
Abstract
Spray pyrolysis was used to deposit CeO2–TiO2 coatings on stainless steel substrates. The addition of CeO2 to TiO2 changes morphology of the coatings, causing the surface to become laced and some CeO2 ions are incorporated into TiO2 crystal lattice. Part of CeO2 is highly dispersed on the surface of TiO2. The XPS showed the simultaneous existence of Ce4+ and Ce3+ on coating surface. As the CeO2 content in composites increases from 0.5 to 20%, the band gap decreases from 3.16 to 2.88 eV, respectively. The photoactivities in the degradation of methyl orange and lindane were significantly influenced by the content of Ce in composite. For methyl orange photodegradation, the highest activity had coating with CeO2 content of 2 wt%. In contrast, the activity of coatings in lindane photodegradation gradually decreases as the CeO2 content increases. However, efficiency of CeO2–TiO2 coatings remained satisfactory for application in process of lindane degradation. Lindane conversion at 6 h of irradiation is in the range from 88% for pure TiO2 to 60% for CeO2–TiO2 coating with CeO2 content of 20 wt%. Photodegradation of lindane was enhanced on pure TiO2 sites, while methyl orange degradation is highest on composite CeO2/TiO2 species with CeO2 content of 2 wt%.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Owing to the growth of industries that discharge hazardous substances, such as dyes, agricultural and medical chemicals, solvents, etc., water contamination has increased significantly in recent decades. Pesticides manufacture and its use produce large quantities of hazardous materials. Unused pesticides and water rinsing equipment contain high levels of pesticides that, if not properly handled, can lead to contamination of soil and eventually water resources [1]. Photocatalysis provides an effective way to clean polluted water from various contaminants, and some semiconductors show satisfactory photocatalytic activity [2]. Besides, photocatalysis is an effective method for converting solar energy into chemical energy [3]. Among the different metal oxides and sulfides such as TiO2 [3], ZnO [4, 5], WO3 [6], CdS [7], and so on, TiO2 is the most widely used and systematically studied photocatalyst. However, the extensive application of semiconductors photocatalysts in industry is still limited by poor quantum yield, low utilization of visible light, and high recombination rate of photogenerated electron (e−)/hole (h+) pairs. The TiO2 is an n-type semiconductor commonly used as photoactive material due to its small band gap under UV light irradiation [8]. Photocatalytically active electron–hole pairs, (e−)/(h+), are produced by absorbing a specific wavelength when the TiO2 surface is exposed to light. These photo-induced electrons (e−) and holes (h+) migrate to the TiO2 surface and can react with compounds that are adsorbed by the semiconductor. Photocatalytic degradation of pollutants from water is primarily considered as the use of TiO2 powder dispersed in polluted solution. However, applying TiO2 powder involves particle separation from the suspension, which is a time-consuming and costly process. While suspended systems are more efficient than those based on immobilized catalysts the issue of particle–fluid separation and catalyst recycling is the predominant motivation for immobilization of photoactive compounds. Immobilization of TiO2 on various supports, such as glass, quartz, ceramics [9], stainless steel foils [10], titanium wire [11], etc., solves the problem of particle separation. Various methods can be used for the deposition of the active materials, such as sol–gel [12], chemical and physical vapor deposition [13, 14], spray pyrolysis [15, 16], sputtering [17], plasma electrolytic oxidation [18], etc. Among the various methods for film deposition the spray pyrolysis method offers the advantages of reduced production cost and great versatility thanks to a large number of adjustable process parameters (substrate temperature; atomization gas flow; type and flow rate of liquid precursor; the geometry of system; etc.) [19].
TiO2 is a preferred material for photoactive coatings due to its beneficial optical properties, non-toxicity, chemical inertness, inexpensiveness, and light sensitivity [20]. The photocatalytic efficiency of coatings can be improved by coupling TiO2 with various dopants, such as SnO2 [21], WO3 [22], ZrO2 [23], and V2O5 [24]. Although TiO2 exists as anatase, rutile, and brookite phases, the primary photocatalytically active polymorphs are rutile and anatase. The band gap of the anatase phase is ~ 3.2 eV and the band gap of the rutile phase is ~ 3.0 eV [25]. Rutile exhibited better irradiation response than anatase but anatase has better photoactivity due to its lower recombination rate of (e−)/(h+) pairs [26]. Therefore, the main strategy for improving the photocatalytic behavior of pure TiO2 is to widen the photoreaction range from ultraviolet to visible spectra and reduce the (e−)/(h+) recombination rate. Our recent study has shown that the photocatalytic degradation of methyl orange (MO) could be performed over rutile TiO2 coating and enhancement of its photocatalytic properties was obtained by rutile doping with CeO2 [27].
In this paper, we have prepared CeO2 doped TiO2 coating intending to investigate which formulation of CeO2–TiO2 composite is optimal for photocatalytic application. A spray pyrolysis technique was used to deposit coatings on the stainless steel (SS) substrate. The degradation of methyl orange and lindane was used to test the photoactivity of these coatings. Methyl orange is a model azo dye and lindane is known as a persistent organic pollutant, chloropesticide with strong immunotoxic and neurotoxic effect [28]. The novelty of this work is to determine the photocatalytic efficiency of immobilized CeO2–TiO2 composite in reaction of lindane degradation. As far as we know, this is the first paper presenting the lindane degradation on these photocatalytic systems. Obtained oxide coatings were characterized in detail, providing an overview of their morphology, physic-chemical properties, and photocatalytic activity.
2 Experimental part
2.1 Materials
Coatings were prepared from TiO2 nanoparticles (Titanium(IV) oxide, mixture of rutile (20%) and anatase (80%)-nanoparticles, < 150 nm particle size, dispersion, 40 wt% in H2O, Alfa Aesar GmbH) and Ce(III) nitrate hexahydrate (99% purity, Sigma-Aldrich).
