
Abstract
A set of D–A–π–A dyes containing a carbazole donor, linker composed of a naphthalimide auxiliary acceptor and spacers such as phenyl, oligothiophene, dithienopyrrole and cyanoacrylic acid acceptor have been synthesized and characterized. Influence of spacers on the optical and photovoltaic properties are analyzed. Nature of the spacer between naphthalimide and cyanoacrylic acid show significant effect on the absorption maximum. Elongation of conjugation and increment in electron richness gradually red shift the absorption. It also helps to tune the oxidation potential of the dyes. All dyes possess high-lying LUMO and low-lying HOMO when compared to CB of TiO2 and reduction potential of iodide electrolyte, which facilitate the electron injection into TiO2, and regeneration of the oxidized dye by electrolyte, respectively. A dye containing dithienopyrrole led to maximum photocurrent density of 3.60 mA cm−2 and an open circuit voltage of 519 mV in the series. This is attributed to the diminished resistance for charge transfer and long-lived electron lifetime when compared to the other dyes. The electrochemical impedance spectroscopy revealed small charge recombination resistance at TiO2/dye/electrolyte interface for all the dyes attesting the low open circuit voltages of the devices.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
After the pioneering work by O’Regan and Grätzel on dye-sensitized solar cells (DSSCs) [1,2,3] using ruthenium-based [4,5,6] organometallic and metal-free organic dyes [7,8,9] they attracted significant research focus due to their comparatively low cost, environmental friendliness, feasible functionalization with facile chemical modifications than the traditional silicon-based solar cells. Despite the superior performance exhibited by ruthenium-based dyes, metal-free organic dyes are considered as the most attractive owing to their ease in synthesis and purification, and superior photophysical properties that can be fine-tuned by simple structural alternations. Organic dyes (D–π–A) are typically constituted with structural units such as donor (D) and acceptor (A) bridged by a suitable aromatic π-conjugation segment [10,11,12]. Organic dyes with D–π–A configuration generally feature donors such as triarylamine [13,14,15], indoline [16,17,18,19], carbazole [20,21,22,23], phenothiazine [24,25,26,27] and dithienopyrrole [28,29,30]. The ease in functionalizing carbazole at the most active C-3 and C-6 position demonstrated it as versatile donor with planarity and rigidity as its structural property [31,32,33,34,35]. The linker also plays a pivotal role in balancing the electronic communication and extend of charge transfer between the donor and acceptor depending on its mode of linkage, electron density and co-planarity [36,37,38,39,40]. In addition to this basic architecture, various organic sensitizers featuring modified structural framework using auxiliary donor and acceptor such as D–D–π–A [41, 42] and D–A–π–A [43,44,45] are explored as an attempt to boost the photophysical and photovoltaic performance of DSSCs. The incorporation of additional auxiliary acceptor has been proved to be the most effective in fine-tuning light harvesting properties and the energy levels of the dyes [46]. Many auxiliary acceptors such as benzothiadiazole [47, 48], diketopyrrolopyrrole [49, 50], quinoxaline [51, 52], phthalimide [53], and benzotriazole [53,54,55,56], have been incorporated successfully to modify the excited state energetics of the dyes.
The benefit of tailoring organic sensitizer with an additional acceptor in configuration of D–A–π–A relies on facilitating electron transfer and improving photo-stability. Zhu et al. [45] demonstrated the advantages of benzothiadiazole auxiliary acceptor in enhancing photo-stability and light harvesting properties of indoline-based dyes. Cui et al. [53] emphasized the role of benzotriazole auxiliary acceptor in facilitating electron transfer from donor to acceptor and improving the performance of DSSCs fabricated with indoline dyes. They found that it helped to suppress charge recombination and enhance open-circuit photovoltage. Li et al. [54] synthesized two D–A–π–A organic sensitizers and studied effect of phthalimide and benzotriazole as auxiliary acceptor on absorption and charge recombination dynamics. The effect of conjugation extension by incorporation of imidic chromophore as a linker was illustrated recently by Bobe et al. [56]. They have synthesized a triphenylamine-based dye containing naphthalenediimide as π-bridge with improved light harvesting, photocurrent injection, electron lifetime and DSSC performance compared to its analog lacking additional π-extender. The use of naphthalimide [57,58,59,60,61,62] acceptor in triphenylamine and indoline-based organic dyes with D–π–A architecture with a carboxylic group as anchoring group was reported by Huang et al. [60]. Margalias et al. [61] reported D–π–A architecture based on triphenylamine donor, naphthalimide-fused benzimidazole linker and carboxylic group acceptor to illustrate the effect of additional electron donating groups on the photophysical and photovoltaic properties of the dyes.
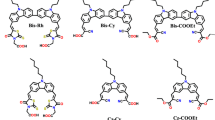
Herein, we use a new design featuring naphthalimide as auxiliary acceptor bridge tethered to carbazole donor at imidic nitrogen cyanoacrylic acid acceptor on C4 via an aromatic spacer. The objective of the work is to elucidate the effect of naphthalimide in the bridging role and further study the effect of variations in the conjugation pathway on the optical and photovoltaic properties. The presence of naphthalimide unit is expected to have advantages such as (a) modulation in the excited state energetics of the dyes (b) suppression of vibrational energy loss in excited state of the dye due to rigid planar structure and (c) ease in fine-tuning the photophysical properties by the nature of aromatic spacer attached at C4. Herein we report four naphthalimide-based D–A–π–A organic sensitizers 6a–6d as shown in Fig. 1. To the surprise we observe that the longest wavelength absorption in the dyes originate from the π–π* electronic excitation confined to the naphthalimide, aromatic spacer and cyanoacrylic acid segment. The carbazole donor at the imidic nitrogen is decoupled from the rest of the molecule thereby resist charge transfer to acceptor segment. Though the dyes showed appreciable absorption, due to the lack intramolecular charge transfer, the dyes could not effectively harvest light energy. In the series, a dye 6d possessing dithienopyrrole as linker showed decent photons to electron conversion efficiency. An overall maximum photocurrent efficiency of 0.92% is observed for device based on 6d due to enhanced short circuit current and open circuit voltage. A structure–function correlation involving the nature of the spacers is established.

