
Abstract
A simple triple resonance NMR experiment that leads to the correlation of the backbone amide resonances of each amino acid residue ‘i’ with that of residues ‘i−1’ and ‘i+1’ in (13C, 15N) labelled intrinsically disordered proteins (IDPs) is presented. The experimental scheme, {HN-NCA heteronuclear TOCSY-NH}, exploits the favourable relaxation properties of IDPs and the presence of 1 J CαN and 2 J CαN couplings to transfer the 15N x magnetisation from amino acid residue ‘i’ to adjacent residues via the application of a band-selective 15N–13Cα heteronuclear cross-polarisation sequence of ~100 ms duration. Employing non-uniform sampling in the indirect dimensions, the efficacy of the approach has been demonstrated by the acquisition of 3D HNN chemical shift correlation spectra of α-synuclein. The experimental performance of the RF pulse sequence has been compared with that of the conventional INEPT-based HN(CA)NH pulse scheme. As the availability of data from both the HCCNH and HNN experiments will make it possible to use the information extracted from one experiment to simplify the analysis of the data of the other and lead to a robust approach for unambiguous backbone and side-chain resonance assignments, a time-saving strategy for the simultaneous collection of HCCNH and HNN data is also described.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Intrinsically disordered proteins (IDPs), dynamic and flexible molecules lacking a stable secondary and tertiary structures under physiological conditions, have been shown to be involved in a variety of cellular processes and the impairment of their functional roles has been implicated in different human diseases (Felli et al. 2012; Rezaei-Ghaleh et al. 2012; Schulenburg and Hilvert 2013; Skrabana et al. 2006; Tompa 2011, 2012; Uversky et al. 2009). As multidimensional NMR spectroscopy provides structural and dynamical information at atomic resolution, it has become the method of choice for the study of IDPs (Bermel et al. 2006, 2013a; Felli and Pierattelli 2014; Gil et al. 2013; Hellman et al. 2011, 2014; Isaksson et al. 2013; Jensen et al. 2013; Konrat 2014; Kosol et al. 2013; Liu and Yang 2013; Mäntylahti et al. 2010, 2011; Motácková et al. 2010; Narayanan et al. 2010; Novácek et al. 2012, 2013, 2014; Pantoja-Uceda and Santoro 2013, 2014; Reddy and Hosur 2014; Sahu et al. 2014; Salmon et al. 2010; Sibille and Bernadó 2012; Szalainé Ágoston et al. 2011; Solyom et al. 2013; Theillet et al. 2011; Waudby et al. 2013; Wen et al. 2011; Zawadzka-Kazimierczuk et al. 2012). Among the different procedures available for resonance assignments, triple resonance experiments such as HNCANH, HNCOCANH and HNCACONH that lead to the establishment of the direct connectivities between the amide groups of sequentially neighbouring residues in a single experiment have received considerable attention in the study of both structured and disordered proteins (Bracken et al. 1997; Chandra et al. 2012; Ikegami et al. 1997; Motácková et al. 2010; Panchal et al. 2001; Shirakawa et al. 1995; Weisemann et al. 1993). Due to the presence of 1 J CαN and 2 J CαN couplings, correlation peaks in these experiments appear via different magnetisation transfer pathways. For example, cross-peaks in the (H)N(CA)NH experiment (Chandra et al. 2012; Ikegami et al. 1997; Panchal et al. 2001; Weisemann et al. 1993) arise via pathways such as:
The resulting correlation of the backbone amide resonances of each amino acid residue ‘i’ with that of residues ‘i−1’ and ‘i+1’ is exploited to achieve protein backbone assignments. The RF pulse scheme in the HNCANH experiment, as implemented currently, involves magnetisation transfer through transverse 13Cα spins using INEPT steps for anti-phase coherence transfers and constant-time periods for mapping 15N chemical-shift evolution frequencies in the indirect dimensions. The high flexibility exhibited by IDPs results in NMR spectra with relatively intense and sharp signals. The long transverse relaxation times typically seen in IDPs can be exploited to acquire correlation data with good spectral resolution. However, the usage of constant-time chemical-shift evolution periods could limit the achievable spectral resolution and the possibility to fully exploit the potential of non-uniform data sampling procedures in the indirect dimensions (Hyberts et al. 2014; Kazimierczuk and Orekhov 2011; Maciejewski et al. 2012; Palmer et al. 2014; Paramasivam et al. 2012; Rovnyak et al. 2011). Considering that IDPs typically exhibit poor chemical shift dispersions and, hence, generation of multidimensional chemical shift correlation data with good spectral resolution is a prerequisite for achieving unambiguous resonance assignments, we have implemented in this study a novel experimental scheme, {HN-NCA heteronuclear TOCSY-NH}, for the correlation of the backbone amide resonances of each amino acid residue ‘i’ with that of residues ‘i−1’ and ‘i+1’ in (13C, 15N) labelled IDPs. The experimental scheme exploits the favourable relaxation properties of IDPs and the presence of 1 J CαN and 2 J CαN couplings to transfer the 15N x magnetisation from amino acid residue ‘i’ to adjacent residues via the application of a band-selective 15N–13Cα heteronuclear cross-polarisation (Glaser and Quant 1996) sequence of ~100 ms duration. Employing non-uniform sampling in the indirect dimensions, the efficacy of the approach has been demonstrated by the acquisition of 3D HNN chemical shift correlation spectrum of α-synuclein. The experimental performance of the {HN-NCA heteronuclear TOCSY-NH} pulse sequence involving free 15N evolution periods has been compared with that of the conventional INEPT-based HN(CA)NH pulse scheme.
