monoanionic silver dichalcogenophosph(in)ate clusters, namely [Ag9(S){S2P(OEt)2}8]− (1) and [Ag9(Se){Se2P(OEt)2}8]− (2) are reported. They exhibit original shapes and topologies, with a highly distorted chalcogenide-centered Ag9 tricapped trigonal prism. The coordination of the encapsulated chalcogenide can be in first approximation considered as hepta-coordinate, although bonding interactions exist with all the metal atoms. Calculations predict that this chalcogenide should move nearly freely around the center of its large cage, which is itself fairly deformable. This peculiar bonding of Se2− in 2 is associated with an extremely up-field 77Se NMR chemical shift (−1702.7 ppm). Both compounds are luminescent and the yellow emission is assigned by TDDFT calculations to be associated with a 3LMCT excited state involving both types of chalcogen atoms.
Electronic Supplementary Information (ESI)
Solution NMR, ESI mass spectra, TGA, and DFT optimized structures. The structures reported herein have been deposited at the Cambridge Crystallographic Data Centre, CCDC 1443865 (1) and 1443866 (2). For ESI and crystallographic data in CIF or other electronic format see doi:10.1039/x0xx00000x.
References
M. C. Gimeno and A. Laguna in J. A. McCleverty and T. J. Meyer (eds.), Comprehensive Coordination Chemistry II, vol. 6 (Elsevier, Oxford, 2004), pp. 911–990.
W. E. van Zyl (2010). Comments Inorg. Chem. 31, 13.
C. Latouche, C. W. Liu, and J.-Y. Saillard (2014). J. Clust. Sci. 25, 147.
X.-Y. Tang, H.-X. Li, J.-X. Chen, Z.-G. Ren, and J.-P. Lang (2008). Coord. Chem. Rev. 252, 2026.
O. Fuhr, S. Dehnen, and D. Fenske (2013). Chem. Soc. Rev. 42, 1871.
G. Li, Z. Lei, and Q.-M. Wang (2010). J. Am. Chem. Soc. 132, 17678.
Y. Shen, S. Liu, P. Sun, C. Tang, and D. Jia (2016). Eur. J. Inorg. Chem. 3380.
R. S. Dhayal, Y.-R. Lin, J.-H. Liao, Y.-J. Chen, Y.-C. Liu, M.-H. Chiang, S. Kahlal, J.-Y. Saillard, and C. W. Liu (2016). Chem. Eur. J. 22, 9943.
R. S. Dhayal, J.-H. Liao, Y.-C. Liu, M.-H. Chiang, S. Kahlal, J.-Y. Saillard, and C. W. Liu (2015). Angew. Chem. Int. Ed. 54, 3702.
A. Panneerselvam, C. Q. Nguyen, M. A. Malik, P. O’Brien, and J. Raftery (2009). J. Mater. Chem. 19, 419.
C. W. Liu, J. T. Pitts, and J. P. Fackler Jr. (1997). Polyhedron 16, 3899.
A. V. Ivanov, S. A. Zinkin, A. V. Gerasimenko, A. N. Antsutkin, and W. Forsling (2007). Koord. Khim 33, 22.
C. W. Liu, I.-J. Shang, C.-M. Hung, J.-C. Wang and T.-C. Keng (2002). J. Chem. Soc., Dalton Trans. 1974.
W. Kuchen and H. Mayatepek (1968). Chem. Ber. 101, 3454.
T. S. Lobana, J.-C. Wang, and C. W. Liu (2007). Coord. Chem. Rev. 251, 91.
C. W. Liu, I.-J. Shang, J.-C. Wang, and T.-C. Keng (1999). Chem. Commun. 995, 129.
K. Matsumoto, R. Tanaka, R. Shimomura, and Y. Nakao (2000). Inorg. Chim. Acta. 304, 293.
J.-H. Liao, H.-W. Chang, Y.-J. Li, C.-S. Fang, B. Sarkar, W. E. van Zyl, and C. W. Liu (2014). Dalton Trans. 43, 12380.
Y.-B. Zhu, S.-F. Lu, X.-Y. Huang, Z.-X. Haung, and Q.-J. Wu (2004). Chin. J. Chem. 22, 673.
M. Shafaei-Fallah, C. E. Anson, D. Fenske, and A. Rothenberger (2005). Dalton Trans. 13, 2300–2304.
C. W. Liu, H.-W. Chang, P.-K. Liao, C.-S. Fang, J.-Y. Saillard, and S. Kahlal (2011). J. Clust. Sci. 22, 381.
K. Bowman-James, A. Bianchi, and E. Garcia-Espana Anion Coordination Chemistry (Wiley, Weinheim, 2011).
D. L. Klayman and T. S. Griffin (1973). J. Am. Chem. Soc. 95, 197.
C. W. Liu, H.-W. Chang, C.-S. Fang, B. Sarkar, and J.-C. Wang (2010). Chem. Commun. 46, 4571.
SMART V4.043: Software for the CCD Detector System, Bruker Analytical X-ray System (Madison, 1995)
SAINT V4.043: Software for the CCD Detector System, Bruker Analytical X-ray System (Madison, 1995)
G. M. Sheldrick SADABS (University of Göttingen, Göttingen, 1996).
SHELXL v6.14 (PC Version): Program Library for Structure Solution and Molecular Graphics, Bruker Analytical X-ray System (Madison, 2001)
A. Desireddy, B. E. Conn, J. Guo, B. Yoon, R. N. Barnett, B. M. Monahan, K. Kirschbaum, W. P. Griffith, R. L. Whetten, U. Landman, and T. P. Bigioni (2013). Nature 501, 399.