Structures of D–A–π–A naphthalimide-based dyes
2 Results and discussion
2.1 Synthesis and characterization
The synthetic protocol of the dyes 6a–6d is displayed in Schemes 1 and 2. The precursor 4-bromo-N-(9-ethyl-9H-carbazol-3-yl)-1,8-naphthalimide (3) was synthesized by following literature procedure [62]. It was subsequently treated with appropriate boronic acid or tin derivatives to yield aldehydes 5a–5c and dithienopyrrole derivative 4. Later 4 was converted to the corresponding aldehyde, 5d by Vilsmeier–Haack formylation reaction with DMF/POCl3. Finally the aldehyde derivatives (5a–5d) were subjected to Knoevenagel condensation [63] with cyanoacetic acid in the presence of ammonium acetate to form the dyes 6a–6d in good yields. All compounds were characterized thoroughly by NMR and mass spectroscopy. The spectral data of all the dyes are consistent with the proposed structures. The dyes assure yellow to red colour and show moderate solubility in common organic solvents such as toluene (Tol), dichloromethane (DCM), tetrahydrofuran (THF) and dimethyl formamide (DMF).

Synthesis of the dyes 6a–6c

Synthesis of the dye, 6d
2.2 Photophysical properties
The absorption spectra of the four sensitizers recorded in DCM are displayed in Fig. 2 and the corresponding photophysical data listed in Table 1. All the dyes exhibit two distinctive absorption bands, one originating from carbazole localized π–π* at shorter wavelength and other assignable to the delocalized π–π* absorption from the conjugated segment comprising naphthalimide, spacer and cyanoacrylic acid units. A progressive shift in absorption wavelength of the dyes is observed as the conjugating length and electron richness within the molecular skeleton is strengthened. It is found that the presence of units such as thiophene, bithiophene and dithienopyrrole in the conjugation pathway red shift the absorption profile due to the coplanar arrangement. Hence, the dye 6b exhibits a bathochromic shift of 41 nm when compared to 6a. Similarly, introduction of additional thiophene, for instance compare 6b and 6c, results in a red shift of ca. 46 nm attributable to the increase in the electron richness of the π-linker. Dye 6d possessing rigid and the most electron rich linker in the series displays absorption band at 481 nm with a bathochromic shift of 125 and 38 nm, respectively, in comparison to 6a and 6c. It also displays a characteristic absorption band corresponding to dithienopyrrole at 300–350 nm [29]. The effect of rigidified and electron rich linker is also reflected in the molar extinction coefficients of the absorption (Fig. 2).