As mentioned earlier, the HNN experiment leads to the correlation of the backbone amide resonances of each amino acid residue ‘i’ with that of residues ‘i−1’ and ‘i+1’. The correct identification of the ‘i−1’ and ‘i+1’ cross-peaks is typically carried out by the acquisition of a second correlation spectrum, such as (H)N(COCA)NH, that leads to the correlation of the backbone amide resonances of amino acid residue ‘i’ with only one of the two adjacent amino acid residues. In addition to (H)N(COCA)NH and (H)N(CACO)NH correlation spectra, it is worth noting that the correct identification of the HNN cross-peaks can also be achieved via the acquisition of correlation spectra from experiments such as HCCNH (Lyons and Montelione 1993; Richardson et al. 1993) and HCCCONH (Clowes et al. 1993; Grzesiek et al. 1993; Logan et al. 1992). In this study, we have made use of the HCCNH experiment involving the magnetisation transfer pathway 1Hsc → 13Csc → 13Cα → 15N → 1HN for this purpose. The HCCNH experiment uses 13C isotropic mixing to achieve correlations of the sidechain aliphatic resonances with the backbone amide groups and, due to the presence of 1 J CαN and 2 J CαN couplings, amide proton correlation peaks involving intra- and inter-residue sidechain aliphatic carbons can appear via different magnetisation transfer pathways. For example, correlation peaks in the (H)C(C)NH experiment can arise via pathways such as:
The HCCNH experiment leads only to the correlation of the backbone amide resonances of each amino acid residue ‘i’ with the sidechain aliphatic resonances of its own and that of the previous residue. The intra- and inter-residue cross peaks are identified by making use of the fact that the intensities of the correlation peaks arising via 2 J CαN couplings are typically smaller than from that of 1 J CαN couplings. It is worth mentioning that the HCCNH experiment itself can be effectively applied for the sequential resonance assignment of sidechain as well as backbone nuclei in proteins. Starting with a cross-peak in the [1H, 15N]-HSQC spectrum, corresponding to the amino acid residue ‘i’, this would involve identifying first the sidechain intra- and inter-residue resonances of ‘i’ in the HCCNH experiment. The sidechain resonances observed for the remaining cross-peaks in the [1H, 15N]-HSQC spectrum are then compared with that of the residue ‘i’. This permits the identification of the amide cross-peak corresponding that of residue ‘i−1’, as the intra-residue sidechain resonances of amino acid residue ‘i−1’ would appear as inter-residue sidechain resonances of residue ‘i’. Once the cross-peak/amino acid residue ‘i−1’ has been identified, the process can be repeated to identify the residues ‘i−2’, ‘i−3’ and so on. Making use of the amino acid type information obtained from the sidechain chemical shifts, the sequentially linked residues can then be mapped onto the primary amino acid sequence. Such an approach to sequential resonance assignments is considerably simplified by significantly reducing the number of peaks in the [1H, 15N]-HSQC spectrum whose sidechain resonances have to be compared with that of the residue ‘i’ for the identification of the amide cross-peak ‘i−1’. Here we have made use of the HNN data to simplify the analysis of HCCNH spectra. Considering that the availability of data from both the HCCNH and HNN experiments would allow using the information extracted from one experiment to simplify the analysis of the data from the other and lead to a robust approach for unambiguous backbone and sidechain resonance assignments, a time-saving strategy involving sequential data acquisitions (Bellstedt et al. 2014; Goradia et al. 2015; Wiedemann et al. 2014a, b) has also been demonstrated for an ‘one-shot’ collection of HCCNH and HNN data.
Materials and methods
NMR experiments to assess the performance of the RF pulse schemes were carried out on a 600 MHz narrow-bore Avance III NMR spectrometer equipped with pulse field gradient accessories, pulse shaping units and a triple-resonance cryo-probe and with the sample temperature kept at 288 K. Homonuclear 13C–13C TOCSY mixing was achieved using the amplitude and phase-modulated sequence (AK2-JCC) reported in the literature (Kirschstein et al. 2008a). 15N–13C anisotropic cross polarisation transfers were carried out via the AK2-JCHaniso1 sequence (Kirschstein et al. 2008b) using appropriate scaling of the RF field strength and duration of the mixing period. The States procedure was used for phase-sensitive detection in the indirect dimensions (States et al. 1982). Unless mentioned otherwise, DSS was used for direct 1H chemical shift referencing and 13C and 15N chemical shifts were indirectly referenced. Frequency switching in the 13C and 1H channels were carried out, where needed, when the relevant magnetisation is along the z axis. Uniformly (13C, 15N) labelled sample of human α-synuclein (pH 6.2), an IDP that has been extensively studied via solution state NMR (Bermel et al. 2006, 2013a, b; Hsu et al. 2009; Piai et al. 2014; Reddy and Hosur 2014), was prepared as per established procedures (Hoyer et al. 2002). The efficacy of the RF pulse scheme involving sequential HCCNH and HNN data acquisitions was also experimentally assessed using (13C, 15N) labelled samples of the N-terminal part of the A-type voltage gated potassium channel Kv1.4 (Pep61; 63 amino acids) (Sahoo et al. 2013) and the C-terminal winged helix (WH) domain (82 amino acids) protein of the minichromosome maintenance (MCM) complex of Sulfolobus solfataricus (Wiedemann et al. 2015). The non-uniformly sampled multidimensional data sets were collected employing random sampling in the indirect dimensions and processed either with the multi-dimensional decomposition (Jaravine et al. 2006; Luan et al. 2005; Orekhov et al. 2003; Tugarinov et al. 2005) or compressed sensing (Kazimierczuk and Orekhov 2011) processing protocols available in the Bruker Topspin version 3.2 software.
Results and discussion
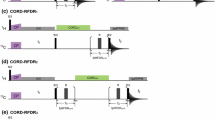
The RF pulse scheme {HN-NCA heteronuclear TOCSY-NH} implemented in this study is shown in Fig. 1a. The initial transverse 1H magnetisation generated by the first 90° pulse is allowed to evolve under the one-bond heteronuclear 15N–1H scalar couplings during the period 2Δ0 to generate the relevant anti-phase 1H magnetisations. The anti-phase 1H magnetisation is converted into the corresponding anti-phase nitrogen magnetisation by the 90° pulses applied to the different nuclei. The anti-phase 15N magnetisation is chemical shift labelled in t 1 and allowed to refocus via 15N–1H scalar coupling evolution during the interval 2τ1. The refocused 15N magnetisation is flipped to the z axis. After z-filtering, the 15N magnetisation is brought to the transverse plane and subjected to a period (τmix) of 15N ↔ 13Cα magnetisation exchange via the application of a band-selective het-TOCSY mixing sequence. Although the pulse scheme was experimentally tested using the AK2-JCHaniso1 heteronuclear Hartmann–Hahn (HEHAHA) mixing scheme, other HEHAHA sequences that have been reported in the literature (Glaser and Quant 1996) can also be effectively employed. At the end of the het-TOCSY mixing step, the transverse 15N magnetisation is flipped to the z axis. After z-filtering, the 15N magnetisation is brought to the transverse plane, chemical shift labelled during t 2 and then transferred to the attached proton via an INEPT-type transfer for detection (t 3 ) using the WATERGATE sequence for water suppression. An implementation of the {HN-NCA heteronuclear TOCSY-NH} experiment using the het-TOCSY approach for the 1H ↔ 15N transfers (Supplementary material, Figure S1a) also provides equally satisfactory performance. Numerical simulations (Fig. 2a) carried out considering a N1–C1–N2–C2–N3 heteronuclear spin system with 1 J N1C1 = 1 J N2C2 = 11 Hz and 2 J C1N2 = 2 J C2N3 = 7 Hz indicate that application of a heteronuclear TOCSY mixing sequence for a period of ~100 ms leads to a substantial transfer of the starting (15N2)x magnetisation to the adjacent N1 and N3 nuclei. This was also found to be essentially in agreement with experimental data (Supplementary material). The {HN-NCA heteronuclear TOCSY-NH} data presented below were generated with τmix = 100 ms employing both uniform and non-uniform sampling procedures.

