
Abstract
Different nanoparticles (NPs) including S1, S2, S3, S4, and S5 are prepared, using the co-precipitation route. The substitution of Co and Ni into the SnO2 matrix brought considerable modification in the physical attributes of all co-doped samples. The XRD data reveal that the calculated crystallite size is reduced from 10.54 to 1.65 nm with piling up oxygen vacancies (OVs) concentration when the Co content varies from 2.2 to 11 mM. The insertion of Co and Ni in SnO2 (lattice) produces ample alteration in the material traits of all samples. The dielectric properties strongly exhibit doping dependence. The dielectric measures including (dielectric loss ε′, dielectric loss factor tan δ, and AC conductivity σac ) are varied with growing Co content and achieved the utmost values for S4 NPs. The tanδ shows the dispersive nature, and relaxation peaks are identified, in which intensities are enhanced with growing Co content. The dielectric behavior and σac variations show that the dispersion is induced by polarization at the interfaces due to OVs and hopping processes. All these materials (samples) are interesting for device applications due to their large dielectric constant. The appearance of hysteresis loops show ferromagnetic nature with enhancement in saturation magnetization arises from a substantial measure of induced OVs (defects) by co-doping in the system (samples). The current work shows the doping-induced variations in dielectric and magnetic attributes. The OVs produce ferromagnetic behavior in (Ni, Co) co-doped SnO2 NPs applicable in high-frequency dielectric devices, and spintronics.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The diluted magnetic semiconductors (DMS’s) are the key components of proposed spintronic devices and, thus, are the focus of researchers including [1,2,3,4]. Ferromagnetism at 300 K (FRT) in Mn-doped GaN and ZnO somewhere else in [5], enabled other research groups to focus on doped and undoped metal oxides including ZnO, TiO2, and SnO2 [6,7,8,9,10,11,12]. The DMSs can be formed by substituting a specific amount of magnetic dopants to the host particles (within the lattice). Typically, it is represented as N(1−y) My O(n−δ)) where M is magnetic dopant, δ shows the concentration of OVs that depends on the growth conditions and formation process, while n would be 1 or 2. The term “N” shows a non-magnetic cation. The DMS’s magnetic properties depend on (for spintronic applications) carrier densities, the number of magnetic dopants and the nature of the crystals. Although metal oxides (MOs) are frequently studied in detail, there is ambiguities about the ferromagnetism origin in these MO-based DMSs. Recently due to numerous studies and scientific work on nanostructures, doped MOs in bulk, and thin films, different research groups are agreed upon that defects in these MOs show an important role in the ferromagnetism origin and dielectric behavior [7, 11,12,13,14,15,16,17]. The dielectric properties (including σac, tan δ, and εr) of metal oxides like SnO2 are very important to be studied for the device applications in microelectronics. The dielectric nature furnishes the free band conduction, ac conductivity, and internal position of the specimen with proportionately less conductivity [18]. The defects (intrinsic OVs) and dopants play an important role to modify the behavior of metal oxides especially SnO2 (n-type) with a band gap of 3.60 eV at 27 °C. The presence of more defects (OVs) in SnO2 (in its rutile tetragonal shape) is responsible for its high n-type conductivity. The transparency in visible light, the large conductivity, and the high infrared reflectance enable it a suitable candidate for technological point of view [19,20,21]. To modify its properties, the formation and crystal growth of SnO2 nanostructures with a doping agent are required. The introduction of suitable dopants to SnO2 lattice resulting to modify its structure, defect density, its dielectric, optical, and magnetic properties. To further improve its electrical and magnetic properties, researchers proposed the substitution of a couple of magnetic dopants (co-doping) rather than a single dopant. The saturation magnetization is increased as a consequence of Iron, Cobalt co-doping in SnO2 [22]. The electric and magnetic characteristics of (Fe, Co):SnO2 is given by Wang et al. [23], where the exchange interaction between Iron and Cobalt ions reduces by enhancing the (Fe, Co) concentrations, and thus, no increase in Curie- temperature TC takes place.
This research work describes the structural, magnetic, and dielectric properties of S1, S2, S3, S4, and S5 NPs formed by the co-precipitation technique. The Ni concentration (11 mM) is fixed and the Co- concentration is varied from 2.2, 6.6, to 11mM. The introduction of Cobalt in the SnO2 matrix prompts a decline in particle size while an uptick in defects (OVs). The increment in OVs in (Co, Ni) co-doped SnO2 NPs contributes to the facilitation to improve both the magnetic and dielectric features.