2.2 Preparation of thin films
The spray pyrolysis process was used to obtain TiO2-CeO2 composite coatings on stainless steel (SS) foils (Sandvik-Sweden, type-OC 404, thickness 35 μm; composition: 20.0 wt% Cr, 5.0 wt% Al, 0.02 wt% C, balance Fe). A homemade spray pyrolysis apparatus is described in detail in our previous study [29]. A 0.02 M TiO2 colloidal solution and 0.005 M Ce(NO3)3⋅6H2O were used as precursors. The ratio of these precursors in spraying solution was adjusted to obtain CeO2 concentration in TiO2–CeO2 in the range from 0 to 20 wt%. The final precursor solution was prepared by adding the TiO2 colloidal solution into the Ce(NO3)3⋅6H2O aqueous solution which was then further ultrasonically mixed for 60 min. Prior to the depositions of the oxide layer, the SS foil substrates, that are 10 cm long and 1.5 cm wide, were subjected to standard procedures of degreasing with hot alkaline solutions (15% solution of NaOH, at 50 °C during 30 min). Subsequently, the SS foils were ultrasonically cleaned in distilled water for 45 min. The precursor solution was atomized by air using a two-fluid nozzle, 0.2 mm in diameter, which was moved by a controlled speed and direction over SS substrate. The initial temperature of the SS substrate during the spraying was kept at 480 ± 10 °C by resistive heating. Thesubstrate temperature was measured by a non-contact pyrometer with an error within a range of ± 10 °C. Due to high airflow, the cyclical motion of the nozzle over the substrate surface cools down the substrate to a temperature of 120 ± 10 °C. When the spraying spot left the targeted area the temperature of the substrate quickly returned to the initial value. During the spraying, with each pass of the nozzle, this situation was continuously repeated over the substrate.
The main parameters of the spray pyrolysis are presented in Table 1.
The samples were named: TiO2, 0.5 CeO2/TiO2, 1 CeO2/TiO2, 2 CeO2/TiO2, 5 CeO2/TiO2, 10 CeO2/TiO2, and 20 CeO2/TiO2, depending on the CeO2 (wt%) content.
2.3 Characterization
The morphology and structure of coatings were examined using field emission scanning electron microscopy (FE–SEM; Tescan Mira 3 XMU), equipped with an energy-dispersive X-ray spectrometer (EDS) for compositional analysis. Beam current of 50 pA and operating voltage of 10 kV were used for ETD detector. Prior to analysis, the samples were gold-coated which was necessary as a conducting material for SEM imaging. The phase composition of coatings was determined using XRD patterns recorded by RigakuUltima IV diffractometer in the range of 2θ from 20° to 70°, employing Ni-filtered CuKα radiation having a wavelength of 1.54178 Å. The X-ray photoelectron spectroscopy (XPS) measurement was performed using a Kratos AXIS Supra photoelectron spectrometer with a monochromatic Al Kα X-ray source with an energy of 1486.6 eV. The base pressure in the XPS analyzer chamber was 5 × 10−8 Pa. A UV–Vis diffuse reflectance spectrophotometer (Shimadzu UV-3600) was used to analyze diffuse reflectance spectra (DRS). The optical band gap value (Eg) is determined based on the Kubelka–Munk theory combined with the Tauc relation: (αhν)m = A(hν − Eg); where hν is photon energy, A is a material dependent constant, m is constant that depends on the type of the transitions and α is the energy-dependent absorption coefficient. By plotting [F(R∞)hυ]m vs. (hυ), where R∞ is diffuse reflectance, we can determine the direct (m = 1/2) and indirect (m = 2) energy band gap (Eg). The best linearity is obtained for m = 1/2, and therefore direct allowed transition was used to determination of Eg.
2.4 Photocatalytic tests
The photoactivity of CeO2–TiO2 coatings was determined by monitoring the degradation of Methyl Orange (MO) and lindane. MO and lindane were chosen as model compounds representing azo dyes and persistent chloropesticide, respectively. The experiments were carried out at 20 °C in an open cylindrical thermostated pyrex glass reactor, 6.8 cm in diameter and a capacity of 250 cm3. The SS foils coated with oxide film were placed at the inert steeled wire holder, 5 mm above the glass. The solutions were stirred with a rotating magnet located under the holder (500 rpm). The volume of test solutions in all experiments was 100 cm3. The area of photocatalytic samples was 15 cm2 in all the experiments. Initial concentrations of MO and lindane were 8 mg/L and 300 ppb (0.3 mg/L), respectively. A simulated solar radiation lamp (Solimed BH Quarzlampen) with a power consumption of 300 W, housed 25 cm above the top surface of the test solution, was used as a light source.
Before the photocatalytic measurements, both pollutants were tested for adsorption in the dark for 30 min, in the presence of the photocatalytic foil. Also, both pollutants were tested for photolysis under simulated solar light in the absence of the photocatalyst.
Photocatalytic activities were determined by measuring the decomposition of pollutants after an appropriate light exposure time. The concentration of MO was measured by a spectrophotometer (UV–Vis Thermo Electron Nicolet Evolution 500) using the most intense MO absorption peak at 464 nm. All Lindane samples have been analyzed on a gas chromatograph using an Agilent 7890A GC system, equipped with a split-splitless injector and an electron capture detector (ECD) system, and a TG-5MT capillary column, 30 m × 0.25 mm × 0.25 µm, was used. The GC parameters were as follows: 50 °C for 3 min, then increased at a rate of 30 °C min−1 to 210 °C and held at this temperature for 20 min. The mode of injection was splitless. Hydrogen was used as the carrier gas with a flow rate 60 mL min−1.