a RF pulse scheme {HN-NCA heteronuclear TOCSY-NH} using the INEPT approach for 1H ↔ 15N magnetisation transfers. Delay durations are as follows: 2Δ0 = 4.76 ms, τNCα = 100 ms. Phase cycling is as follows: ϕ1 = 4(x), 4(−x); ϕ2 = y, −y; ϕ3 = 2(y), 2(−y); ϕ4 = 8(x), 8(−x); ϕ5 = x, -x; ϕ6 = 2(y), 2(−y); ϕ R = x, 2(−x), x, −x, 2(x), 2(−x), 2(x), −x, x, 2(−x), x. Gradients with a sine bell amplitude profile were used. Durations and strengths (with respect to the maximum strength of 50 G/cm) are G 1,2,3 = 1 (50 %), 1 (80 %), 1 (70 %) ms. Open and filled rectangles represent 180° and 90° hard pulses, respectively. b RF pulse scheme for the acquisition of 3D HNN spectra via the transfer pathway {…(15N) x → (13Cα) x → (13Cα) z → (13Cα) x → (15N) x …} involving a (13Cα) z intermediate state. Delay durations are as follows: 2Δ0 = 4.76 ms, τNCα = 50 ms. Phase cycling is as follows: ϕ1 = 4(x), 4(−x); ϕ2 = y, −y; ϕ3 = 8(y), 8(−y); ϕ4 = 2(y), 2(−y); ϕ5 = x, −x; ϕ6 = 2(y), 2(−y); ϕ R = x, 2(−x), x, -x, 2(x), 2(−x), 2(x), −x, x, 2(−x), x. Gradients with a sine bell amplitude profile were used. Durations and strengths (with respect to the maximum strength of 50 G/cm) are G 1,2,3 = 1 (−50 %), 1 (80 %), 1 (70 %) ms. Open and filled rectangles represent 180° and 90° hard pulses, respectively. c RF pulse schemes for the simultaneous acquisition of 3D {(H)C(C)NH and (H)N(CA)NH} chemical shift correlation spectra with dual sequential 1H acquisitions in the direct dimension. Delay durations are as follows: Δ0,1,2 = 2.576, 1.788, 0.788 ms, 2τ1 = 4.576 ms, 2τ2 = 3.0 ms, τCC = 9.6 ms, τNCα = 57.6 ms. Phase cycling is as follows: a ϕ1 = x, −x; ϕ2 = 8(y), 8(−y); ϕ3 = 2(y), 2(−y); ϕ4 = 8(y), 8(−y); ϕ5 = 4(y), 4(−y); ϕ6 = y, −y; ϕ7 = x, −x; ϕ8 = 8(x), 8(−x); ϕ R1 = ϕ R2 = x, 2(−x), x, −x, 2(x), 2(−x), 2(x), −x, x, 2(−x), x. Gradients with a sine bell amplitude profile were used. Durations and strengths (with respect to the maximum strength of 50 G/cm are G 1,2,3,4 = 1 (60 %), 1 (80 %), 1 (65 %), 1 (−70 %) ms. Open and filled rectangles represent 180° and 90° hard pulses, respectively. Where required frequency switching in the 13C and 1H channels were carried out when the relevant magnetisation is along the z axis. Arrow pointing downwards indicates the positions in the RF pulse sequence at which 13C frequency switching was effected

Simulated magnetisation transfer characteristics observed in a N1–C1–N2–C2–N3 heteronuclear spin system with scalar couplings of 1 J N1C1 = 1 J N2C2 = 11 Hz and 2 J C1N2 = 2 J C2N3 = 7 Hz, starting (15N2)x magnetisation and at the end of the application of 15N–13Cα het-TOCSY mixing of duration τmix. High power AK2-JCHaniso1 mixing sequence was used in the simulations considering all nuclei on resonance. These plots were obtained (a) following the application of a single 15N–13Cα het-TOCSY mixing sequence and (b) following 15N–13Cα het-TOCSY mixing that has been split into two parts so as to achieve a magnetisation transfer pathway {(15N) x → (13Cα) x } → (13Cα) z → {(13Cα) x → (15N) x } involving a (13Cα) z intermediate state
In the context of acquiring both the HCCNH and {HN-NCA heteronuclear TOCSY-NH} data sets in ‘one-shot’, we have also examined the performance of the RF pulse scheme shown in Fig. 1b for the acquisition of 3D HNN spectra. This pulse scheme is essentially the same as that given in Fig. 1a except that the 15N–13Cα het-TOCSY mixing period has been split into two parts so as to achieve a magnetisation transfer pathway {(15N) x → (13Cα) x } → (13Cα) z → {(13Cα) x → (15N) x } involving a (13Cα) z intermediate state. Although numerical simulations (Fig. 2b) indicate that the transfer of the (15N) x magnetisation from amino acid ‘i’ to the ‘i+1’ and ‘i−1’ residues gets substantially reduced compared to that seen via the previous scheme, it should still lead to measurable cross-peak intensities. This has been confirmed by experimental measurements (see below) and permitted the implementation of the RF pulse scheme (Fig. 1c) for the sequential acquisition of 3D HCCNH and HNN data sets.
In the RF pulse scheme for the sequential acquisition of 3D (H)C(C)NH and (H)N(CA)NH data sets (Fig. 1c) the initial transverse magnetisation is generated from both 15N and 13C attached protons by the first 90° pulse. It is allowed to evolve under the one-bond heteronuclear 15N–1H and 13C–1H scalar couplings during the periods 2Δ0 and (Δ0 + Δ1−Δ2), respectively, to generate the relevant anti-phase 1H magnetisations. The anti-phase 1H magnetisations are converted into the corresponding anti-phase nitrogen and carbon magnetisations by the 90° pulses applied to the different nuclei. The anti-phase 15N and 13C polarisations are allowed to refocus during the interval 2τ1 and 2τ2 and flipped to the z axis to generate (15N/13C)z magnetisation. The (15N)z magnetisation is first brought to the transverse plane and is allowed to undergo chemical evolution during the t 1 period. At the end of the t 1 evolution, the 15N transverse magnetisation is flipped to the z axis. The (13Cs)z magnetisation is then brought to the transverse plane and is allowed to undergo chemical shift evolution during the \( {t_{1}^{{\prime }} }\) period. At the end of the \( {t_{1}^{{\prime }} }\) period, the 13Cs transverse magnetisation is flipped to the z axis and subjected to a period of 13C–13C TOCSY mixing (τcc), keeping the 13C RF carrier at the middle of the aliphatics spectral range (~40 ppm), to generate 13Cα magnetisation and the 13C RF carrier is switched to ~55 ppm at the end of the TOCSY mixing. The 13Cα and 15N magnetisations are brought to the transverse plane and subjected to a period of 13Cα ↔ 15N magnetisation exchange via the application of a band-selective het-TOCSY mixing sequence. The transverse 15N and 13Cα magnetisation arising via 13Cα → 15N and 15N → 13Cα transfers at the end of the het-TOCSY mixing step are flipped to the z axis. The 15N magnetisation generated by the 13Cα → 15N transfer is first allowed to evolve under its chemical shift during the \( {t_{2}^{{\prime }} }\) period and transferred to the attached proton via an INEPT-type transfer for detection \( ({t_{3}^{{\prime }} })\) using the WATERGATE sequence for water suppression. After the completion of the first 1H acquisition, the 13Cα magnetisation arising from the first 15N → 13Cα transfer is brought to the transverse plane and subjected to a period of 13Cα → 15N magnetisation exchange via the application of a second band-selective het-TOCSY mixing sequence. The transverse 15N and 13Cα magnetisation at the end of the het-TOCSY mixing step are flipped to the z axis. The 15N magnetisation generated by the 13Cα → 15N transfer is allowed to evolve under its chemical shift during the t 2 period and transferred, as before, to the attached proton via an INEPT-type transfer for detection (t 3 ) using the WATERGATE sequence for water suppression. Signals arising from different pathways in the two sequentially collected data sets are selected by standard phase cycling procedures and by application of appropriate field gradient pulses. The spectral widths in the corresponding indirect \( {t_{1}} /{t_{1}^{{\prime }}}\) dimensions in the two data sets can be independently adjusted by appropriate scaling of the increments and spectral folding in one data set does not lead to resonance overlaps in the other, as two independent data sets are being acquired. Other details are given in the figure captions. The RF pulse scheme (Fig. 1c) was also modified so as to achieve the simultaneous collection of (a) 3D H(CC)NH and 3D H(NCA)NH data sets leading to the sidechain aliphatic proton and backbone amide proton chemical shifts in t 1 , instead of the corresponding 15N and 13C nuclei and (b) 3D C(C)NH and 3D (H)N(CA)NH data sets for studies on 13C-, 15N- and 2H-labelled protein systems.