2 Experimental
2.1 Samples
SnCl2·2H2O (99%), CoCl2 (99%), NiCl2 (99%), and NH3·H2O (25–28%) are obtained from Sigma Aldrich. Distilled water (18.3MΩ) is obtained and utilized for the analyses.
2.2 Formation of Sn (1−x−y) NixCoyO2 NPs
All chemicals are utilized without further purification (As of analytical grade). (0.22 M) of SnCl2·2H2O into 50 mL of ultrapure water and stirring was maintained until SnCl2·2H2O were dissolved solely. In the next step, NH3 (37%) of 5 mL is combined with distilled water (25 mL) and was stirred for 5 min. The above sedimentary ammonia suspension is then poured into the prepared precursor suspension by ongoing stirring dropwise 3 mL/minute. The sediments were then consolidated via centrifugation followed by consequent rinsing with purified water and were parched at 60 °C and annealed at a temperature of 400 °C in the furnace for 2 h to accomplish SnO2 NPs [24]. Sn (1−x−y)NixCoyO2 NPs were synthesized by adding x = 0.011 M ( fixed) NiCl2, and (y = 2.2 mM, 6.6 mM, and 11 mM ) of CoCl2·6H2O to the 50 mL solution comprising SnCl2·2H2O. The rest of the process is the same as that of pure SnO2 NPs. The SnO2, and (xNi, yCo) co-doped SnO2 NPs with (x = 11mM ( fixed ) and y = 2.2, 6.6 and 11 mM) are labeled as S1, S2, S3, S4, and S5 respectively.
2.3 Materials characterizations
The specimens after synthesis are undergone through different techniques including XRD, SEM, EDX, and FTIR. The structural properties are investigated through (XRD) using a JEOL-3532 running at 40 kV and 30 mA having CuKα (λ = 1.54056 Å) source. The morphological peculiarities of the samples are recorded by JSM-5910 JEOL (Japan) SEM. The elemental arrangement is determined EDX linked to the SEM. The FTIR spectra were recorded by a Perkin Elmer Spectrum with a twin FTIR unit, outfitted with a Universal Attenuated Total Reflectance accomplice. The dielectric behavior and AC conductivity are obtained on a Gold coated pellet by a specific Impedance analyzer (40 Hz–7 M Hz). The dc magnetization room-temperature calculations are taken via the Quantum Design system (MPMS).
3 Results and discussion
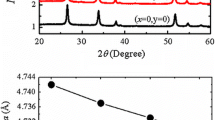
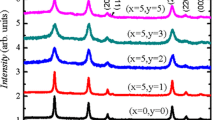
The XRD patterns of all samples are depicted in Fig. 1a. All of the X-ray reflections are in agreement with the rutile tetragonal structure of SnO2 as justified by JCPDS data (41-1445). The tetragonal structure of SnO2 does not alter with doping. Also no evidence of impurity phases of Co and Ni declares that Co and Ni are doped in the SnO2 matrix. The increase in Co content enables the position of peaks to shift slightly towards larger angles with reducing intensities and enhancement in peak widths, which could be due to the small ionic radius of Co2+ (0.065 nm) that replaces the larger Sn4+ (0.069 nm) ions as shown in Fig. 1b. XRD peaks look wider, indicating modification in the crystallite size and/or strain. The crystallite size and microstrain are determined by employing the Williamson-Hall (W-H) technique, utilizing the relation:
In the W-H plot, let the crystallite size and microstrain widening are extra parts of the integral of a Bragg peak as mentioned in the above equation. The broadening in strain arises from crystal defects and distortion which is given by
The crystallite size and lattice strain (dependent broadening) fit by a curve for all the peak widths (function of diffracting angle θ) and FWHM for XRD peak. The W-H graphs of the samples provide the equation of a straight line, y = mx + c with slope “m” equal to micro-structural strain, and the average crystallite size is calculated from intercept “c” depicted in Fig. 2. The estimated crystallite sizes and tensile strains for S1, S2, S3, S4, and S5 nanoparticles are indicated in Fig. 2. The maximum tensile strain and minimum crystallite size are reported for the S4 sample [25]. This clearly shows that higher concentrations of “Co” not only provide OVs for facilitating the densification but also help in the grain growth inhibitor role [21, 22]. For the highly doped samples, the grain size reduces due to the segregation at the grain boundaries.