The reproducibility of the photocatalytic measurements was verified by performing each test several times.
3 Results and discussions
3.1 Morphology of coatings
TiO2 and cerium oxide-doped TiO2 films have been deposited on one side of SS substrates. The amount of deposited coatings was determined by the mass difference before and after the deposition of oxides on the SS substrate. Assuming that the coatings have an ideal uniform thickness, according to a simple calculation based on apparent density and film loading, the thicknesses of TiO2 and 20 CeO2/TiO2 coatings are 1.06 to 1.12 µm, respectively. The thicknesses of all the other composite coatings are between these limiting values.
The strong adhesion of the coating to the support is necessary for its efficient application. Under real working conditions, the immobilized coating can quickly detach from the SS foil which makes the immobilization technique meaningless. To test the stability of coating all samples were subjected to the same conditions that were used in photocatalytic experiments (T = 20 °C, water volume V = 100 mL, stirring = 500 rpm). After 8 h of this test, no detachment of the films was detected, indicating good adhesion of the coatings with no visible defects such as attrition and cracking.
The influence of CeO2 addition on the surface morphology of TiO2 coatings is shown on SEM micrographs, Fig. 1.

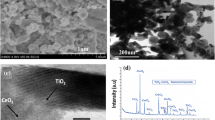
A FE-SEM images of TiO2 and CeO2–TiO2 with different content of CeO2 at magnifications ×10,000; a TiO2, b 0.5 CeO2/TiO2, c 1 CeO2/TiO2, d 2 CeO2/TiO2, e 5 CeO2/TiO2, f 10 CeO2/TiO2, g 20 CeO2/TiO2; B FE-SEM images of pure TiO2 (h) and 2 CeO2/TiO2 i at magnifications ×65,000; and C EDS spectra: j pure TiO2, k 2 CeO2/TiO2
The TiO2 surface consists of densely packed grains and exhibits an open-porous structure. These lace-like structures were composed of a network of interconnected regions, similar to hierarchical-porosity systems. There are no evident fractures or cracks on its surface. As indicated in Fig. 1B structures of both, pure TiO2 and 2 CeO2/TiO2, were consisted of small spherical aggregates ranging from 20 to 40 nm in size. This image has also shown that the addition of CeO2 to TiO2 does not change substantially the primary nanoparticles structure of the films. The particle size in the 2 CeO2/TiO2 composite remained almost unchanged. This effect is probably caused by the proper distribution of CeO2 on TiO2.
An introduction of CeO2 influences the structure of the coatings and their surface becomes laced with elevated and lowered regions. Besides, with increasing CeO2 content, pores and voids on the surface become more pronounced. The creation of such a complex structure has been explained in aspects of the rapid decomposition and hydrolysis of precursors. As the precursor droplet (TiO2 sol along with a liquid cerium precursor) contacts the heated substrate the fast vaporization of water induces an acceleration of the sol–gel process [30]. The transition of gel into a solid layer is accompanied by further vaporization of precursors leaving a more porous structure, thus creating an additional internal porosity [31]. It is a consequence of the release of additional gases, such as nitrogen oxides, produced by the thermal decomposition of cerium precursor, Ce(NO3)3⋅6H2O [32]. Then, due to an increase in the participation of cerium precursors and the high amount of gaseous materials, the creation of a large open porous structure network was promoted. The numbers and depth of voids are related to the ceria precursor concentration. These coatings with microcavities, open porosity, and structure imperfection, are beneficial for potential use in photocatalysis. EDS spectra of pure TiO2 and 2 CeO2/TiO2 coatings show expected elements without any surface contamination. Carbon originates from both grid holder and surface adsorbed carbon-containing species from the air. Pure TiO2 coating is composed of Ti and O, while the 2 CeO2/TiO2 composite is consisted of Ti, Ce, and O, indicating the existence of oxide-like species of Ce. The Ti:Ce weight ratio correlates well with the predetermined content of Ti and Ce in the spraying solution. According to EDS analysis, the content of Ce, Ti and O in the 2 CeO2/TiO2 composite is 1.25 wt%, 58.18 wt% and 40.57 wt%, respectively.
3.2 Structural properties
The phase composition of sole TiO2 and cerium oxide doped TiO2 coatings was analyzed by X-ray diffraction (XRD). The XRD patterns of all samples are presented in Fig. 2.

XRD patterns of: a TiO2, b 0.5 CeO2/TiO2, c 1 CeO2/TiO2, d 2 CeO2/TiO2, e 5 CeO2/TiO2, f 10 CeO2/TiO2, g 20 CeO2/TiO2
It can be seen that coatings are polycrystalline, composed of anatase and rutile phases. According to Joint Committee on Powder Diffraction Standards (JCPDS# 21-1272) the 2θ peaks located at 25.33°, 37.78°, 38.49°, and 48.10° are assigned to the (101), (103), (004), and (200) planes of tetragonal anatase TiO2 phase, respectively. The 2θ diffraction peaks at 27.50°, 36.40°, 41.30°, and 53.95° are intrinsic peaks of tetragonal rutile TiO2 (110), (101), (111), and (211) planes, respectively (JCPDS# 21-1276). Peaks observed at 2θ of 44.50, and 64.65° were attributed to the (110) and (200) planes of α-Fe (JCPDS# 87-0721). This is expected due to the thickness of the coatings (about 1.1 μm) and the fact that the SS substrate mainly contains Fe. It can be also observed that the anatase and rutile peaks intensity slightly decreases with the increase of cerium content, indicating covering of TiO2 by cerium compounds.