Representative ω1 spectral cross-sections indicating the 15N chemical shifts of the diagonal (‘i’) and cross-peaks (‘i+1’ and ‘i−1’) and taken from the ω13 planes of 3D HNN spectra collected via different approaches are given in Fig. 3. The ω13 planes were taken at the 15N chemical shift values (ω2) corresponding to that of the diagonal peak. The 3D spectra were collected for the same total acquisition time using the {HN-NCA heteronuclear TOCSY-NH} pulse schemes with uniform and random non-uniform sampling (25 %) and via the INEPT-based conventional HN(CA)NH approach with uniform sampling. Spectra collected using the RF pulse schemes described in Fig. 1a, b are given in Fig. 3a–f and h–m, respectively. 3D spectra with non-uniform sampling were collected with twice the number of points in the indirect dimensions compared to those acquired with uniform sampling. The 3D spectrum via the conventional INEPT-based HN(CA)NH pulse scheme was collected with constant-time 15N evolution periods in the indirect dimensions and using the WATERGATE sequence for water suppression (Supplementary material, Figure S1b). In the HN(CA)NH experiment, the total delay period used for the transfer of the 13Cα magnetisation to the 15N nuclei was kept at 50 ms so as to maximize the cross-peak and minimise the diagonal peak intensities and to avoid potential cancellation of overlapping diagonal and cross-peaks with opposite phases that can arise under short delay conditions (Chandra et al. 2012). The 1D spectral cross-sections given in Fig. 3a–f clearly show that the cross peak intensities seen in the uniformly sampled 3D HNN spectrum generated using the 15N–13Cα het-TOCSY approach (τmix = 100 ms) is essentially comparable to that obtained via the conventional approach. However, significantly improved spectral resolution and sensitivity can be realised through the het-TOCSY approach by extending the evolution times in the indirect dimensions via non-uniform sampling. From the cross-sections shown in Fig. 3h–m it is observed that, compared to the conventional INEPT-based approach, the het-TOCSY approach involving a (13Cα) z intermediate state leads to cross-peaks with reduced signal intensities in the uniformly sampled 3D spectrum. However, extending the evolution times in the indirect dimensions via non-uniform sampling, as in the earlier case, leads to spectra with significantly improved spectral resolution and sensitivity. In most of the cases cross-peaks with intensities comparable to that obtained by the conventional approach was obtained.

Comparison of the ω1 spectral cross-sections taken from the ω13 planes of 3D HNN spectra collected via different approaches employing the same total data acquisition time. 15N chemical shifts of the diagonal (‘i’) and cross-peaks (‘i+1’ and ‘i−1’) are indicated in these spectra and the ω13 planes were taken at the 15N chemical shift values (ω2) corresponding to that of the diagonal peak. The data sets were collected with 16 transients per t1 increment, spectral widths in the indirect dimensions of 1519 Hz (15N), a recycle time of 1.0 s and a proton acquisition time of 60 ms. The AK2-JCHaniso1 sequence was used for 15N–13CA heteronuclear cross-polarisation, keeping the RF carriers at the middle of the spectral region and employing 15N/13C peak RF power levels of ~3.6 kHz. The uniformly sample data sets were collected with 32 t 1 and 32 t 2 increments while the data with non-uniform sampling were collected with 64 t 1 and 64 t 2 increments. Cross-sections plotted in blue, red and green in Fig. 3a–f were extracted from 3D HNN spectra generated via the conventional INEPT-based (H)N(CA)NH pulse scheme with uniform sampling, {HN-NCA heteronuclear TOCSY-NH} approach (τmix = 100 ms) with uniform sampling and {HN-NCA heteronuclear TOCSY-NH} approach (τmix = 100 ms) with non-uniform sampling, respectively. The corresponding cross-sections from the 3D HNN spectra generated using the RF pulse scheme involving a (13Cα) z intermediate state during the 15N–13Cα heteronuclear mixing period, Fig. 1b, are given in Fig. 3h–m. Other details are given in the main text
Representative ω13 spectral cross-sections from the 3D {HN-NCA heteronuclear TOCSY-NH} and 3D (H)C(C)NH spectra of α-synuclein showing the sequential connectivities along the backbone residues V82-A90 are given in Fig. 4a, b. The 3D HNN spectral data were acquired using the RF pulse scheme depicted in Fig. 1a with a single 15N–13Cα het-TOCSY mixing period of 100 ms. The availability of both the HNN and HCCNH data sets allows to establish unambiguous sequential connectivities following a simple and robust procedure: (1) A cross peak P1 in the [1H,15N]-HSQC spectrum (Supplementary material), with chemical shifts N1 and H1 and corresponding to an arbitrary amino acid residue ‘i’ is picked. (2) The 3D HNN spectrum at the (15N, 1H) chemical shifts corresponding to the residue ‘i’/peak P1 is then analysed so as to obtain from the ω1 spectral cross-sections the 15N chemical shifts of the backbone amide nitrogens, N2 and N3, adjacent to residue ‘i’ in the protein sequence. (3) All possible cross-peaks with amide nitrogen chemical shifts N2 and N3 in the [1H, 15N]-HSQC spectrum are then identified. (4) The 3D HNN spectrum at all the possible (15N, 1H) chemical shift positions indicated in step 3 is then analysed and the cross-peaks P2 and P3 in the [1H, 15N]-HSQC spectrum that correspond to the amide groups that are adjacent to the residue ‘i’ are then identified. This can be achieved easily as the ω1 spectral cross-sections taken at the (15N, 1H) chemical shift positions corresponding to the peaks P1, P2 and P3 will show characteristic cross-peak connectivities; the diagonal peak in P1 will appear as cross-peaks in P2 and P3 and the diagonal peaks in P2 and P3 will appear as cross-peaks in P1. (5) The 3D (H)C(C)NH spectrum is then analysed only at the (15N, 1H) chemical shift positions corresponding to the peaks P1, P2 and P3 to correctly identify the ‘i−1’ and ‘i+1’ backbone amide resonances. This can easily be achieved due to the fact that the HCCNH experiment leads only to the correlation of the backbone amide resonances of each amino acid residue ‘i’ with the sidechain aliphatic resonances of its own and that of the previous residue. Hence, if cross-peaks P2 and P3, respectively, correspond to the amino acid ‘i−1’ and ‘i+1’, the (H)C(C)NH ω1 spectral cross-sections taken at the (15N, 1H) chemical shift positions corresponding to the peaks P1, P2 and P3 will show characteristic cross-peak connectivities; the intra-residue resonances in P2 will appear as inter-residue cross-peaks in P1 and the intra-residue resonances in P1 will appear as cross-peaks in P3, with