Absorption spectra of the dyes 6a–6d recorded in DCM
It is interesting to compare the optical properties of the dyes 6a–6d with their precursors (Fig. S1 and Table S2). The present dyes exhibit a bathochromic shift of 10–50 nm in absorption profiles when compared to their aldehyde precursors attributed to the elongation of conjugation due to cyanoacrylic acid unit. Precursor 3 displays two absorption bands lying above and below 300 nm attributed to the π–π* transitions arising from carbazole and naphthalimide, respectively. Observation of localized absorptions points the absence of significant interactions between the two chromophores. Similar observations have been observed for N-functionalized naphthalimides before [62]. However, functionalization of 3 to different aryl aldehyde segments result in emergence of a new longer wavelength absorption. This appears at the cost of naphthalimide absorption. On comparing 4 and 5d, the later shows a 20 nm blue shift in absorption suggesting the effect of aldehyde. It is probable in 4, a weak donor acceptor interaction exists between naphthalimide and dithienopyrrole which is disrupted on introduction of aldehyde. It will be interesting to compare the optical properties of naphthalimide-based dyes with their control analogs, which lack naphthalimide. Compared to 2-cyano-3-[4-(9-ethyl-9H-carbazol-3-yl)phenyl]acrylic acid [64] and 2-cyano-3-[5-(9-ethyl-9H-carbazol-3-yl)thiophen-2-yl]acrylic acid [64], dyes 6a and 6b display a blue-shift of 6 and 14 nm in absorption, respectively. However, when compared to the known naphthalimide-based dyes, [60] present dyes 6c and 6d display superior absorption properties with a red shift of ~ 20–50 nm, despite the absence of donor–acceptor interaction.
Organic dyes when anchored on TiO2 display either blue or red shift depending on the nature of interaction among them and with TiO2 [65,66,67]. Deprotonation caused by strong interaction with TiO2 and H-aggregation between the dyes lead to blue shift while the J-aggregation among the dyes give red shift. The dyes except 6a show a broad and a blue shifted spectra when anchored on TiO2 (Fig. 3) than that observed in solution. This may be presumably either due to the formation of H-aggregates or deprotonation of the carboxyl group or large thickness of the TiO2 film. However, the spectral trend for dye 6a remained more or less same which can be related to presence of non-planar phenyl group as linker which prevent aggregation of dye. The formation of H-aggregates can be attributed to the rigid structure of naphthalimide unit which favored stacking interactions between the dyes.

Absorption spectra of the dyes 6a–6d recorded on TiO2 film
The effect of protonation and deprotonation of the carboxyl group is studied by the acid–base equilibria performed by addition of small amount of trifluoroacetic acid (TFA) and triethylamine (TEA) to the dye solutions in DCM (Fig. 4, Table S1). All the dyes displayed ~ 10 nm bathochromic shift for delocalized π–π* absorption band on addition of TFA. While addition of TEA leads to a hypsochromic shift which point towards the deprotonated state of dyes [68] in the solution particularly for 6c and 6d. The interaction of acid and base with the carboxyl group highlights the alteration of delocalization of electron density due to protonation and deprotonation of the dyes. Further to get insight into the solvent–solute interactions and nature of solvent on the photophysical behavior of dyes, solvatochromism [69] of dyes were observed (Fig. 4, S2 and S3). The effect of solvent polarity is less significant. In case of DMF due to the basic nature of solvent, the dyes exist in deprotonated state with a blue shift of absorption band similar to that of absorption profile observed on addition of TEA [70]. Since the solvent induced absorption changes are minimal, we can safely conclude that they originate due to minor alternations in the conjugation due to protonation/deprotonation.