Besides, no diffraction peaks were observed to verify the existence of any crystal phase of compounds containing cerium. Generally, there are three accepted explanations for the absence of noticeable CeO2 diffraction peaks in the CeO2/TiO2 coatings: low content of CeO2 with high dispersion, very small CeO2 crystallites, and CeO2 in the amorphous phase [33]. Due to the relatively high loading of CeO2 in CeO2/TiO2 samples, the explanation that CeO2 species are well dispersed in the coatings is most likely [34]. For samples with high cerium content, the lack of peaks that identify cerium compounds may also indicate its amorphousness. The absence of CeO2 phases in XRD patterns could also be due to the interaction between the cerium and TiO2 lattice species. However, the radius of Ce4+ ion (0.097 nm) is greater than that of Ti4+ (0.061 nm), which hinders cerium ions to enter into the crystal lattice of TiO2 [35]. With the addition of Ce in composites, no shift of the (101) diffraction peaks of the anatase phase was observed to confirm the insertion of Ce ions into the TiO2 crystal lattice and formation of any crystal phase of Ti–Ce-Ox. Taking into account that coatings are deposited on the SS substrate using different precursors, TiO2 as a nanoparticle from colloidal dispersion and Ce from solution, the probability of insertion of some Ce ions into the TiO2 crystal lattice is lower than the formation of highly dispersed amorphous CeO2 agglomerates on the surface of TiO2 particles. The presence of such small and amorphous CeO2 agglomerates at a temperature of coatings deposition (≤ 460 °C) indicates their early and rapid formation by hydrolysis of cerium precursor.
3.3 Surface oxidation state
XPS analysis was performed to investigate the oxidation state of species present in the coatings and their mutual interaction. The XPS spectra of Ce 3d, Ti 2p, and O 1s are shown in Fig. 3.

XPS spectra of Ce 3d, Ti 2p, and O 1s; a TiO2, b 0.5 CeO2/TiO2, c 1 CeO2/TiO2, d 2 CeO2/TiO2, e 5 CeO2/TiO2, f 10 CeO2/TiO2, g 20 CeO2/TiO2
Analyses of Ce 3d spectra are rather complex due to the presence of Ce3+ and Ce4+ ions together on the surface of the coating. On the Ce 3d XPS spectra in Fig. 3 the labels of v and u indicate the spin–orbit coupling 3d5/2 and 3d3/2, respectively. Four 3d5/2 peaks at the binding energies of 883.2 eV, 885.9 eV, 890.6 eV, and 899.2 eV are marked as v, vI, vII, and vIII, respectively. The 3d3/2 peaks at binding energies of 901.5 eV, 904.2 eV, 908.5 eV, and 917.3 eV were marked as u, uI, uII, and uIII, respectively. According to the literature data, the peaks observed in Ce 3d spectra denoted with u, uII, uIII, and v, vII, vIII were related to the spin–orbit coupling of Ce4+ ions [36]. The peaks denoted as uI and vI correspond to the Ce3+ ions in the coatings. The coexistence of Ce in + 4 and + 3 oxidation states is related to the ability of cerium oxide to store and move oxygen depending on the environment [37]. The intensities of Ce 3d peaks reflect the content of Ce in the TiO2–CeO2 coatings. For coatings with CeO2 content lower than 2 wt% the Ce 3d peaks cannot be clearly detected, which can be related to the low content of Ce in composites or too small size of CeO2 particles. In addition, the relatively low intensity of Ce peaks could be a result of the high dispersion of Ce particles in composite or substitutional Ce doping into TiO2 lattice, which is in agreement with XRD data. Detailed analysis of the Ce 3d peaks showed that the intensities of peaks belonging to Ce4+ are much stronger than that peaks belonging to Ce3+. This indicates that the majority of Ce ions are in the + 4 oxidation state. The simultaneous existence of Ce4+ and Ce3+ in close contact is very useful for the photoactivity of ceria containing materials due to its ability to transfer oxygen through Ce4+/Ce3+ redox cycles. Nevertheless, when Ce4+ and TiO2 are in close contact their strong electronic interaction could produce more electronegative Ce3+ that creates oxygen vacancies. This could be advantageous for increasing the number of very active photocatalytic species on the surface of the coating.
Ti 2p spectra of pure TiO2 coating indicate peaks of 458.7 eV and 464.6 eV binding energies, which are attributed to Ti 2p3/2 and Ti 2p1/2 respectively [38]. These values, including the difference between Ti 2p1/2 and Ti 2p3/2 of 5.9 eV in the binding energy, suggest that Ti ions are in the + 4 oxidation state. With the increasing Ce content in composites, a gradual positive shift of the binding energy of Ti 2p levels up to 0.5 eV is detected. This shift of binding energy indicates the intermediate oxidation state of Ti and suggests the incorporation of Ce into TiO2 lattice. Subsequently, the release of oxygen from this Ti–Ce-oxide-like species can create Ce3+ and Ti3+ ions, thus increasing the electron density clouds surrounding Ti4+.
The O 1s spectrum is fitted into two peaks. The first intense peak at 530.2 eV was attributed to lattice oxygen O2−. The second weak peak at about 531.2 eV corresponds to surface oxygen or surface hydroxyl groups [38]. Surface oxygen can be formed from either a non-stoichiometric Ti-Ce oxide or a hydroxyl group. This type of oxygen may be useful for the creation and control of the very active photocatalytic sites.
3.4 Optical properties
The electronic structure changes and optical absorption properties of the pure TiO2 and cerium oxide doped TiO2 coatings were studied using UV–Vis diffuse reflectance spectra. The DRS spectra have been investigated in the range of 300–550 nm, Fig. 4. The measured absorbance spectra (Fig. 4A) have been transformed into Kubelka–Munk function (F(R)) (Fig. 4B). The light absorption is expressed as F(R) = (1 −R)2/2 R = α/S, where R = 10−A is the reflectance and A is the absorbance. The α represents the coefficient of absorption and S denotes the scattering coefficient.