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox Gaussian 09, Revision A.1 (Gaussian Inc., Wallingford, 2009).
C. Adamo and V. Barone (1999). J. Chem. Phys. 110, 6158.
T. H. Dunning Jr. and P. J. Hay in H. F. Schaeffer (ed.), Methods of Electronic Structure Theory (Plenum Press, New York, 1977).
P. J. Hay and W. R. Wadt (1985). J. Chem. Phys. 82, 270.
P. J. Hay and W. R. Wadt (1985). J. Chem. Phys. 82, 284.
P. J. Hay and W. R. Wadt (1985). J. Chem. Phys. 82, 299.
A. Schafer, H. Horn, and R. Ahlrichs (1992). J. Chem. Phys. 97, 2571.
E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, and F. Weinhold, (Theoretical Chemistry Institute, University of Wisconsin, Madison, 2001). http://www.chem.wisc.edu/~nbo5
S. I. Gorelsky, AOMix program. http://www.sg-chem.net/
F. London (1937). J. Phys. Radium 8, 397.
R. McWeeny (1962). Phys. Rev. 126, 1028.
R. Ditchfield (1974). Mol. Phys. 27, 789.
J. K. Dodds, R. McWeeny, and A. J. Sadlej (1977). Mol. Phys. 34, 1779.
K. Wolinski, J. F. Hinton, and P. Pulay (1990). J. Am. Chem. Soc. 112, 8251.
F. Weigend and R. Ahlrichs (2005). Phys. Chem. Chem. Phys. 7, 3297.
J.-H. Liao, H.-W. Chang, H.-C. You, C.-S. Fang, and C. W. Liu (2011). Inorg. Chem. 50, 2070.
H.-W. Chang, J.-H. Liao, B. Li, Y.-J. Chen, and C. W. Liu (2014). J. Struc. Chem. 55, 1426.
H. Duddeck (1995). Selenium-77 nuclear magnetic resonance spectroscopy. Prog. Nucl. Magn. Reson. Spectrosc. 27, 1–323.
X.-J. Wang, T. Langetepe, C. Persau, B.-S. Kang, G. M. Sheldrick, and D. Fenske (2002). Angew. Chem. Int. Ed. 41, 3818.
J.-X. Chen, Q.-F. Xu, Y. Xu, Y. Zhang, Z.-N. Chen, and J.-P. Lang (2004). J. Organomet. Chem. 689, 1071.
C. W. Liu, C. J. McNeal, and J. P. Fackler Jr. (1996). J Clust. Sci. 7, 385.
J.-X. Chen, Q.-F. Xu, Y. Xu, Y. Zhang, Z.-N. Chen, and J.-P. Lang (2004). Eur. J. Inorg. Chem. 21, 4247–4252.
C. E. Anson, A. Eichhöfer, I. Issac, D. Fenske, O. Fuhr, P. Sevillano, C. Persau, D. Stalke, and J. Zhang (2008). Angew. Chem. Int. Ed. 47, 1326.
J. D. Corbett (1997). Struct. Bonding 87, 157.
A. Schnepf (2003). Angew. Chem. Int. Ed. 42, 2624.
H. Schmidbaur and A. Schier (2015). Angew. Chem. Int. Ed. 54, 746.
Y.-J. Li, C. Latouche, S. Kahlal, J.-H. Liao, R. S. Dhayal, J.-Y. Saillard, and C. W. Liu (2012). Inorg. Chem. 51, 7439.
J.-H. Liao, C. Latouche, B. Li, S. Kahlal, J.-Y. Saillard, and C. W. Liu (2014). Inorg. Chem. 53, 2260.
S. I. Gorelsky, SWizard program, revision 4.5. http://www.sg-chem.net
Acknowledgments
This research was supported by Ministry of Science and Technology of Taiwan (MOST 103-2113-M-259-003). The French national computer centers GENCI-CINES and GENCI-IDRISS are acknowledged for computational resources (Grant c2015087367).
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Chang, HW., Shiu, RY., Fang, CS. et al. A Sulfide (Selenide)-Centered Nonanuclear Silver Cluster: A Distorted and Flexible Tricapped Trigonal Prismatic Ag9 Framework. J Clust Sci 28, 679–694 (2017). https://doi.org/10.1007/s10876-016-1076-x
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10876-016-1076-x