
Abstract
In our attempts to identify probable intermediates in the preparation of anionic silver hydride cluster [Ag8(H){S2CC(CN)2}6]5− under the condition of excess NaBH4, we isolated and analyzed a new cluster compound, namely, [Bu4N]6[Ag7(H){S2CC(CN)2}6]. We report here the synthesis, crystal structure, characterizations by various spectroscopic (UV–Vis, IR, multinuclear NMR, elemental analyses) techniques and the luminescent properties of this cluster. Its X-ray crystal structure reveals that the cluster contains a distorted; tricapped tetrahedral silver core enclosed an interstitial hydride anion and is surrounded by six 1,1-dicyanoethylene-2,2-dithiolate ([S2CC(CN)2]2−, i-MNT) ligands. The presence of hydride inside the heptanuclear silver cluster is unequivocally certified by both 1H and 109Ag NMR spectroscopies. The title compound represents the first atomic precisely anionic Ag7-H skeleton with a non-disordered, tricapped tetrahedral silver framework.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Coinage metal clusters are of great interests for their structural versatility and broad applications [1]. Typically, coinage metal-hydride clusters [2,3,4], polynuclear clusters [5, 6], anion-templated high-nuclearity clusters [7], alloy clusters [8,9,10], as well as nanoclusters [9, 11] are remarkable architectures in this area. Among them coinage metal-hydride clusters are important intermediates prior to the formation of metal nanoclusters or nanoparticles under wet chemical reductions methods and are far from being accurately determined due to the difficulties of their preparation, analysis and characterizations.
With the development of efficient means (notably by neutron diffraction) and more refined theoretical advances, molecular structures of coinage metal-hydride clusters (MnHxLy, M = Cu/Ag/Au, L = ligand, n ≥ 3, x ≥ 1, y ≥ 1) have been possible to precisely unravel although it is still quite challenging. In contrast to silver and gold, copper hydride chemistry is the fastest developed province and copper hydride clusters have been structurally studied extensively from the trimeric [Cu3(μ3-H)(dcpm)3]2+ (dcpm = 1,1-bis(dicyclohexylphosphino)methane) [12], the tetrahedral [Cu4(μ4-H)(μ2-X)2(PPh2Py)4]+ clusters (X = Cl, Br; Py = pyridyl) [13] and the hexameric [CuH(PR3)]6 {R = C6H5, p-MeC6H4, O-i-Pr} clusters to a series of dichalcogen (S or Se) donor ligand protected polyhydrido copper clusters [3, 4] or (phosphine)copper polyhydrides [14]. For gold hydride chemistry, theoretical advances proposed that Au atoms at a low-coordination site of Au clusters can be replaced with a H atom while retaining the structural motif and electronic structure [15]. Till recently, only one hydride-doped gold superatom (Au9H)2+ was observed by mass spectrometry and NMR spectroscopy with its structure predicted by theoretical calculation [15, 16]. With advances in copper and gold hydride clusters, researchers began to design and synthesis of ligand-stabilized silver hydride clusters due to the potential for silver hydrides to mediate important transformations [2] and to the key intermediates for the fabrication of silver nanoparticles [17, 18]. In general, Ag-H clusters mainly fall into two categories: (phosphine)silver hydrides and clusters supported by dichalcogen-based donor ligands. Isolable (phosphine)silver hydride cluster [Ag3(µ3-H)(Cl)(dppm)3]BF4 was first reported in 2013 by O’Hair team under the guidance of the mass spectrometry results [19]. In the following, they reported and structurally described the di-cationic [Ag3(μ3-H)(dppm)3](BF4)2 [20] and the hydride- and borohydride-bridged trinuclear silver cluster cation, [Ag3(μ3-H)(μ3-BH4)(dppm)3]+, through the treatment of AgBF4 with dppm and excess borohydride in CH3CN, at or below −10 °C [21]. Although several Ag polyhydride clusters, such as [Ag18H16(PPh3)10]2+, [Ag25H22(DPPE)8]3+ (DPPE = 1,2-bis(diphenylphosphino)ethane), and [Ag26H22(TFPP)13]2+ (TFPP = tris(4-fluorophenyl)phosphine), were confirmed by mass spectrometry and NMR spectroscopy [22], only one polyhydrido silver cluster; namely [Ag6H4(dppm)4(OAc)2], was structurally characterized by X-ray diffraction analysis [23]. On the other hand, our group successfully isolated a number of AgI–hydride clusters stabilized by bridging dichalcogen-based donor ligands in the last decade. These include cationic [Ag8(µ4-H){E2P(OR)2}6]+ (E = S, Se) [24], [Ag11(H)(S2CNR2)9]+ [25], neutral [Ag7(H){E2P(OR)2}6] (E = Se, S) [17] and anionic [Ag8(H){S2CC(CN)2}6]5− [26] clusters. The latter can be synthesized from an empty silver cubic cluster precursor [Ag8{S2CC(CN)2}6]4− [28] by hydride insertion reaction. It seems that cationic silver clusters are the easiest in the hydride insertion, followed by the neutral ones, with the anionic silver clusters the least possible. From the point of view of ionic bonding it is disfavored and synthetically unachievable. Although we successfully realized the H−insertion reaction of the empty anionic Ag clusters[Ag8{S2CC(CN)2}6]4−, it is bigger challenging for inserting H−into more negative ones, let alone getting their structures described.