a XRD patterns of all the samples, b Magnified XRD indicating shift in peaks

W-H graphs of all samples, tensile strain, and crystallite are given table
The SEM images of all samples (including rough surfaces) are depicted in Fig. 3. The spherical-shaped nanoparticles with increasing agglomeration are observed upon increasing the Co concentration. The elements of “Sn” and “O” and “Sn,” “O,” “Ni,” and “Co” are observed for the pure doped and co-doped samples. In S4, the oxygen content is minimum revealing a high concentration of defects (number of oxygen vacancies) in the sample as shown in Fig. 4a. It is observed from quantitative analysis of the EDX that increasing content of Co decreases Ni too, indicates that Co replaces Sn as well as Ni in the SnO2 lattice as shown in Fig. 4b.

SEM micrographs and EDX of a S1, b S2, c S3, d S4, e S5 samples

a Oxygen and Tin contents (at%), b Ni and Co contents (at%) of all samples
The FTIR of all synthesized samples is depicted in Fig. 5. From the spectrum of S1, the modes arise at 533 and 609 cm−1 holding the Sn–O asymmetric stretching vibrational mode and Sn–O–Sn to symmetric stretching vibrational mode, respectively. For S2, S3, S4, and S5 samples, the Sn–O asymmetric stretching vibration and Sn–O–Sn symmetric stretching vibrations are found at 547, 630 cm−1, 554, 623 cm−1 and 547, 602 cm−1 and 553, 612 cm−1, respectively. The higher frequency absorption peaks center arose at 3482 and 1628 cm−1 are assigned to O–H stretching vibrations and H2O adsorption (O–H bending vibrations) on SnO2 surface. The peaks shift slightly due to a decrease in crystallite size and the difference in the bond length that occurs when Co and Ni ions replace Sn ions, thus, confirming the incorporation of Co and Ni ion in SnO2 lattice.

FTIR spectra of all samples
UV–Vis absorption spectra are depicted in Fig. 6. By growing dopants content, the absorbance increases and its optimum value is obtained for the S4 sample exhibiting a comprehensive amount of OVs introduced by dopants into SnO2 lattice. The estimated band-gap energies are 3.28 eV, 3.01 eV, 2.83 eV, 2.52 eV, and 2.94 eV for S1, S2, S3, S4, and S5, respectively by Applying Tauc’s plot. The band-gap energies of the samples are reduced as a result of the existence of an enormous number of defects in them.

UV–Vis absorbance spectra and the estimated band-gap energies of all samples using Tauc’s plot
The room-temperature PL spectra of all samples are shown in Fig. 7. The enhanced intensity of the PL emission S3, S4, and S5 samples is pretty obvious. The PL emission centered at 560 nm might be from defect electronic states as a result of co-doping as well as due to the inhomogeneous strain present in these nanocrystals. Such defect states might also be possible by “bridging” OVs. In such a situation, electron is excited to the bulk shallow energy levels accompanied by a return to the intragap level of SnO2 corresponding to oxygen surface vacancies via radiative transitions might occur [26]. This strong luminescence was related to the bridging and in-plane OVs, which form a series of metastable energy levels within the band gap [27]. The nature of the transition causing these emissions was generally thought to be Sn or O vacancies formed during the doping process, inducing deeper trapping states within the band gap. The maximum intensity of S4 is due to small grain size (large number of grain boundaries and OVs).

Photoluminescence Spectra of all synthesized samples
The XPS of all S1, S2, S3, S4, and S5 NPs were carried out to find the elemental composition, and oxygen vacancies as shown in Fig. 8. Figure 8a shows the full-scan survey of all five samples indicating the most obvious peaks of Oxygen, Tin, Ni, Co, and C having no impurity peaks. For investigation of OVs in the all five samples, the high-resolution XPS spectra for O1s are shown in Fig. 10b. This peak is deconvoluted with three Gaussian peaks at 530.15 eV, 531.25 eV, and 532.4 eV, respectively. The peak-I at 530.15 eV is ascribed to the O2− ions in the structure. The peak-III at 532.4 eV is accredited to the loosely bound oxygen on the surface of SnO2. The Gaussian peak-II located at 531.25 eV is associated with O2− in the oxygen-deficient regions in S1, S2, S3, S4, and S5 NPs [28,29,30]. The intensity of this peak most probably causes variations in the concentration of OVs. Therefore, changes in the intensity of this peak might be due to variations in the number of OVs. From the O1s spectra, the intensity of peak located 531.25 eV is maximum in S4 sample revealing large concentration of OVs. This result is in firm agreement with UV–Vis, EDX, and PL spectra.