Absorption spectra of the dye 6d recorded in different solvents
2.3 Electrochemical properties
In order to investigate the effect of linker on the redox characteristics of the dyes, cyclic voltammetry (CV) and differential pulse voltammetry (DPV) in DCM were performed and the data compiled in Table 1. All the dyes show irreversible oxidation couple at ~ 0.8 V attributed to removal of an electron from the carbazole. Multiple irreversible oxidations were observed on increasing the electron richness of the conjugation pathway (6c and 6d). These oxidation potentials are dependent on the nature of linker in the dyes. It is interesting to note that the dyes showed high oxidation potentials when compared to that of the aldehyde precursors owing to the comparatively electron-deficient cyanoacrylic acid unit. Further, in order to use these dyes as sensitizers in DSSCs, they should possess suitable energy levels for efficient electron injection from excited state of dye to TiO2 (− 4.2 eV/− 0.5 V) and regeneration of dye from I−/I3− electrolyte redox couple (− 5.1 eV/0.4 V) [70]. The HOMO of the dyes except 6c and 6d are estimated to be sufficiently lower than the redox potential of electrolyte redox couple which promotes efficient regeneration of oxidized dyes. This may be one of the reasons for inferior performance of DSSC devices by 6c and 6d although they possess better light harvesting and optical property among the dyes. It has been noticed that the replacement of phenyl with other linkers displayed significant changes in electrochemical properties of the dyes. The excited state oxidation potentials (Eox*) of the sensitizers are calculated to be more negative than the conduction band of TiO2. This allowed feasible electron injection from the excited state of the dyes into the conduction band of the TiO2. Among the dyes, dyes possessing electron rich linker (6b, 6c and 6d) exhibited low lying Eox* when compared to phenyl analog (6a) attributed to favourable electronic communication induced by relatively co-planar linker. This also attested the favourable kinetic over thermodynamic feasibility for the efficient electron injection (ΔGinj) into the conduction band of TiO2 by former set of dyes (Fig. 5).

Energy level diagram of the dyes
2.4 Theoretical analysis
In order gain insight on the electronic coupling between the various chromophoric segments and their role on optical and related properties, DFT calculations were performed using M06-2x/6-31G(d,p) method. The optimized structures show that the naphthalimide is significantly tilted from carbazole and spacer aromatic (Fig. 6). This retards electronic interaction between the donor–acceptor chromophores.

Optimized geometries and computed interplanar angles between different aryl groups of the dyes
The frontier molecular orbitals of the selected dyes are displayed in Fig. 7. It is obvious to mention that the HOMO orbital of the dyes is delocalized over carbazole unit except 6d where HOMO is contributed by electron rich dithienopyrrole unit. Also, HOMO-1 is found to be localized on carbazole unit except for 6c where it is seen on the naphthalimide, bithiophene and cyanoacrylic acid units. For the dyes, 6a and 6b HOMO-2 is composed of naphthalimide, aryl and cyanoacrylic acid units. Considering the composition of contributing orbitals, the prominent electronic absorption (Table 2) may be described as a delocalized π–π* transitions influenced by elongation of conjugation, electron richness and co-planar nature of spacers. From this analysis, it is evident that the longer wavelength absorption does not lead to migration of charge from donor to acceptor. Thus, a charge separation is not evident on electronic excitation. The dyes displayed a similar trend of optical properties as calculated experimentally.

Frontier molecular orbitals and their contribution to major vertical excitation in dyes 6a–6d
Also, in order to investigate the utility of these dyes for DSSCs, free energy change for the electron injection process (∆Ginj) was calculated as described by Preat et al. [71] (∆Ginj = Eox* − ECB (TiO2), where ECB (TiO2) is the redox potential of the conduction potential of TiO2 and Eox* is oxidation potential of dye in excited state). Our attempts to correlate the effect of linker on optical properties are presented in Fig. 8 as relationship between oscillator strength (f) and ∆Ginj with band gap (Eg). Interestingly, ∆Ginj increases with band gap of the dyes while the reverse trend is observed for the oscillator strength. It is reasonable to believe that the choice of linkers is crucial to tune the band gap, oscillator strength and injection efficiency.

Dependence of oscillator strength, and ∆Ginj on band gap of the dyes
2.5 Photovoltaic properties
To investigate the effect of different π-linkers on photovoltaic performance of the dyes, DSSCs were fabricated using these sensitizers. The device parameters are summarized in Table 3. The incident photon-to-current conversion efficiencies (IPCE) spectra of the DSSCs are displayed in Fig. 9. Among the dyes, 6d displays a broader spectral response attributed to its better absorption and comparatively high molar extinction coefficients. The broadening of IPCE spectra is favorable for enhancing the light harvesting efficiency, efficient photocurrent and efficiency of the DSSCs [70]. On the contrary, dye 6a shows poor IPCE response ascribed to its weak absorption above 400 nm and low dye loading. Relatively low dye-loading of 6a when compared to 6b may be due to the phenyl linker in the former which reduce the acidity of the dye and its interaction with TiO2. All the dyes displayed an IPCE below 50%, which hampered the efficiency of DSSCs.