As part of our continuous work, we attempted to isolate the possible new products in the preparation of [Ag8(H){S2CC(CN)2}6]5− [26] by using excess amount of NaBH4. Herein we report the synthesis and detailed characterizations of hexa-anionic Ag7 hydride cluster supported by i-MNT ligand (i-MNT = [{S2CC(CN)2}6]2−). The presence of hydride inside the heptanuclear silver cluster is unequivocally confirmed by both 1H and 109Ag NMR spectroscopies. To the best of our knowledge, this work represents the first anionic Ag7-H skeleton with a non-disordered, tricapped tetrahedral silver framework.
Experimental
All chemicals were purchased from commercial sources and used as received. Solvents were purified following standard protocols. All the reactions were performed in oven-dried Schlenk glassware and carried out under a N2 atmosphere by using standard inert atmosphere techniques. [Bu4N]5[Ag8(H){S2CC(CN)2}6] (2H) [26] was prepared as previously reported. The elemental analyses were done using a PerkinElmer 2400 CHN analyzer. NMR spectra were recorded on a Bruker Advance DPX300 FT-NMR spectrometer that operates at 300 MHz while recording 1H, 46.1 MHz for 2H, and 75.5 MHz for 13C NMR. The 109Ag NMR spectra were recorded on a Bruker Advance II 600NMR that operates at 27.9 MHz. The chemical shift (δ) and coupling constant (J) are reported in parts per million and hertz, respectively. Melting points were measured by using a Fargo MP-2D melting point apparatus. All infrared spectra were recorded on a Bruker Optics FTIR TENSOR 27 spectrometer (180–4000 cm−1) at 20 °C using CsI plates. UV–visible spectra were measured on a PerkinElmer Lambda 750 spectrophotometer.
Synthesis of [Bu4N]6[Ag7(H){S2CC(CN)2}6], 3H
Method (a): To a solution of 2H (0.3 g, 0.103 mmol) in THF (50 mL) was added (Bu4N)(BH4) (0.027 g, 0.103 mmol), and the resulting pale-green solution was stirred at -20 °C for 3 h. The solution was filtered to get rid of any solids, and the filtrate was evaporated to dryness under vacuum. It was washed with MeOH and then the solid was dried under vacuum to give 3H as a yellow-green powder. Yield: 0.213 g (68%).
Method (b): AgNO3 (0.095 g, 0.56 mmol) and [Bu4N]2[S2CC(CN)2] (0.3 g, 0.48 mmol) were charged in a 100 mL flask and 40 mL of THF was added to it. The solution was stirred at RT for 1 h to form the yellow solution. (Bu4N)(BH4) (0.021 g, 0.08 mmol) was added to this solution and stirred for 3 h. Then it was filtered to get rid of any solids, and the filtrate was evaporated to dryness under vacuum. It was washed with MeOH and then the solid was dried under vacuum to give 3H as a yellow-green powder. Yield: 0.188 g (77%). Mp: 157 °C decomposed. Anal calcd for Ag7H217C120N18S12: C 47.22; H 7.17; N 8.26. Found: C 47.22; H 7.18; N 8.18%. 1H NMR (300 MHz, acetone-d6): 1.00 (t, 3JHH = 7.4 Hz, 60H, CH3), 1.47 (m, 40H, CH2), 1.82(m, 40H, CH2), 3.39(t, 3JHH = 7.4 Hz, 40H, CH2), 9.88(octet, JHAg = 37.32 Hz, 1H). 13C NMR (75.5 MHz, acetone-d6): 13.3, 19.8, 23.9, 59.0, 74.9, 120.0, 211.5 ppm. IR, cm−1: υ(CN), 2195(s); υ(C = C), 1470 (s); υ(C-S), 935(s); υ(Ag–S), 226(m), 191 (m). UV–Vis [λmax in nm, (ε in M−1cm−1)], 292(55,000), 368(191,000).