A UV–Vis DRS spectra and B Kulbeka-Munk function vs. photon energy (hν); a TiO2, b 0.5 CeO2/TiO2, c 1 CeO2/TiO2, d 2 CeO2/TiO2, e 5 CeO2/TiO2, f 10 CeO2/TiO2, g 20 CeO2/TiO2
The band gap values obtained from Fig. 4B are presented in Table 2.
For pure TiO2 coating, the band gap occurs at 3.18 eV, which is related to electron transfer from the valence to the conduction band due to their photo-excitation. For all the CeO2–TiO2 composites the band gaps have been observed to gradually decrease with the increase of cerium content, which indicates electron interaction between TiO2 and cerium compounds. The obtained band gap energies were observed to be in the range from 3.16 eV, for the sample with 0.5 wt% of CeO2, to 2.88 eV, for a composite with 20 wt% of CeO2. The observed red shift toward higher wavelengths and narrowed optical band gap of the composites may have improved light collection capability which increases photoactivities of coatings in the visible-light region.
3.5 Photocatalytic activity
Properties of pure TiO2 and TiO2–CeO2 composite coatings in photocatalytic processes were examined by measuring the degradation of MO and Lindane under UV–Vis light illumination. Figure 5A, B shows the time-dependent conversion rate of MO and lindane degradation over coatings illuminated during 12 h. For clear visualization of the impact of Ce on the photoactivity of composites, the conversions at 6 h of MO and lindane degradation as a function of CeO2 content are shown in Fig. 5C, D.

Photocatalytic performance of coatings; A-MO degradation vs. time of irradiation; B-Lindane degradation vs. time of irradiation; C-MO conversion at 6 h of irradiation vs. content of CeO2; D-Lindane conversion at 6 h of irradiation vs. content of CeO2; a TiO2, b 0.5 CeO2/TiO2, c 1 CeO2/TiO2, d 2 CeO2/TiO2, e 5 CeO2/TiO2, f 10 CeO2/TiO2, g 20 CeO2/TiO2
The kinetics of photocatalytic reactions is described by pseudo first-order kinetic model:
where Co is the initial concentration, Ct is the concentration at time t, and the kapp is apparent first-order rate constant. Apparent reaction rate constants are summarized in Table 3.
Before photocatalytic tests, degradation of the MO and lindane was monitored in the absence of photocatalysts using the same illumination as in photocatalytic experiments. The results of the photolysis test showed that, after 12 h of irradiation, the photodegradation of MO and lindane was negligible, below the limit of an experimental error (< 1%). In addition, tests of MO and lindane degradation in the dark were performed. The results of these tests showed that the concentration of MO and lindane remained unchanged after 30 min, suggesting that the adsorption of both pollutants on photoactive coatings was insignificant. In other words, light is required for the catalytic degradation of MO and Lindane.
In the presence of UV–Vis light, the initial concentration of both reactants gradually decreases with a time of irradiation. Pure TiO2 coating has a satisfactory photoactivity in MO degradation, Fig. 5A. The photoactivity of MO degradation increases with increasing CeO2 content, reaching the maximum value at CeO2 content of 2 wt% (Fig. 5C). A further increase of CeO2 content in composites decreases its MO degradation efficiency. When the content of CeO2 reaches 10 wt% the conversion of MO becomes almost equal to the conversion of pure TiO2. For composite with 20 wt% CeO2 photoactivity in MO degradation is even lower compared to photoactivity of pure TiO2.
In comparison to their properties in the degradation of MO, composite coatings have different photocatalytic behavior in the reaction with lindane (Fig. 5B). It should be noted that pure TiO2 has very good photocatalytic activity in lindane degradation, although lindane is well known as a persistent organic compound. The addition of CeO2 to the coating had a detrimental effect on the photodegradation of lindane. As the content of CeO2 in coatings increases, the photoactivity gradually decreases, as shown in Fig. 5D. However, even for the largest drop in the photoactivity, observed for the sample with CeO2 content of 20 wt%, photoefficiency of composite remained satisfactory for application in process of lindane removal from real agroindustrial wastewater.
Different behavior during photocatalytic degradation of MO and lindane can be described using a widely accepted mechanism for the removal of pollutants on composite photocatalysts [39]. By this mechanism, the first step of the reaction is an illumination of the surface by light which causes the adsorption of photons (hν). For CeO2–TiO2 composites, the conduction and valence bands of CeO2 are located slightly higher than the conduction and valence bands of TiO2. After adsorption of photons, photoexcited surfaces of CeO2 and TiO2 generate electrons in conduction bands while leaving the holes in the valence bands. The photogenerated electrons in the conduction band of CeO2 are then moved to the conduction band of TiO2. Simultaneously, the holes transfer occurs in the opposite direction, from the valence band of TiO2 to the valence band of CeO2. These photo-induced electrons and holes generate electron–hole pairs, which work as a redox media for hydroxyl (HO⋅) and peroxyl (O2⋅−) radicals formation. When reacting with organic compounds, these radicals are strong oxidizers. The amount of photogenerated electron–hole pairs and their lifetime in the separated state determines photoactivity.