the intensities of inter-residue cross-peaks typically weaker than that of the intra-residue resonances. The sequential linking is further confirmed by making use of the protein sequence information and the amino acid type obtained from sidechain chemical shifts. Thus, starting with a peak P1, corresponding to the residue ‘i’, the analysis of the HCCNH spectrum for locating the peak P2, corresponding to the amino acid ‘i−1’ in the sequence, gets considerably simplified due to the availability of the HNN spectrum. Similarly, starting with a peak P1, the availability of the HCCNH data set makes it possible to correctly identify the ‘i−1’ and ‘i+1’ peaks in the HNN spectrum. Once a triplet of residues…–(‘i−1’)–(‘i’)–(‘i+1’)–… is correctly identified, the above procedure is repeated again starting with the peak P3 or P2 so as to conveniently extend the fragment of sequentially linked residues…–(‘i−2’)–(‘i−1’)–(‘i’)–(‘i+1’)–(‘i + 2’)–… (6) The sequentially linked residues are then mapped onto the primary amino acid sequence. It has to be mentioned, as a note of caution, that a variety of factors, e.g. the presence of proline residues, poor signal to noise ratios due to fast exchange of solvent-exposed labile protons with water protons leading to significant line broadening of the amide proton resonances or significant relaxation losses, e.g. 13Cα relaxation in glycine residues during the long 15N–13C mixing periods, can lead to breaks in the sequential linking of amino acid residues. This could result in difficulties in sequentially linking all the amino acid residues in one stretch and instead, result in several fragments of sequentially linked amino acids. However, as shorter fragments of connected residues can be linked to form extended fragments using the amino acid sequence information, this may not pose any serious problem. In summary, the availability of both the HCCNH and HNN data sets should lead to a reliable backbone and sidechain resonance assignment in proteins. It is worth pointing out that, due to 1 J CN and 2 J CN couplings, the application of 15N–13Cα het-TOCSY mixing sequence in a N1–C1–N2–C2–N3 heteronuclear spin system leads to the transfer of the starting (15N2) x magnetisation to the N1 and N3 nuclei via the C1 and C2 nuclei. The plot shown in Fig. 2a suggests that starting with (15N2) x magnetisation and after the application of a 15N–13Cα het-TOCSY mixing sequence, the amount of magnetisation residing on the C2 and C1 nuclei should be sufficient enough, even for large τmix values of ~100 ms, to lead to measurable cross-peak intensities in a {HN-NCA heteronuclear TOCSY-CAHA} experiment. The 1HN-1Hα spectral cross-sections, Fig. 4c, taken at the 15N chemical shift positions of the backbone amide nitrogens indicated and showing the sequential walk along the residues V82-A90 of α-synuclein, are essentially in agreement with this and consistent with the sequential connectivities generated via the HNN and HCCNH data.

ω13 spectral cross-sections from a 3D HNN, b 3D (H)C(C)NH and c 3D HN(CA)HA spectra of α-synuclein showing sequential connectivities along the backbone residues V82-A90. The HNN and HN(CA)HA experiments were carried out via the {HN-NCA heteronuclear TOCSY-NH} and {HN-NCA heteronuclear TOCSY-CAHA} pulse schemes, respectively, employing τmix = 100 ms and with non-uniform sampling (25 %) in the indirect dimensions. The AK2-JCHaniso1 sequence was used for the 15N ↔ 13C band-selective anisotropic mixing, keeping the 13C RF carrier at the positions mentioned in the text, employing 15N/13C peak RF power level of ~3.6 kHz and for a duration of 100 ms by repeating the basic sequence four times (25 ms * 4). The (H)C(C)NH data was acquired with uniform sampling in the indirect dimensions and using a standard RF pulse scheme. Data acquisition parameters: a 16 transients per t 1 increment, 64 t 1 increments, 64 t 2 increments, spectral widths in the indirect dimensions of 1519 Hz (15N), a recycle time of 1.0 s and a proton acquisition in the direct dimension of 125 ms; b 8 transients per t 1 increment, 80 t 1 increments, 48 t 2 increments, spectral widths in the indirect dimensions of 1519 Hz (15N) and 10,556 Hz (13C), a recycle time of 1.0 s and a proton acquisition in the direct dimension of 125 ms; c 8 transients per t 1 increment, 64 t 1 increments, 128 t 2 increments, spectral widths in the indirect dimensions of 600 Hz (1HN) and 1519 Hz (15N), a recycle time of 1.0 s and a proton acquisition (1Hα) in the direct dimension of 125 ms
The possibility to achieve simultaneous acquisition of HNN and HCCNH data sets was assessed using the RF pulse scheme given in Fig. 1c and its variants with uniform sampling in the indirect dimensions. These studies were carried out on two different 13C and 15N labelled IDPs (α-synuclein and N-terminal part of the A-type voltage gated potassium channel Kv1.4 (Pep61; 63 amino acids)) as well as on a moderately sized globular protein [the C-terminal winged helix (WH) domain (82 residues) protein of the MCM complex of S. solfataricus (Sso)]. Representative first results are presented below and in the supplementary material. Spectral cross-sections from the sequentially acquired 3D (H)C(C)NH and 3D (H)N(CA)NH spectra showing the sequential connectivities along the backbone residues E4-S12 of Pep61 are given in Fig. 5. The RF pulse scheme in Fig. 1c was modified so as to achieve the simultaneous collection of 3D H(CC)NH and 3D H(NCA)NH data sets leading to the sidechain aliphatic proton and backbone amide proton chemical shifts in t 1 . For studies on 13C-, 15N- and 2H-labelled protein systems, the RF pulse scheme was also modified, e.g. with direct 13C excitation, so as to acquire 3D C(C)NH and 3D (H)N(CA)NH data sets. These pulse schemes have been tested on the WH domain of the Sso-MCM complex and representative spectral cross-sections showing the sequential connectivities along the backbone residues spanning the region Y676-K685 are given in the Supplementary material. These results suggest that the simultaneous collection of HCCNH and HNCANH spectra is a very efficient and robust approach for achieving in one-shot sequential backbone and sidechain assignments without taking recourse to information from other experiments such as HNCOCANH and HCCCONH. Considering that these initial data sets were generated using conventional uniform sampling, one can envisage the possibility for achieving further improvements in sensitivity and resolution, without substantially increasing the experimental time, by implementing efficient non-uniform sampling protocols in the indirect dimensions coupled with sequential data acquisitions in the direct dimension. Such studies are planned.