Synthesis of [Bu4N]6[Ag7(D){S2CC(CN)2}6], 3D
This was synthesized in a similar procedure described for 3H by replacing using (Bu4N)(BH4) with (Bu4N)(BD4). Yield: 0.173 g (71%). Mp: 157 °C decomposed. Anal calcd for Ag7H216D1C120N18S12: C 47.21; H 7.20; N 8.26. Found: C 47.01; H 7.23; N 8.21%. 1H NMR (300 MHz, acetone-d6): 1.00 (t, 3JHH = 7.4 Hz, 60H, CH3), 1.47 (m, 40H, CH2), 1.82(m, 40H, CH2), 3.39(t, 3JHH = 7.4 Hz, 40H, CH2). 2H NMR (acetone, ppm): 9.87 (bs, 1D). 13C NMR (75.5 MHz, acetone-d6): 13.3, 19.8, 23.9, 59.0, 74.9, 120.0, 211.5 ppm. IR, cm−1: υ(CN), 2195(s); υ(C=C), 14670 (s); υ(C–S), 933(s); υ(Ag–S), 226(m), 192 (m). UV–Vis [λmax in nm, (ε in M−1cm−1)], 292(55,000), 368(191,000).
X-ray Structure Determination
Crystal of 3H suitable for X-ray diffraction experiment was obtained by diffusing hexane into an acetone solution of the compound. Crystal was mounted on the tips of glass fibers with epoxy resin. Data was collected on a Bruker APEXII CCD diffractometer using graphite monochromated Mo kα radiation (κ = 0.71073 Å). Absorption corrections for the area detector was performed with the program SADABS. Structure was solved by direct methods and was refined against the least-squares methods on F2 with the SHELXL-97 package, incorporated in SHELXTL/PC V5.10. Several carbon atoms of the tetrabutylammonium cations in 3H were found disordered and thus were refined isotropically. Restraints to the alkyl side chains were applied in some cases, and they were described in the cif. H atoms on the tetrabutylammonium cations were added at idealized positions except for the disorder carbon atoms of the alkyl side chains. The central hydride was located from the different Fourier map and refined isotropically. Crystallographic detail for compound 3H is given in Table 1 and selected bond lengths in Table 2.
Results and Discussions
Synthesis and Spectroscopy
The reaction of [Bu4N]5[Ag8(X){S2CC(CN)2}6] (X = H, 2H; D, 2D) and [Bu4N][BH4] ([Bu4N][BD4]) in an 1:1 molar ratio at − 20 °C for 3 h, produced [Bu4N]6[Ag7(H)(i-MNT)6], 3H, and [Bu4N]6[Ag7(D)(i-MNT)6], 3D, respectively. It can also be prepared by the reaction of AgNO3, [Bu4N]2[S2CC(CN)2], and [Bu4N][BH4] in a 7:6:1 molar ratio in THF solution (Scheme 1).

Reactivity of hydride–centered silver clusters supported by i-MNT ligands (Note: Here we use C1, C2-H and C3H to represent the anionic clusters of [Bu4N]4[Ag8(i-MNT)6], 1, [Bu4N]5[Ag8(H)(i-MNT)6], 2H and [Bu4N]6[Ag7(H)(i-MNT)6], 3H, respectively)
The UV–Vis spectrum of 3H shows two absorption bands at 368(191,000 M−1 cm−1) and 292(55,000 M−1 cm−1) in CH2Cl2 that are almost the same as those displayed in the compound 2H. The IR spectrum of 3H displays [v(CN)], [v(C=C)], and [v(C–S)] stretching bands at 2195, 1470, and 935 cm−1, respectively, which are similar in comparison with the hydride-centered Ag8 cluster, 2H (vide supra). Compound 3H shows two Ag–S bands at 226 and 191 cm−1, almost identical to those in the compound 2H. However, we have not been able to obtain evidences for the μ4-H (or μ4-D) ligand in two silver hydride clusters in the IR spectrum. The M–H stretching frequency of those clusters may be either too weak or in a frequency region below 1500 cm−1 where IR bands from the n-butylammoniun salts mask it.