Therefore, CeO2–TiO2 coatings could have improved photoactivity than pure TiO2 because of the larger photoreaction spectral range and lower recombination rate of electron–hole pairs. The second elementary step of the photocatalytic reaction is the adsorption of the organic reactants on the coating surface. Then, adsorbed organic compounds are oxidized and degraded by the hydroxyl and peroxyl radicals that already exist on the coating surface. Two important effects should be considered when comparing MO and lindane photocatalytic degradation: adsorption properties and compounds persistency. Since lindane is a stronger electron donor than MO, it should be more strongly adsorbed on the coating surface than MO. If an increase of the CeO2 content in the composite leads to a decrease of the available sites for lindane adsorption, then lindane reactivity decrease. Besides, self-poisoning could inhibit lindane photodegradation; strongly adsorbed lindane permanently blocks the active centers of the composite. The surface of composites has various types of active centers such as pure TiO2 and CeO2, Ce ions in + 3 oxidation states, composite CeO2-TiO2 in which some of Ce ions penetrate TiO2 lattice, indicating that the reaction is governed by different adsorption properties of MO and lindane on these centers. Each of these types of active centers contributes to the overall photocatalytic activity. Pure TiO2 active centers have higher activity in lindane degradation than TiO2 modified by CeO2. The fact that pure TiO2 centers are modified or covered by excess CeO2 is the most reasonable explanation for such photocatalytic behaviors. Obviously, some TiO2 centers are blocked for lindane adsorption, resulting in lower lindane activation and reduced photoactivity. On the other hand, the sample containing 2 wt% CeO2 has the highest photoactivity in MO degradation, suggesting that there is a suitable content of CeO2 in coatings for optimal MO adsorption and activation. As shown by XPS measurements, the existence of Ce in the close contact with TiO2 lattice play the role of Ce4+/Ce3+ redox couples. The capture of electrons by redox couples increases the number of holes in the valence band and making electron–hole recombination more difficult [40]. This effect enhances photocatalytic MO degradation. This also explains the detrimental impact of exceeding the optimal content of CeO2 because highly active CeO2–TiO2 centers for MO degradation are overlaid by surplus CeO2. Generally, lindane adsorption and activation are favored on pure TiO2, while MO is better adsorbed and activated on CeO2–TiO2 composite sites. It is well known that the overall rate of a reaction is controlled by the rate of the slowest step. It is generally accepted that the rate-limiting steps of photocatalytic reaction on semiconductors are electron–hole pair rate and electron transport from semiconductors surface to adsorbed O2 molecules [41]. When TiO2 is modified by Ce, the excited electrons in the conduction band can be captured by dopant, which acts as an electron capture center and significantly suppresses the electron–hole recombination rate.
The reaction rate of hydroxyl (HO⋅) and peroxyl (O2⋅−) radicals with the adsorbed pollutant is faster than the rate of pollutants adsorption, which indicates that the rate-determining step of the overall photocatalytic reaction is the adsorption of MO or lindane. These results also suggest that photogenerated electron–hole pairs behave similarly regardless of which active centers they are produced and the rupture of the weakest C–C bonds of adsorbed pollutants determine the overall reaction rate.
4 Conclusions
Spray pyrolysis was used to deposit pure TiO2 and CeO2–TiO2 composites on an SS substrate. The surface of coatings is created of dense grains that produce a lace-like texture that becomes more pronounced as the CeO2 content increases. The lack of CeO2 phases in XRD patterns show that CeO2 is highly dispersed on the surface of TiO2 particles or that Ce ions are incorporated into the TiO2 lattice. The simultaneous existence of Ce4+ and Ce3+ on the coating surface, as well as strong interaction between Ce ions and TiO2 particles, is also confirmed by XPS analysis. DRS analyses revealed a decrease in band gaps as cerium content increased, supporting the strong electron transfer between TiO2 and cerium compounds.
Pure TiO2 coatings, as well as CeO2–TiO2 composites, have satisfactory photoactivity in the degradation of both pollutants, MO and lindane. For MO degradation, photoactivity increases with increasing CeO2 content, reaching a maximum at 2 wt% CeO2 in coatings. Further increase of CeO2 content led to a decrease in MO degradation. On the other side, for lindane degradation, the addition of CeO2 decreases the photoactivity of pure TiO2. Various MO and lindane adsorption characteristics on different active sites are responsible for the difference in photoactivity. Generally, lindane photodegradation is favored on pure TiO2 sites, while MO degradation is promoted on CeO2–TiO2 species. Besides, important benefits for degradation of both pollutants originate from the close contact of CeO2 and TiO2 which creates additional very reactive radicals.
Although composite CeO2–TiO2 active sites have improved charge separation, preventing the recombination of photogenerated electron–hole pairs, the rate-determining steps of overall photocatalytic reactions are MO and lindane adsorption over various types of active centers.
Data availability
The data and material were available.