a Full-survey scans XPS, b HR-XPS of O1s for S1, S2, S3, S4, and S5 nanoparticles
Figure 9a shows relative permittivity (εr) (a function of frequency) for S1, S2, S3, S4, and S5 nanoparticles. The εr decreases with increasing frequency, so that the dispersion behavior is exhibited by all the samples. The decrease in εr at low frequencies is very sharp and reduces with increasing frequencies, and at very high frequencies, it becomes constant. The dispersion in εr is explained on the basis of the Maxwell–Wagner model and Koops theory [31, 32]. It describes that the volume of grain boundaries (poorly conducting) play a major role to control the variation in εr at small frequencies. The explanation on the variation in εr can also be based on hopping model, describing that at small frequencies, the variation in εr is due to charge carriers hopping in the field direction up to grain boundaries, where the accumulation of all charge carriers takes place and, hence, generates large polarization. However, the decreasing εr at higher frequencies is due to lagging of the changing direction of the carriers behind that of the applied field. Resulting in the molecular dipoles (which take more time) to change their direction with applied field, which is causing to decrease their probability of reaching grain boundaries and, hence, polarization. The dielectric modification in nanomaterials can also be described by rotation direction polarization (RDP) and the space charge polarization (SCP) [33, 34]. The contribution of RDP is very important for higher εr of the Sn(1−x−y) NixCoyO2 NPs. There are various defects in the case of n-type semiconductor (oxides), acting as shallow donors. The OVs contribute to RDP and, in nanomaterials, large specific surface area contributes to SCP [35, 36]. Due to the presence of external electric field, charges are attracted to opposite sides of electric field and ensnared by defects (oxygen vacancies) rendering SCP. In nanomaterials due to large surface to volume ratio, there is significant contribution of RDP and SCP in comparison to bulk, which enhances εr at low frequencies. At high frequencies, the alterations in electric field direction are so abrupt that RDP and SCP cannot keep up, and as a result, εr decreases. The reduction in crystallite size with enhancement in Co concentration causes to increase the interfaces, and hence, εr increases due to collective contribution of RDP and SCP.
The utmost value of εr is reported for S4 nanoparticles due to minimum crystallite size and maximum strain having large number of grain boundaries and large surface area accommodating maximum defects (OVs).
The dielectric loss factor (tan δ) vs. frequency for S1, S2, S3, S4, and S5 nanoparticles is manifested in Fig. 9b. Figure 9b exhibits the manifestation of greater tan δ at smaller frequencies that are evincing from SCP. The SCP elicits as a consequence of the ensnaring of the surface electron by the dopants in bulk [36]. This implies that an abundance of impurity ions contributes to substantial SCP at the surface. Relaxation peaks are ascertained for all samples but the utmost value is achieved in the case of S4 nanoparticles as exhibited in Fig. 9b. Relaxation peaks originate when the hopping frequencies of the charge carriers and applied field match [37].
Figure 9c presents the variation in conductivity (σac) as a function of frequency. In the case of small frequencies, ac conductivity is inadequate as a result of scattering of charge carriers through infinite paths. Improvement in conductivity is observed at higher frequencies, ascribed to the increasing applied field, which compels the conduction of more and more charge carriers along its direction. At high enough frequencies, the enhancement in conductivity is abrupt, which is owing to the hopping of the charge carriers between the Sn4+/Sn2+, and Co2+/Co3+ ions causing an enhancement in the conduction of charges, and therefore, increases the σac. The utmost value of σac is reported for S4 as illustrated in Fig. 9c. It is most likely due to the incorporation of Co2+ in SnO2 accompanied by oxygen vacancies (OVs), and at high enough frequencies, the charges unleashed from oxygen vacancies are accompanied by conduction charge carriers to boost conductivity.