The 1H NMR spectrum of 3H displays chemical shifts corresponding to the butyl group, and an octet of octets resonance centered at 9.88 ppm that integrate to 1H relative to 72 methyl protons of a total of six tetrabutylammonium cations. Thus, the peak can be reasonably assigned to the hydride resonance. Surprisingly, at room temperature this peak splits into an octet of octets peak, compared to the compound 2H, which the nonet peak split into nonet of nonets at 263 K [26]. Its line shape did not change upon a further decrease of the temperature to 243 K (Fig. 1a). It then became broad until 213 K. Overall, there is about a ~ 0.1 ppm upfield shift for the hydride resonance in the variable-temperature 1H NMR spectra. Compound 3D exhibits a peak at 9.87 ppm in the 2H NMR spectrum that originate from the entrapped deuteride (Fig. 1b). A band of 15 lines (1J1H–107Ag = 34.5 Hz) in the 1H{109Ag} spectrum is observed, where the intensities of four outermost peaks are too low to be detected (Fig. 1c). A doublet peak at 1105.2 ppm (1J1H–109Ag = 39.7 Hz) in the 109Ag NMR spectrum (Fig. 1d), slightly upfield shift by the comparison with the corresponding resonance of the Ag8H cluster (1136.1 ppm) [26], and somewhat downfield shift by the contrast with the parallel resonance of Ag8 cube (1056.1 ppm), was detected. The NMR spectroscopic data clearly suggest that the hydirde is coupled to seven, magnetically equivalent silver nuclei on the NMR time scale. Finally, the octet of octets split referred to the superposition of the spectra of the different isotopomers was confirmed by the simulated spectrum of an AX7X’7 (X, 107Ag; X’, 109Ag) spin system (Fig. 2). Coupling constants of J1H–107Ag (34.5 Hz) and J1H–109Ag (39.7 Hz), respectively, were assumed. Presumably, the fast interchange between the capping and vertex silver atoms in compound 3H has all seven silver atoms to become equivalent on the NMR time scale [26]. This sort of dynamical behavior has been theoretically proved by metadynamics simulation [27]. The 13C NMR spectrum of 3H shows one resonance at 211.5 ppm, which is assigned to the chemical shifts of CS2 moiety and slightly downfield shift compared to the corresponding resonances of compound 2H (207.0 ppm) and compound 1 (197 ppm), respectively.

a VT (293 K ~ 213 K) 1H NMR spectra of 3H; b2H NMR spectrum of 3D; c1H{109Ag} NMR spectrum of 3H; d109Ag NMR of 3H with the magnified spectrum shown in the inset

Experimental 1H NMR spectrum of 3H in d-acetone at RT (bottom), and Simulated 1H NMR spectrum of 3H (top)
It is worthwhile to mention that the hydride–centered (or deuteride–centered) Ag8 clusters (2H and 2D) surrounded by six i-MNT can be isolated from the reaction of empty cubic Ag8 cluster with (Bu4N)(BH4) [or (Bu4N)(BD4)] within 3 h, respectively. Further reaction of compound 2H (or 2D) with one equiv. of BH4– (or BD4–) anion excised a silver atom to produce the Ag7 cluster, 3H (or 3D) (Scheme 1). Compound 3H (or 3D) can be easily converted to 2H (or 2D) by adding 1 equiv of AgI salt to the Ag7 cluster in solution. Moreover, compound 3H is chemically stable since it can be stored in solid state for months at room temperature; however, when compound 3H is dissolved in solution, such as acetone or dichloromethane, it is easily converted to 2H via capture of an AgI ion, which can be monitored by 1H NMR spectroscopy. That suggested that compound 3H is highly reactive towards availability of AgI ion to convert into compound 2H. Besides, we have also test the reaction of compound 3H with CuI salt in order to form a hypothetic, heterometallic cluster, [Ag7Cu(H)(i-MNT)6]5–. Unfortunately, after addition of CuI salts into the acetone solution of compound 3H, the solution turns to brown color immediately, and there is only partially decomposed the compound 3H, which is revealed from the monitored experiment via 1H NMR spectroscopy.