1HΝ–15N and 1HN–13C spectral cross-sections from the (H)N(CA)NH (a) and (H)C(C)NH (b) spectra showing the sequential walk along the backbone residues spanning the region E4-S12 of Pep61. The cross-sections were taken at the 15N chemical shift values corresponding to that of the backbone amide nitrogens indicated. The simultaneously acquired spectra were recorded at 600 MHz with sample temperature kept at 283 K, 32 transients per t 1 increment, 40 t 1 increments, 48 t 2 increments, spectral widths in the indirect dimensions of 1458 Hz (15N) and 10,206 Hz (13C), a recycle time of 1.0 s and a proton acquisition in the direct dimension of 60 ms. The AK2-JCHaniso1 sequence was used for the 15N ↔ 13C band-selective anisotropic mixing, keeping the 13C RF carrier at the positions mentioned in the text, employing 15N/13C peak RF power level of ~3.125 kHz and for a duration of 57.6 ms by repeating the basic sequence twice (28.8 ms * 2). Longitudinal 13C–13C mixing in the aliphatic region was carried out employing AK2-JCC sequence, with a peak 13C RF power level of 10 kHz and for a duration of 9.6 ms by repeating two times the basic cycle of duration 4.8 ms (4.8 ms *2), keeping the RF carrier at 41 ppm
In conclusion, the results presented here demonstrate that although the overall duration of the {HN-NCA heteronuclear TOCSY-NH} pulse scheme is significantly longer than the conventional INEPT-based HN(CA)NH scheme, the high flexibility leading to long transverse relaxation times in IDPs permits the acquisition of HNN spectra of high quality. Implementation of RF pulse schemes with free evolution in the indirect dimensions and application of non-uniform sampling procedures allow the generation of HNN spectra with improved resolution and sensitivity without substantially increasing the data acquisition time and makes the approach outlined here a possible alternative to the existing RF pulse scheme. Making use of the 15N–13Cα het-TOCSY based approach, it is possible to achieve unambiguous backbone and sidechain resonance assignments in ‘one-shot’ by the simultaneous collection of HCCNH and HNN data sets. The basic strategy demonstrated here can be easily extended to implement other RF pulse schemes such as {HNCON-NCA heteronuclear TOCSY-NH} and {HACACON-NCA heteronuclear TOCSY-NH} to achieve sequential HN correlations via the generation of (15N, 13CO) and (15N, 13CO, 13Cα) resolved multidimensional chemical shift correlation spectra. These studies are currently in progress.
References
Bellstedt P, Ihle Y, Wiedemann C, Kirschstein A, Herbst C, Görlach M, Ramachandran R (2014) Sequential acquisition of multi-dimensional heteronuclear chemical shift correlation spectra with 1H detection. Sci Rep 4:4490
Bermel W, Bertini I, Felli IC, Lee YM, Luchinat C, Pierattelli R (2006) Protonless NMR experiments for sequence-specific assignment of backbone nuclei in unfolded proteins. J Am Chem Soc 128:3918–3919
Bermel W, Bruix M, Felli IC, Kumar MVV, Pierattelli R, Serrano S (2013a) Improving the chemical shift dispersion of multidimensional NMR spectra of intrinsically disordered proteins. J Biomol NMR 55:231–237
Bermel W, Felli IC, Gonnelli L, Koźmiński W, Piai A, Pierattelli R, Zawadzka-Kazimierczuk A (2013b) High-dimensionality 13C direct-detected NMR experiments for the automatic assignment of intrinsically disordered proteins. J Biomol NMR 57:353–361
Bracken C, Palmer AG 3rd, Cavanagh J (1997) (H)N(COCA)NH and HN(COCA)NH experiments for 1H–15N backbone assignments in 13C/15N-labeled proteins. J Biomol NMR 9:94–100
Chandra K, Jaipuria G, Shet D, Atreya HS (2012) Efficient sequential assignments in proteins with reduced dimensionality 3D HN(CA)NH. J Biomol NMR 52:115–126
Clowes RT, Boucher W, Hardman CH, Domaille PJ, Laue ED (1993) A 4D HCC(CO)NNH experiment for the correlation of aliphatic side-chain and backbone resonances in 13C/15N-labelled proteins. J Biomol NMR 3:349–354
Felli IC, Pierattelli R (2014) Novel methods based on 13C detection to study intrinsically disordered proteins. J Magn Reson 241:115–125
Felli IC, Pierattelli R, Tompa P (2012) Intrinsically Disordered Proteins. In: Bertini I, McGreevy KS, Parigi G (eds) NMR of biomolecules: towards mechanistic systems biology, First Edition edn. Wiley-VCH Verlag & Co. KGaA, Weinheim, pp 137–152
Gil S, Hošek T, Solyom Z, Kümmerle R, Brutscher B, Pierattelli R, Felli IC (2013) NMR spectroscopic studies of intrinsically disordered proteins at near-physiological conditions. Angew Chem Int Ed Engl 52:11808–11812
Glaser SJ, Quant JJ (1996) Homonuclear and heteronuclear Hartmann–Hahn transfer in isotropic liquids. Adv Magn Reson 19:59–252
Goradia N, Wiedemann C, Herbst C, Görlach M, Heinemann SH, Ohlenschläger O, Ramachandran R (2015) An approach to NMR assignment of intrinsically disordered proteins. ChemPhysChem 16:739–746
Grzesiek S, Anglister J, Bax A (1993) Correlation of backbone amide and aliphatic side-chain resonances in 13C/15N-enriched proteins by isotropic mixing of 13C magnetization. J Magn Reson B 101:114–119
Hellman M, Tossavainen H, Rappu P, Heino J, Permi P (2011) Characterization of intrinsically disordered prostate associated gene (PAGE5) at single residue resolution by NMR spectroscopy. PLoS One 6:e26633
Hellman M, Piirainen H, Jaakola VP, Permi P (2014) Bridge over troubled proline: assignment of intrinsically disordered proteins using (HCA)CON(CAN)H and (HCA)N(CA)CO(N)H experiments concomitantly with HNCO and i(HCA)CO(CA)NH. J Biomol NMR 58:49–60
Hoyer W, Antony T, Cherny D, Heim G, Jovin TM, Subramaniam V (2002) Dependence of alpha-synuclein aggregate morphology on solution conditions. J Mol Biol 322:383–393