References
A. Singh, N. Dhiman, A.K. Kar, D. Singh, M.P. Purohit, D. Ghosh, S. Patnaik, Advances in controlled release pesticide formulations: prospects to safer integrated pest management and sustainable agriculture. J. Hazard. Mater. 385, 121525 (2020)
J.J. Rueda-Marquez, I. Levchuk, P. Fernandez Ibanez, M. Sillanpaa, A critical review on application of photocatalysis for toxicity reduction of real wastewaters. J. Clean. Prod. 258, 120694 (2020)
M. Pelaez, N.T. Nolan, S.C. Pillai, M.K. Seery, P. Falaras, A.G. Kontos, P.S.M. Dunlop, J.W.J. Hamilton, J.A. Byrne, K. O’Shea, M.H. Entezari, D.D. Dionysiou, A review on the visible light active titanium dioxide photocatalysts for environmental applications. Appl. Catal. B. 125, 331–349 (2012)
I. Ahmad, S. Shukrullah, M.Y. Naz, M.A. Rasheed, M. Ahmad, E. Ahmed, M.S. Akhtar, N.R. Khalid, A. Hussain, S. Khalid, Boosted hydrogen evolution activity from Sr doped ZnO/CNTs nanocomposite as visible light driven photocatalyst. Int. J. Hydrogen Energy 46, 26711–26724 (2021)
I. Ahmad, S. Shukrullah, M.Y. Naz, M. Ahmad, E. Ahmed, M.S. Akhtar, S.U. Rehman, M.M. Makhlouf, Efficient hydrogen evolution by liquid phase plasma irradiation over Sn doped ZnO/CNTs photocatalyst. Int. J. Hydrogen Energy 46, 30019–30030 (2021)
L. Hromadko, M. Motola, V. Cicmancova, R. Bulanek, J. Macak, Facile synthesis of WO3 fibers via centrifugal spinning as an efficient UV and VIS-light-driven photocatalyst. Ceram. Int. 47, 35361–35365 (2021)
Y. Yuan, R. Guo, L. Hong, X. Ji, Z. Lin, Z. Li, W. Pan, A review of metal oxide-based Z-scheme heterojunction photocatalysts: actualities and developments. Mater. Today Energy 21, 100829 (2021)
S. Bera, D. Won, S.B. Rawal, H.J. Kang, W.I. Lee, Design of visible-light photocatalysts by coupling of inorganic semiconductors. Catal. Today 335, 3–19 (2019)
A. Fernandez, G. Lassaletta, V.M. Jimenez, A. Justo, A.R. Gonzalez-Elipe, J.M. Herrmann, H. Tahiri, Y. Ait-Ichou, Preparation and characterization of TiO2 photocatalysts supported on various rigid supports (glass, quartz and stainless steel). Comparative studies of photocatalytic activity in water purification. Appl. Catal. B. 7, 49–63 (1995)
J. Dostanic, B. Grbic, N. Radic, P. Stefanov, Z. Saponjic, J. Buha, D. Mijin, Photodegradation of an azo pyridone dye using TiO2 films prepared by the spray pyrolysis method. Chem. Eng. J. 180, 57–65 (2012)
G. Ali, M. Maqbool, Field emission properties of TiO2 nanotubes fabricated on Ti wire. Mater. Chem. Phys. 233, 21–26 (2019)
M.D. Hernandez-Alonso, I. Tejedor-Tejedor, J.M. Coronado, J. Soria, M.A. Anderson, Sol–gel preparation of TiO2–ZrO2 thin films supported on glass rings: influence of phase composition on photocatalytic activity. Thin Solid Films 502, 125–131 (2006)
Z. Ding, X. Hu, P.L. Yue, G.Q. Lu, P.F. Greenfield, Synthesis of anatase TiO2 supported on porous solids by chemical vapor deposition. Catal. Today 68, 173–182 (2001)
S. Varnagiris, D. Girdzevicius, M. Urbonavicius, D. Milcius, Incorporation of SiO2 and TiO2 additives into expanded polystyrene foam using physical vapour deposition technique. Energy Procedia 128, 525–532 (2017)
B. Grbić, N. Radić, S. Stojadinović, R. Vasilić, Z.D. Mitrović, Z. Šaponjić, P. Stefanov, TiO2/WO3 photocatalytic composite coatings prepared by spray pyrolysis. Surf. Coat. Technol. 258, 763–771 (2014)
I. OjaAcik, A. Junolainen, V. Mikli, M. Danilson, M. Krunks, Growth of ultra-thin TiO2 films by spray pyrolysis on different substrates. Appl. Surf. Sci. 256, 1391–1394 (2009)
G. Žerjav, G. Scandura, C. Garlisi, G. Palmisano, A. Pintar, Sputtered vs. sol-gel TiO2-doped films: characterization and assessment of aqueous bisphenol A oxidation under UV and visible light radiation. Catal. Today 357, 380–391 (2020)
S. Stojadinovic, N. Tadic, N. Radic, B. Grbic, R. Vasilic, TiO2/SnO2 photocatalyst formed by plasma electrolytic oxidation. Mater. Lett. 196, 292–295 (2017)
S.K. Shinde, S.M. Mohite, A.A. Kadam, H.M. Yadav, G.S. Ghodake, K.Y. Rajpure, D.S. Lee, D.-Y. Kim, Effect of deposition parameters on spray pyrolysis synthesized CuO nanoparticle thin films for higher supercapacitor performance. J. Electroanal. Chem. 850, 113433 (2019)
R. Daghrir, P. Drogui, D. Robert, Modified TiO2 for environmental photocatalytic applications: a review. Ind. Eng. Chem. Res. 52(10), 3581–3599 (2013)
A. Enesca, L. Isac, L. Andronic, D. Perniu, A. Duta, Tuning SnO2–TiO2 tandem systems for dyes mineralization. Appl. Catal. B. 147, 175–184 (2014)
Z. Balta, E.B. Simsek, Insights into the photocatalytic behavior of carbon-rich shungite-based WO3/TiO2 catalysts for enhanced dye and pharmaceutical degradation. New Carbon Mater. 35(4), 371–383 (2020)
C.F. Carbuloni, J.E. Savoia, J.S.P. Santos, C.A.A. Pereira, R.G. Marques, V.A.S. Ribeiro, A.M. Ferrari, Degradation of metformin in water by TiO2–ZrO2 photocatalysis. J. Environ. Manag. 262, 110347 (2020)
M. Mondal, H. Dutta, S.K. Pradhan, Enhanced photocatalysis performance of mechano-synthesized V2O5–TiO2 nanocomposite for wastewater treatment: correlation of structure with photocatalytic performance. Mater. Chem. Phys. 248, 122947 (2020)
I. Tanabe, T. Ryoki, Y. Ozaki, Significant enhancement of photocatalytic activity of rutile TiO2 compared with anatase TiO2 upon Pt nanoparticle deposition studied by far-ultraviolet spectroscopy. Phys. Chem. Chem. Phys. 16, 7749–7753 (2014)