Since the reaction of compound 2H with BH4– anion can yield 3H via loss a AgI ion, thus it would be interesting to know the further reaction products of cluster 3H with more equiv. of borohydrides. Is it the case that one more AgI ion will be lost through the addition of borohydrides? However, we have failed to isolate any new clusters from the reaction with borohydrides, but only yielded black precipitates instead. This work is similar to those of [Ag7(H){E2P(OR)2}6], which was suggested as an intermediate for silver nanoparticle growth, reported by our group in 2013 [17].
X-ray Crystallography
[Bu4N]6[Ag7(H){S2CC(CN)2}6], 3H
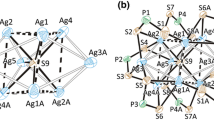
The hexa-anionic, heptanuclear silver complex 3H crystallizes in triclinic P(-)1 space group, with one cluster and six tetrabutylammonium cations per asymmetric unit. The cluster contains a distorted; tricapped tetrahedral silver core centered about an interstitial hydride anion and is surrounded by six i-MNT ligands (Fig. 3). Compared with the tetracapped tetrahedral silver skeleton of 2H, this tricapped tetrahedral silver skeleton 3H (Fig. 3) appears to arise from losing one capping silver atom of 2H. The central tetrahedron, which approaches a triangular pyramid arising from a pseudo-C3 elongation like those observed in [Cu7(H){S2C(aza-15-crown-5)}6] [29]. The edge of Ag4 triangular pyramid comprises Ag1, Ag3, Ag4, and Ag7 atoms (abbreviated as Agv), and the three capping atoms are Ag2, Ag5, and Ag6 (abbreviated as Agcap). The basal plane of atom Ag1, Ag3, and Ag4 lacks a capping atom. Although the elongated edge of the pyramidal range from 3.9425(11) to 4.019(1) Å, the average length of the basal plane edge is 2.9604(10) Å, which is shorter than the sum of the van der Waals radii for two silver atoms, 3.4 Å, and is longer than Agv–Agcap [av. 2.8973(10) Å]. Of the six i-MNT ligands only three achieve a tetrametallic, tetraconnective (μ2–S; μ2–S) bonding mode and are located on the top of Ag4 butterflies where hinge positions are elongated edges of the triangular pyramid and wingtips are capping Ag atoms. The other three i-MNT ligands exist in the trimetallic, triconnective (μ2-S; μ1-S) bonding mode, which are located about the uncapped Ag3 basal plane linking two vertices and one capping Ag atom. Thus Ag atoms in the basal plane present a distorted tetrahedral S3H coordination. The Agv–S distances in 3H [av. 2.637(3) Å] are slightly longer than Agcap–S [av. 2.528(2) Å]. Both distance are comparable with those observed in the hydride-centered octanuclear silver cluster [Ag8(H)(i-MNT)6]5– [26]. Furthermore, the average S…S bite distance in 3H [av. 3.062(3) Å] is also comparable to those in compound 2H [av. 3.096(4) Å] [26]. Distance between the interstitial hydride and three, directly linked silver atoms are in the range of 1.91(5)–2.11(6) Å. It is noteworthy that although six ammonium cations are around one C3H, there are no remarkable anion-cation contacts observed via checking the short contacts between Bu4N+ and C3H.