Hsu ST, Bertoncini CW, Dobson CM (2009) Use of protonless NMR spectroscopy to alleviate the loss of information resulting from exchange-broadening. J Am Chem Soc 131:7222–7223
Hyberts SG, Arthanari H, Robson SA, Wagner G (2014) Perspectives in magnetic resonance: NMR in the post-FFT era. J Magn Reson 241:60–73
Ikegami T, Sato S, Wälchli M, Kyogoku Y, Shirakawa M (1997) An efficient HN(CA)NH pluse scheme for triple-resonance 4D correlation of sequential amide protons and nitrogens-15 in deuterated proteins. J Magn Reson 124:214–217
Isaksson L, Mayzel M, Saline M, Pedersen A, Rosenlöw J, Brutscher B, Karlsson BG, Orekhov VY (2013) Highly efficient NMR assignment of intrinsically disordered proteins: application to B- and T cell receptor domains. PLoS One 8:e62947
Jaravine V, Ibraghimov I, Orekhov VY (2006) Removal of a time barrier for high-resolution multi-dimensional NMR spectroscopy. Nat Methods 3:605–607
Jensen MR, Ruigrok RW, Blackledge M (2013) Describing intrinsically disordered proteins at atomic resolution by NMR. Curr Opin Struct Biol 23:426–435
Kazimierczuk K, Orekhov VY (2011) Accelerated NMR spectroscopy by using compressed sensing. Angew Chem Int Ed Engl 50:5556–5559
Kirschstein A, Herbst C, Riedel K, Carella M, Leppert J, Ohlenschläger O, Görlach M, Ramachandran R (2008a) Broadband homonuclear TOCSY with amplitude and phase-modulated RF mixing schemes. J Biomol NMR 40:227–237
Kirschstein A, Herbst C, Riedel K, Carella M, Leppert J, Ohlenschläger O, Görlach M, Ramachandran R (2008b) Heteronuclear J cross-polarisation in liquids using amplitude and phase modulated mixing sequences. J Biomol NMR 40:277–283
Konrat R (2014) NMR contributions to structural dynamics studies of intrinsically disordered proteins. J Magn Reson 241:74–85
Kosol S, Contreras-Martos S, Cedeño C, Tompa P (2013) Structural characterization of intrinsically disordered proteins by NMR spectroscopy. Molecules 18:10802–10828
Liu X, Yang D (2013) HN(CA)N and HN(COCA)N experiments for assignment of large disordered proteins. J Biomol NMR 57:83–89
Logan TM, Olejniczak ET, Xu RX, Fesik SW (1992) Side chain and backbone assignments in isotropically labeled proteins from two heteronuclear triple resonance experiments. FEBS Lett 314:413–418
Luan T, Jaravine V, Yee A, Arrowsmith CH, Orekhov VY (2005) Optimization of resolution and sensitivity of 4D NOESY using multidimensional decomposition. J Biomol NMR 33:1–14
Lyons BA, Montelione GT (1993) An HCCNH triple-resonance experiment using carbon-13 isotropic mixing for correlating backbone amide and side-chain aliphatic resonances in isotopically enriched proteins. J Magn Reson B 101:206–209
Maciejewski MW, Mobli M, Schuyler AD, Stern AS, Hoch JC (2012) Data sampling in multidimensional NMR: fundamentals and strategies. Top Curr Chem 316:49–77
Mäntylahti S, Aitio O, Hellman M, Permi P (2010) HA-detected experiments for the backbone assignment of intrinsically disordered proteins. J Biomol NMR 47:171–181
Mäntylahti S, Hellman M, Permi P (2011) Extension of the HA-detection based approach: (HCA)CON(CA)H and (HCA)NCO(CA)H experiments for the main-chain assignment of intrinsically disordered proteins. J Biomol NMR 49:99–109
Motáčková V, Nováček J, Zawadzka-Kazimierczuk A, Kazimierczuk K, Žídek L, Šanderová H, Krásný L, Koźmiński W, Sklenář V (2010) Strategy for complete NMR assignment of disordered proteins with highly repetitive sequences based on resolution-enhanced 5D experiments. J Biomol NMR 48:169–177
Narayanan RL, Dürr UH, Bibow S, Biernat J, Mandelkow E, Zweckstetter M (2010) Automatic assignment of the intrinsically disordered protein Tau with 441-residues. J Am Chem Soc 132:11906–11907
Nováček J, Haba NY, Chill JH, Židek L, Sklenář V (2012) 4D non-uniformly sampled HCBCACON and 1JNCα-selective HCBCANCO experiments for the sequential assignment and chemical shift analysis of intrinsically disordered proteins. J Biomol NMR 53:139–148
Nováček J, Janda L, Dopitová R, Židek L, Sklenář V (2013) Efficient protocol for backbone and side-chain assignments of large, intrinsically disordered proteins: transient secondary structure analysis of 49.2 kDa microtubule associated protein 2c. J Biomol NMR 56:291–301
Nováček J, Židek L, Sklenář V (2014) Toward optimal-resolution NMR of intrinsically disordered proteins. J Magn Reson 241:41–52
Orekhov VY, Ibragimov I, Billeter M (2003) Optimizing resolution in multidimensional NMR by threeway decomposition. J Biomol NMR 27:165–173
Palmer MR, Wenrich BR, Stahlfeld P, Rovnyak D (2014) Performance tuning non-uniform sampling for sensitivity enhancement of signal-limited biological NMR. J Biomol NMR 58:303–314
Panchal SC, Bhavesh NS, Hosur RV (2001) Improved 3D triple resonance experiments, HNN and HN(C)N, for HN and 15N sequential correlations in (13C, 15N) labeled proteins: application to unfolded proteins. J Biomol NMR 20:135–147
Pantoja-Uceda D, Santoro J (2013) Direct correlation of consecutive C’–N groups in proteins: a method for the assignment of intrinsically disordered proteins. J Biomol NMR 57:57–63
Pantoja-Uceda D, Santoro J (2014) New 13C-detected experiments for the assignment of intrinsically disordered proteins. J Biomol NMR 59:43–50
Paramasivam S, Suiter CL, Hou G, Sun S, Palmer M, Hoch JC, Rovnyak D, Polenova T (2012) Enhanced sensitivity by nonuniform sampling enables multidimensional MAS NMR spectroscopy of protein assemblies. J Phys Chem B 116:7416–7427
Piai A, Hošek T, Gonnelli L, Zawadzka-Kazimierczuk A, Koźmiński W, Brutscher B, Bermel W, Pierattelli R, Felli IC (2014) “CON-CON” assignment strategy for highly flexible intrinsically disordered proteins. J Biomol NMR 60:209–218
Reddy JG, Hosur RV (2014) A reduced dimensionality NMR pulse sequence and an efficient protocol for unambiguous assignment in intrinsically disordered proteins. J Biomol NMR 59:199–210