N.S. Allen, N. Mahdjoub, V. Vishnyakov, P.J. Kelly, R.J. Kriek, The effect of crystalline phase (anatase, brookite and rutile) and size on the photocatalytic activity of calcined polymorphic titanium dioxide (TiO2). Polym. Degrad. Stab. 150, 31–36 (2018)
N. Radic, B. Grbic, S. Petrovic, S. Stojadinovic, N. Tadic, P. Stefanov, Effect of cerium oxide doping on the photocatalytic properties of rutile TiO2 films prepared by spray pyrolysis. Physica B 599, 412544 (2020)
R. Madaj, E. Sobiecka, H. Kalinowska, Lindane, kepone and pentachlorobenzene: chloropesticides banned by Stockholm convention. Int. J. Environ. Sci. Technol. 15, 471–480 (2018)
T. Novakovic, N. Radic, B. Grbic, T. Marinova, P. Stefanov, D. Stoychev, Oxidation of n-hexane over Pt and Cu–Co oxide catalysts supported on a thin-film zirconia/stainless steel carrier. Catal. Commun. 9, 1111–1118 (2008)
H. Imai, H. Hirashima, Preparation of porous anatase coating from sol-gel derived titanium dioxide and titanium dioxide silica by water vapor exposure. J. Am. Ceram. Soc. 82, 2301–2304 (1999)
N. Arconada, A. Duran, S. Suarez, R. Portela, J.M. Coronado, B. Sanchez, Y. Castro, Synthesis and photocatalytic properties of dense and porous TiO2-anatase thin films prepared by sol–gel. Appl. Catal. B. 86, 1–7 (2009)
C.A. Strydom, C.P.J. Van Vuuren, The thermal decomposition of cerium (III) nitrate. J. Therm. Anal. 32, 157–160 (1987)
L. Jun, W. Liang-liang, F. Zhao-yang, C. Xian, T. Ji-hai, C. Mi-fen, Q. Xu, Structure and properties of amorphous CeO2@TiO2 catalyst and its performance in the selective catalytic reduction of NO with NH3. J. Fuel Chem. Technol. 44(8), 954–960 (2016)
Y. Wang, J. Zhao, T. Wang, Y. Li, X. Li, J. Yin, C. Wang, CO2 photoreduction with H2O vapor on highly dispersed CeO2/TiO2 catalysts: surface species and their reactivity. J. Catal. 337, 293–302 (2016)
H. Liu, M. Wang, Y. Wang, Y. Liang, W. Cao, Y. Su, Ionic liquid-templated synthesis of mesoporous CeO2–TiO2 nanoparticles and their enhanced photocatalytic activities under UV or visible light. J. Photochem. Photobiol. A 223, 157–164 (2011)
B. Huang, D. Yu, Z. Sheng, L. Yang, Novel CeO2@TiO2 core–shell nanostructure catalyst for selective catalytic reduction of NOx with NH3. J. Environ. Sci. 55, 129–136 (2017)
J. Cheng, L. Song, R. Wu, S. Li, Y. Sun, H. Zhu, W. Qiu, H. He, Promoting effect of microwave irradiation on CeO2–TiO2 catalyst for selective catalytic reduction of NO by NH3. J. Rare Earths 38, 59–69 (2020)
M.H. Basha, N.O. Gopal, Solution combustion synthesis and characterization of phosphorus doped TiO2–CeO2 nanocomposite for photocatalytic applications. Mater. Sci. Eng. B 236–237, 43–47 (2018)
R. Gusain, K. Gupta, P. Joshi, O.P. Khatri, Adsorptive removal and photocatalytic degradation of organic pollutants using metal oxides and their composites: a comprehensive review. Adv. Colloid Interface Sci. 272, 102009 (2019)
I. Ahmad, M.S. Akhtar, M.F. Manzoor, M. Wajid, M. Noman, E. Ahmed, M. Ahmad, W.Q. Khan, A.M. Rana, Synthesis of yttrium and cerium doped ZnO nanoparticles as highly inexpensive and stable photocatalysts for hydrogen evolution. J. Rare Earths 39, 440–445 (2021)
I. Ahmad, Inexpensive and quick photocatalytic activity of rare earth (Er, Yb) co-doped ZnO nanoparticles for degradation of methyl orange dye. Sep. Purif. Technol. 227, 115726 (2019)
Acknowledgements
This work was supported by the Ministry of Education, Science and Technological Development of the Republic of Serbia (Grant No. 451-03-9/2021-14/200026) and by the bilateral cooperation between the Bulgarian Academy of Sciences and the Serbian Academy of Sciences and Fine Arts (project ‘‘Heterogeneous catalytical and photocatalytical destruction of organic and pharmaceutical contaminants in the nature by multicomponent systems’’).
Author information
Authors and Affiliations
Contributions
NR: Supervision; Conceptualization; Methodology; Formal analysis; Investigation; Writing–Original Draft; Visualization. BG: Conceptualization; Investigation; Writing–Original Draft; Supervision. SS: Methodology; Investigation; Formal analysis; Visualization. MI: Methodology; Resources; Investigation. OD: Methodology; Investigation. PS: Methodology; Resources; Investigation; Conceptualization.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest. We declare that this submission complies to the ethical standards of the journal and that there is no conflict of interest. We declare that this manuscript is original, has not been published before and is not currently being considered for publication elsewhere. The article has been written by the stated authors who are all aware of its content and approve its submission. All authors have endorsed the publication of this research. If accepted, the article will not be published elsewhere in the same form, in any language, without the written consent of the publisher.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Radić, N., Grbić, B., Stojadinović, S. et al. TiO2–CeO2 composite coatings for photocatalytic degradation of chloropesticide and organic dye. J Mater Sci: Mater Electron 33, 5073–5086 (2022). https://doi.org/10.1007/s10854-022-07698-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10854-022-07698-9