Dielectric properties a Dielectric constant (ϵr), b Dielectric loss factor (tan δ), c AC conductivity, (σac) of all samples
Figure 10a reveals (H) loops at 300 K for S2, S3, S4, and S5 nanoparticles. Figure 10a suggests that with growing Co content, room-temperature ferromagnetism is improved. This improvement is attributable to the creation of large defects (OVs) with increasing Co concentration. This enrichment is a result of a large concentration of oxygen vacancies (OVs) with scaling up Co concentration. The creation of oxygen vacancies (OVs) through the Co and Ni (dopants) is being described as immaculate SnO2 contains Sn4+ ions and O2− ions, and the introduction of dopants (Ni2+ and Co2+/Co3+ ions) to SnO2 lattice disrupts the overall charge balance of the arrangement through excessive negative charge. To maintain charge balance, certain proportion of O2− ions escapes from the matrix leaving behind OVs in the vicinity of dopants (Co and Ni ions) [38]. These oxygen vacancies (OVs) capture conduction charge carriers and the spin–spin interaction of these captured carriers and localized Co ions renders magnetization via the establishment of bound magnetic polarons (BMPs) [39]. The substantial amount of BMPs constitutes magnetic domains and, consequently, ferromagnetism. The boost in magnetization with improving dopant(Co concentration) is ascribed to preferring parallel spin–spin pairing (between Co ions and proximate oxygen atoms contribute to hybridization and carrier swapping between OVs and Co ions) [40]. The furtherance of Ms, Mr, and Hc with the growing amount of dopant(Co) as manifested in Fig. 10b is ascribed to tiny nanoparticles and copious oxygen vacancies.

a M(H) loops of all samples and b Saturation magnetization (Ms) remanence Magnetization (Mr) Coercive field (Hc) vs. Co concentration
4 Conclusions
In a nutshell, we prepared S1, S2, S3, S4, and S5 nanoparticles through the co-precipitation route. The introduction of dopants (Ni, Co) to the SnO2 matrix created substantial modifications in the properties of all the samples. The crystallite size was reduced and the micro-tensile strain enhanced. The absorbance spectra revealed a large number of OVs. The dielectric study reveals a doping dependency. The reduced crystallite size, a large number of grain boundaries, and a substantial amount of defects (OVs) contribute to the scaling up of ϵr, σac, and strong ferromagnetism (large Ms, Mr, and Hc). The current investigations present OVs induced ferromagnetism in (Ni, Co) co-doped SnO2 nanoparticles. Exquisite dielectric and magnetic characteristics of (Ni, Co) co-doped SnO2 make them, prospective suitors, for dielectric materials applications, high-frequency equipment, and applications in spintronics.
References
S. Mehraj, M.S. Ansar, Alimuddin, Structural, electrical and magnetic properties of (Fe,Co)co-dopedSnO2 diluted magnetic semiconductor nanostructures. Phys. E 65, 84–92 (2015)
Y. Zulfiqar, J. Yuan, W. Yang, Z. Wang, Ye, J. Lu, Structural, dielectric and ferromagnetic behavior of (Zn,Co) co-doped SnO2 nanoparticles. Ceram. Int. 42, 17128–17136 (2016)
Zulfiqar, M. Zubair, A. Khan, T. Hua, N. Ilyas, S. Fashu, A.M. Afzal, M.A. Safeen, R. Khan, Oxygen vacancies inducedroom temperature ferromagnetism and enhanced dielectric properties in Co and Mnco-doped ZnO nanoparticles. Mater. Sci. Mater. Electron. 32, 9463–9474 (2021)