Rezaei-Ghaleh N, Blackledge M, Zweckstetter M (2012) Intrinsically disordered proteins: from sequence and conformational properties toward drug discovery. ChemBioChem 13:930–950
Richardson JM, Clowes RT, Boucher W, Domaille PJ, Hardman CH, Keeler J, Laue ED (1993) The use of heteronuclear cross polarization to enhance the sensitivity of triple-resonance NMR experiments. Improved 4D HCNNH pulse sequences. J Magn Reson B 101:223–227
Rovnyak D, Sarcone M, Jiang Z (2011) Sensitivity enhancement for maximally resolved two-dimensional NMR by nonuniform sampling. Magn Reson Chem 49:483–491
Sahoo N, Goradia N, Ohlenschläger O, Schönherr R, Friedrich M, Plass W, Kappl R, Hoshi T, Heinemann SH (2013) Heme impairs the ball-and-chain inactivation of potassium channels. Proc Natl Acad Sci USA 110:E4036–E4044
Sahu D, Bastidas M, Showalter SA (2014) Generating NMR chemical shift assignments of intrinsically disordered proteins using carbon-detected NMR methods. Anal Biochem 449:17–25
Salmon L, Nodet G, Ozenne V, Yin G, Jensen MR, Zweckstetter M, Blackledge M (2010) NMR characterization of long-range order in intrinsically disordered proteins. J Am Chem Soc 132:8407–8418
Schulenburg C, Hilvert D (2013) Protein conformational disorder and enzyme catalysis. Top Curr Chem 337:41–67
Shirakawa M, Wälchli M, Shimizu M, Kyogoku Y (1995) The use of heteronuclear cross-polarization for backbone assignment of 2H-, 15N- and 13C-labeled proteins: a pulse scheme for triple-resonance 4D correlation of sequential amide protons and 15N. J Biomol NMR 5:323–326
Sibille N, Bernadó P (2012) Structural characterization of intrinsically disordered proteins by the combined use of NMR and SAXS. Biochem Soc Trans 40:955–962
Skrabana R, Sevcik J, Novak M (2006) Intrinsically disordered proteins in the neurodegenerative processes: formation of tau protein paired helical filaments and their analysis. Cell Mol Neurobiol 26:1085–1097
Solyom Z, Schwarten M, Geist L, Konrat R, Willbold D, Brutscher B (2013) BEST-TROSY experiments for time-efficient sequential resonance assignment of large disordered proteins. J Biomol NMR 55:311–321
States DJ, Haberkorn RA, Ruben DJ (1982) A two-dimensional nuclear overhauser experiment with pure absorption phase in four quadrants. J Magn Reson 48:286–292
Szalainé Ágoston B, Kovács D, Tompa P, Perczel A (2011) Full backbone assignment and dynamics of the intrinsically disordered dehydrin ERD14. Biomol NMR Assign 5:189–193
Theillet FX, Binolfi A, Liokatis S, Verzini S, Selenko P (2011) Paramagnetic relaxation enhancement to improve sensitivity of fast NMR methods: application to intrinsically disordered proteins. J Biomol NMR 51:487–495
Tompa P (2011) Unstructural biology coming of age. Curr Opin Struct Biol 21:419–425
Tompa P (2012) Intrinsically disordered proteins: a 10-year recap. Trends Biochem Sci 37:509–516
Tugarinov V, Kay LE, Ibraghimov I, Orekhov VY (2005) High-resolution four-dimensional 1H–13C NOE spectroscopy using methyl-TROSY, sparce data acquisition, and multidimensional decomposition. J Am Chem Soc 127:2767–2775
Uversky VN, Oldfield CJ, Midic U, Xie H, Xue B, Vucetic S, Iakoucheva LM, Obradovic Z, Dunker AK (2009) Unfoldomics of human diseases: linking protein intrinsic disorder with diseases. BMC Genom 10(Suppl 1):S7
Waudby CA, Camilloni C, Fitzpatrick AW, Cabrita LD, Dobson CM, Vendruscolo M, Christodoulou J (2013) In-cell NMR characterization of the secondary structure populations of a disordered conformation of alpha-synuclein within E. coli cells. PLoS One 8:e72286
Weisemann R, Rüterjans H, Bermel W (1993) 3D triple-resonance NMR techniques for the sequential assignment of NH and 15N resonances in 15N- and 13C-labelled proteins. J Biomol NMR 3:113–120
Wen J, Wu J, Zhou P (2011) Sparsely sampled high-resolution 4-D experiments for efficient backbone resonance assignment of disordered proteins. J Magn Reson 209:94–100
Wiedemann C, Bellstedt P, Kirschstein A, Häfner S, Herbst C, Görlach M, Ramachandran R (2014a) Sequential protein NMR assignments in the liquid state via sequential data acquisition. J Magn Reson 239:23–28
Wiedemann C, Bellstedt P, Herbst C, Görlach M, Ramachandran R (2014b) An approach to sequential NMR assignments of proteins: application to chemical shift restraint-based structure prediction. J Biomol NMR 59:211–217
Wiedemann C, Szambowska A, Häfner S, Ohlenschläger O, Gührs KH, Görlach M (2015) Structure and regulatory role of the C-terminal winged helix domain of the archaeal minichromosome maintenance complex. Nucleic Acids Res 43:2958–2967
Zawadzka-Kazimierczuk A, Koźmiński W, Šanderová H, Krásný L (2012) High dimensional and high resolution pulse sequences for backbone resonance assignment of intrinsically disordered proteins. J Biomol NMR 52:329–337
Acknowledgments
The FLI is a member of the Science Association ‘Gottfried Wilhelm Leibniz’ (WGL) and is financially supported by the Federal Government of Germany and the State of Thuringia. We are deeply grateful to Dr. Lisa D. Cabrita and Prof. Dr. John Christodoulou (Insitute of Structural and Molecular Biology, University College London and Birkbeck College, London, United Kingdom) for providing us with the plasmid and purification protocol for the α-synuclein sample used in this study. Financial support by the Deutsche Forschungsgemeinschaft (DFG) within FOR 1738 (to O.O. and N.G.) is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wiedemann, C., Goradia, N., Häfner, S. et al. HN-NCA heteronuclear TOCSY-NH experiment for 1HN and 15N sequential correlations in (13C, 15N) labelled intrinsically disordered proteins. J Biomol NMR 63, 201–212 (2015). https://doi.org/10.1007/s10858-015-9976-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10858-015-9976-x