a A perspective view of the cluster anion [Ag4(μ4-H)(μ3-Ag)3{S2CC(CN)2}6]6− in 3H (30% probability ellipsoids). b Schematic drawing of 3H. c The Ag7H core in 3H
Absorption Spectra and Luminescence Properties
The electronic spectra of these three silver clusters, 1, 2H and 3H, were measured in dichloromethane solution, and the data are given in Table 3. The UV–Vis absorption spectra of the 1, 2H and 3H in CH2Cl2 at 298 K are in the range 250–600 nm. In general, the electronic absorption spectrum of these three clusters are quite similar to those of [Ag4(μ-dppm)(μ4-i-MNT)2] [30] and [{Au(PPh3)}2(μ-i-MNT)] [31]. The absorption maxima of 1 ~ 3 at ca. 363–367 nm, having extinction coefficients of 133,000–191,000 dm3mol−1cm−1, quite match with that of [{Au(PPh3)}2(μ-i-MNT)], which are mainly mnt2– ligand–centered transition [30]. The high energy absorptions at ca. 270–294 nm for 1 ~ 3 in dichloromethane, are suggestive of metal–perturbed intraligand transitions. All three silver clusters show luminescence in degassed solid samples at 77 K. The excitation and emission spectral data are summarized at Table 3. Excitation of solid samples of 1 ~ 2 with λ > 330 nm resulted in emission, with lifetimes of 0.5 ~ 5.5 μs, which is suggested that it is from a spin-forbidden triplet emissive state. The emission spectrum of 2H shows a structureless band at 520 nm in solid state at 77 K. In 3H, the emission spectrum displays a structureless band at 561 nm in solid state at 77 K, which is red-shift about ~ 40 nm compared to the emission spectrum of 2H (Fig. 4). It is also worthy to note that the empty cubic Ag8 cluster exhibits an intense peak centered at 589 nm, which is red-shift about ~ 30 nm compared with the emission spectrum of 3H. With reference to the previous spectroscopic studies on the polynuclear AgI/CuI clusters, the emission bands at 520 ~ 589 nm in 1 ~ 3 are tentatively assigned as derived from excited states of S → Ag ligand-to-metal charge-transfer (LMCT), and also originated from excited states of a mixture of a ligand-to-metal–metal charge-transfer (LMMCT) and metal-centered (MC) characters. The emission spectra of these two hydride–centered silver clusters, 2 and 3, are also comparable to that of [Ag7(H){S2P(OR)2}6] [32], which exhibits orange emission in solid state (λmax = 581 nm). However, the energies of the solid-state luminescence of the clusters at 77 K follow the order 2H (520 nm) > 3H (561 nm) > 1 (589 nm), which are not clearly understood the emission mechanism, but the possibility of a mixture of ligand-to-metal charge-transfer (LMCT) and metal-centered (ds/sp) could play important roles for the polynuclear d10 metal clusters.

Normalized emission spectra of compound 1 (blue curve, a), 2H (green curve, b), and 3H (red curve, c) in solid state at 77 K (Color figure online)
Conclusions
In conclusion, we describe herein the synthesis of a new Ag7 hydride cluster (3H) supported by dianionic sulfur based ligand. The compound has been characterized by various spectroscopic techniques and single crystal x-ray crystallography. The silver clusters show luminescence in degassed solid samples at 77 K. It is noteworthy that the 3H exhibits an orange emission with ~ 30 nm blue-shift compared to the empty cubic Ag8 cluster in the emission spectrum. To the best of our knowledge,3Hrepresents the first anionic Ag7-H skeleton with a non-disordered, tricapped tetrahedral silver framework.
Electronic Supplementary Information (ESI)
The structures reported herein have been deposited at the Cambridge Crystallographic Data Centre, CCDC 1895700 (3H). For ESI and crystallographic data in CIF.
References
I. Chakraborty and T. Pradeep (2017). Chem. Rev. 117, 8208–8271.
A. J. Jordan, G. Lalic, and J. P. Sadighi (2016). Chem. Rev. 116, 8318–8372.
R. S. Dhayal, W. E. van Zyl, and C. W. Liu (2016). Acc. Chem. Res. 49, 86–95.
R. S. Dhayal, W. E. van Zyl, and C. W. Liu (2019). Dalton Trans. 48, 3531–3538.
P. C. Ford, E. Cariati, and J. Bourassa (1999). Chem. Rev. 99, 3625–3648.
V. W.-W. Yam, K. K.-W. Lo, W. K. Fung, and C. Wang (1998). Coord. Chem. Rev. 171, 17–41.
Q.-M. Wang, Y.-M. Lin, and K.-G. Liu (2015). Acc. Chem. Res. 48, 1570–1579.
S. Hossain, Y. Niihori, L. V. Nair, B. Kumar, W. Kurashige, and Y. Negishi (2018). Acc. Chem. Res. 51, 3114–3124.
S. Sharma, K. K. Chakrahari, J. Saillard, and C. W. Liu (2018). Acc. Chem. Res. 51, 2475–2483.
A. Ghosh, O. F. Mohammed, and O. M. Bakr (2018). Acc. Chem. Res. 51, 3094–3103.
J. Yan, B. K. Teo, and N. F. Zheng (2018). Acc. Chem. Res. 51, 3084–3093.
Z. Mao, J. Huang, C. M. Che, N. Y. Zhu, L. K. Sarana, and Z. Y. Zhou (2005). J. Am. Chem. Soc. 127, 4562–4563.
H.-H. Nie, Y.-Z. Han, Z.-C. Tang, S.-Y. Yang, and B. K. Teo (2018). J. Clust. Sci. 29, 837–846.