Y. Zulfiqar, Q. Yuan, J. Jiang, L. Yang, W. Feng, Z. Wang, J. Ye, Lu, Variation in luminescence and bandgap of Zn-doped SnO2 nanoparticles with thermal decomposition. J. Mater. Sci. Mater. Electron. (2016). https://doi.org/10.1007/s10854-016-5006-3
T. Dietl, H. Ohno, F. Matsukura, J. Cibert, D. Ferrand, Zener model description of ferromagnetism in Zinc-blende magnetic semiconductors. Science 287, 1019–1022 (2000)
P. Sharma, A. Gupta, K.V. Rao, F.J. Owens, R. Sharma, R. Ahuja, J.M.O. Guillen, B. Johansson, G.A. Gehring, Ferromagnetism above room temperature in bulk and transparent thin films of Mn-doped ZnO. Nat. Mater. 2, 673–677 (2003)
D.A. Schwartz, N.S. Norberg, O.P. Nguyen, J.M. Parker, D.R. Gamelin, Magnetic quantum dots: synthesis, spectroscopy, and magnetism of Co2+ and Ni2+ doped ZnO nanocrystals. J. Am. Chem. Soc. 125, 13205–13218 (2003)
P.V. Radovanovic, D.R. Gamelin, High-temperature ferromagnetism in Ni2+doped ZnO aggregates prepared from colloidal diluted magnetic semiconductor quantum dots. Phys. Rev. Lett. 91, 1–4 (2003)
L.F. Liu, J.F. Kang, Y. Wang, H. Tang, L.G. Kong, X. Zhang, R.Q. Han, The role of oxygen vacancies in magnetism of Co x Ti 1– x O 2–δ films on Si(001) prepared by sol–gel method. Solid State Commun. 139, 263–267 (2006)
R. Suryanarayana, V.M. Naik, P. Kharel, P. Talanga, R. Naik, Room temperature ferromagnetism in spin-coated anatase- and rutile-Ti0.95Fe0.05O2 films. Phys. Cond. Matter. 17, 755–762 (2005)
J. Bryan Daniel, S.A. Santagdo, S.C. Keveren, D.R. Gamelin, Activation of high TC ferromagnetism in Co2+:TiO2 and Cr3+:TiO2, nanorods and nanocrystals by grain boundary defects. J. Am. Chem. Soc. 127, 15568–15574 (2005)
P.I. Archer, P.V. Rodovanovic, S.M. Heald, D.R. Gamelin, Low temperature activation and deactivation of high TC ferromagnetism in a new diluted magnetic semiconductor: Ni2+ doped SnO2. J. Am. Chem. Soc. 127, 14479–14487 (2005)
G. Peleckis, X. Wang, S.H. Dou, High temperature ferromagnetism in Ni-doped In2O3 and indium-tin oxide. Appl. Phys. Lett. 89, 022501–022503 (2006)
N.H. Hong, N. Poirot, J. Sakai, Evidence for magnetism due to oxygen vacancies in Fe-doped HfO2 thin films Appl. Phys. Lett. 89, 042503–042503 (2006)
A. Tiwari, V.M. Bhosle, S. Ramachandran, N. Sudakar, J. Narayan, S. Budak, A. Gupta, Ferromagnetism in Co doped CeO2: observation of a giant magnetic moment with a high Curie temperature. Appl. Phys. Lett. 88, 142511–142513 (2006)
J.M.D. Coey, M. Venkatesan, C.B. Fitzgerald, Donor impurity band exchange in dilute ferromagnetic oxides. Nat. Mater. 4, 173–179 (2005)
J.M.D. Coey, A.P. Douvalis, C.B. Fitzgerald, M. Venkatesan, Ferromagnetism in Fe-doped SnO2 thin films. Appl. Phys. Lett. 84, 1332–1334 (2004)
P.P. Sahay, R.K. Mishra, S.N. Pandey, S. Jha, M. Shamsuddin, Structural, dielectric and photoluminescence properties of co-precipitated Zn-doped SnO2 nanoparticles. Curr. Appl. Phys. 13, 479–486 (2013)
A.K. Sinha, P.K. Manna, M. Pradhan, C. Mondal, S.M. Yusuf, T. Pal, Tin oxide with a p–n heterojunction ensures both UV and visible light photocatalytic activity. RSC Adv. 4, 208–211 (2014)
V.B. Kamble, A.M. Umarji, Defect induced optical bandgap narrowing in undoped SnO2 nanocrystals. AIP Adv. 3, 082120–082125 (2013)
D.S. Ginley, C. Bright, Transparent conducting oxides. MRS Bull. 25, 15–18 (2000)
K. Nomura, J. Okabayashi, K. Okamura, Y. Yamada, Magnetic properties of Fe and Co codoped SnO2 prepared by sol–gel method. J. Appl. Phys. 110, 083901–083903 (2011)
X.L. Wang, Z. Zeng, X.H. Zheng, H.Q. Lin, First-principles investigations of Co and Fe doped SnO2. J. Appl. Phys. 101, 09 H104 (2007)
Y. Zulfiqar, Q. Yuan, J. Jiang, L. Yang, W. Feng, Z. Wang, J. Ye Lu, Structural and optical properties of (Zn, Co) co-doped SnO2 nanoparticles. J. Mater. Sci. Mater. Electron. (2016). https://doi.org/10.1007/s10854-016-5364-x
N. Hussain, S. Zulfiqar, T. Khan, R. Khan, S.A. Khattak, S. Ali, G. Khan, Investigation of structural, optical, dielectric and magnetic properties of Sno2 nanorods and nanospheres. Mater. Chem. Phys. 241, 122382 (2020)
X.S. Fang, C.H. Ye, L.D. Zhang, T. Xie, Twinning mediated growth of Al2O3 nanobelts and their enhanced dielectric responses. Adv. Mater. 17, 1661–1665 (2005)
J.G. Han, Z.Y. Zhu, S. Ray, A.K. Azad, W.L. Zhang, M.X. He, S.H. Li, Y.P. Zhao, Optical and dielectric properties of ZnO tetrapod structures at terahertz frequencies. Appl. Phys. Lett. 89, 031107–031103 (2006)
S. Mehraj, M.S. Ansari, Alimuddin, Rutile-type Co doped SnO2 diluted magnetic semiconductor nanoparticles: structural, dielectric and ferromagnetic behavior. Phys. B 430, 106–113 (2013)
A.M. Feroz, M.B. Khalid, C. Indrajeet, G.M. Bhat, Preparation and ac electrical characterizations of Cd doped SnO2 nanoparticles. J. Mater. Sci. Mater. Electron. 25, 1564–1570 (2014)
Zulfiqar, S.A. Khan, N. Rahman et al., Oxygen vacancies induced variations in structural, optical and dielectric properties of SnO2/graphite nanocomposite. J. Mater. Sci. Mater. Electron. 32, 1402–1412 (2021)
S. Gnanam, V. Rajendran, Preparation of Cd-doped SnO2 nanoparticles by sol–gel route and their optical properties. J. Sol–Gel. Sci. Technol. 56, 128–133 (2010)
J.C. Maxwell, Electricity and Magnetism (Clarendon, Oxford, 1892)
C.G. Koops, On the dispersion of resistivity and dielectric constant of some semiconductors at audio frequencies. Phys. Rev 83, 121 (1951)
S.M. Zhou, Y.S. Feng, L.D. Zhang, A physical evaporation synthetic route to large-scale GaN nanowires and their dielectric properties. Chem. Phys. Lett. 369, 610 (2003)
H.R. Ricardo, P. Hidalgo, H.E.M. Perez, F.J. Ramirez, D. Gouvea, Relationship between surface segregation and rapid propane electrical response in Cd-doped SnO2 nanomaterials Sens. Actuators B 133, 263–269 (2008)
A. Bouaine, N. Brihi, G. Schmerber, C. Ulhaq-Bouillet, S. Colis, A. Dinia, Structural, optical, and magnetic properties of Co-doped SnO2 powders synthesized by the co-precipitation technique. J. Phys. Chem. C 111, 2924–2928 (2007)
W. Shochley, W.T. Read, Statistics of the recombinations of holes and electrons. Phys. Rev. 87, 835–842 (1952)
N. Rezlescu, E. Rezlescu, Dielectric properties of copper containing ferrites. Phys. Status Solidi A 59, 323 (1980)
R. Khan, Zulfiqar, S. Fashu, M.U. Rahman, Effects of Ni co-doping concentrations on dielectric and magnetic properties of (Co, Ni) co-doped SnO2 nanoparticles. J. Mater. Sci. Mater. Electron. (2016). https://doi.org/10.1007/s10854-016-4759-z
H. Jiang, X.F. Liu, Z.Y. Zou, Z.B. Wu, B. He, R.H. Yu, The effect of surfactants on the magnetic and optical properties of Co-doped SnO2 nanoparticles. Appl. Surf. Sci. 258, 236–241 (2011)
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zulfiqar, Rahman, S.u., khan, T. et al. Dielectric and ferromagnetic properties of (Ni, Co) co-doped SnO2 nanoparticles. J Mater Sci: Mater Electron 32, 19859–19870 (2021). https://doi.org/10.1007/s10854-021-06510-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10854-021-06510-4