J. Li, H. Z. Ma, G. E. Reid, A. J. Edwards, Y. Hong, M. J. White, R. J. Mulder, and A. J. R. O’Hair (2018). Chem. Eur. J. 24, 2070–2074.
S. Takano, S. Hasegawa, M. Suyama, and T. Tsukuda (2018). Acc. Chem. Res. 51, 3074–3083.
S. Takano, H. Hirai, S. Muramatsu, and T. Tsukuda (2018). J. Am. Chem. Soc. 140, 8380–8383.
C. W. Liu, Y. R. Lin, C. Fang, C. Latouche, S. Kahlal, and J. Saillard (2013). Inorg. Chem. 52, 2070–2077.
C. Latouche, C. W. Liu, and J. Saillard (2014). J. Clust. Sci. 25, 147–171.
A. Zavras, G. N. Khairallah, T. U. Connell, J. M. White, A. J. Edwards, P. S. Donnelly, and A. J. R. O’Hair (2013). Angew. Chem. Int. Ed. 52, 8391–8394.
A. Zavras, G. N. Khairallah, T. U. Connell, J. M. White, A. J. Edwards, P. S. Donnelly, and A. J. R. O’Hair (2014). Inorg. Chem. 53, 7429–7437.
A. Zavras, A. Ariafard, G. N. Khairallah, J. M. White, R. J. Mulder, A. J. Canty, and A. J. R. O’Hair (2015). Nanoscale 7, 18129–18137.
M. S. Bootharaju, R. Dey, L. E. Gevers, M. N. Hedhili, J. Basset, and O. M. Bakr (2016). J. Am. Chem. Soc. 138, 13770–13773.
A. W. Cook, T. D. Nguyen, W. R. Buratto, G. Wu, and T. W. Hayton (2016). Inorg. Chem. 55, 12435–12440.
C. W. Liu, H. W. Chang, B. Sarkar, J. Y. Saillard, S. Kahlal, and Y. Y. Wu (2010). Inorg. Chem. 49, 468–475.
C. W. Liu, P. K. Liao, C. S. Fang, J. Y. Saillard, S. Kahlal, and J. C. Wang (2011). Chem. Commun. 47, 5831–5833.
P. K. Liao, K. G. Liu, C. S. Fang, C. W. Liu, J. P. Fackler, and Y. Y. Wu (2011). Inorg. Chem. 50, 8410–8417.
C. Latouche, S. Kahlal, E. Furet, P. K. Liao, Y.-R. Lin, C.-S. Fang, J. Cuny, C. W. Liu, and J.-Y. Saillard (2013). Inorg. Chem. 52, 7752–7765.
P. J. Birker, G. C. Verschoor, (1981) J. Chem. Soc., Chem. Commun. 322-324.
P. K. Liao, C. S. Fang, A. J. Edwards, S. Kahlal, J. Y. Saillard, and C. W. Liu (2012). Inorg. Chem. 51, 6577–6591.
W. Su, M. Hong, R. Cao, J. Chen, D. Wu, H. Liu, and J. Lu (1998). Inorg. Chim. Acta. 267, 313–317.
R. M. Davila, A. Elduque, T. Grant, R. J. Staples, and J. P. Fackler (1993). Inorg. Chem. 32, 1749–1755.
C. W. Liu, H. Chang, C. S. Fang, B. Sarkar, and J. C. Wang (2010). Chem. Commun. 46, 4571–4573.
Acknowledgements
This research was supported by Ministry of Science and Technology of Taiwan (MOST 106-2113-M-259-010). This work was also supported by the National Natural Science Foundation of China (Grand. No. 21601097) and the National First-rate Discipline Construction Project of Ningxia (Chemical Engineering & Technology, NXYLXK2017A04).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Liao, PK., Liu, KG., Fang, CS. et al. [Ag7(H){S2CC(CN)2}6]6−: An Anionic Heptanuclear Silver Hydride Cluster Compound Stabilized by Dithiolate Ligands. J Clust Sci 30, 1185–1193 (2019). https://doi.org/10.1007/s10876-019-01538-3
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10876-019-01538-3