IPCE plots of the DSSCs fabricated using the dyes
The short-circuit photocurrent density (JSC), open-circuit photovoltage (VOC), and fill factor (ff) of the cell are estimated from current–voltage (J–V) characteristics plot measured under 1 SUN (Fig. 10). The better JSC observed for dyes 6c and 6d is attributed to its superior light harvesting properties contrary to 6a and 6b. Dyes 6a and 6b display a poor efficiency (η) of 0.13% due to low JSC of 1.37 and 1.54 mA cm−2 and small VOC of 265 and 236 mV, respectively. This is attributed to their inferior IPCE and hindered electronic coupling with the TiO2 and lack of charge separation in the dyes [72]. However, improved dye loading and IPCE response, higher JSC of 3.80 mA cm−2 for DSSC based on 6c compared to other analogs enhanced the efficiency to 0.57%. Device based on 6d displays relatively high efficiency of 0.92% attributable to its photovoltaic parameters such as JSC (3.60 mA cm−2) and VOC (519 mV).

J–V characteristics of the DSSCs fabricated using the dyes
2.6 Electrochemical impedance spectroscopy
The electrochemical impedance spectroscopy (EIS) measurements under dark and one sun illumination conditions were performed to understand the effect of different π-linkers on the fate of electrons at the interfaces of the DSSCs. The Nyquist plots for DSSCs obtained under dark and illuminated conditions are displayed in Fig. 11. The respective plots display electron recombination resistances (Rrec) and charge-transport resistance (Rct2) at the interfaces of TiO2/dye/electrolyte corresponding to the large semicircle of the plots. The dyes containing phenyl and thiophene linker (6a and 6b) possess relatively large Rct2 values. This probably is due to the relatively high-lying excited state energy level which led to unfavorable thermodynamic condition for electron injection [2, 7]. The comparatively smaller Rct2 values for dyes 6c and 6d indicate effective charge injection. The elongated conjugation pathway in 6d resulted in comparatively improved electronic interaction and thus slightly better performance in DSSC when compared to other dyes. In addition, the higher light harvesting ability of 6d compared to other dyes presumably increases the electron density in the TiO2 conduction band that upwardly shifts the Fermi energy and improves VOC of the cell. However, the largest Rrec value obtained for the dye 6a suggests that phenyl linker effectively suppresses recombination of electrons with the oxidized dye, although it display poor performance attributed to low dye loading. A poor electron injection efficiency observed for all the dyes may be a result of low dye loading, faster recombination between the photoinjected electrons in TiO2 with the oxidized dye or lack of separation of charges in the dye. The rate of charge recombination affects the electron lifetime in devices. From the Bode phase plots (Fig. 12), the electron lifetime can be extracted from the angular frequency (ωmin) at the mid-frequency peak using τ = 1/ωmin [73]. The longer electron lifetime of 6d (1.0 ms) than other dyes help to suppress back reaction of the injected electrons with the I3− in the electrolyte resulting in a high VOC. Despite of enhanced lifetime for 6a and 6b compared to 6c based DSSCs; the lower efficiency is attributed to enhanced Rct2 which decrease electron injection efficiency.

Nyquist plots observed for the DSSCs fabricated using dyes under a dark and b illumination conditions

Bode phase plots measured under illumination of the DSSCs fabricated using dyes
3 Conclusions
We have synthesized four D–A–π–A naphthalimide-based dyes 6a–6d possessing different linkers to study the effect of rigidification and electron richness of linker for DSSCs. Carbazole unit is incorporated as donor at imidic position of naphthalimide. The introduction of different linkers such as phenyl, thienyl, bithienyl and dithienopyrrole resulted in elongation of π-conjugating system in that order and resulted in a large bathochromic shift of Δλ = 125 nm in absorption spectra for DTP dye. The electronic richness of the dyes imparted by linkers is reflected in the ease of oxidation. The major electronic absorption results from a local excitation comprising naphthalimide, linker and cyanoacrylic acid units in the dye. Therefore, the prominent absorption of the dyes does not result in charge migration from donor to acceptor. However, due to the elongation of conjugation rendered by electron rich DTP in dye 6d lead to a maximum photocurrent density of 3.60 mA cm−2 and an open circuit voltage of 519 mV in the series. The higher electron lifetime (1 ms) for 6d facilitated suppression of back reaction of the injected electron with I3− in electrolyte resulting in improved VOC and enhanced JSC among the set of dyes. The low charge recombination resistance (Rrec) at TiO2/dye/electrolyte interface for all the dyes corroborates well with the lack of intramolecular charge transfer in the dyes and poor performance of the devices. However, a monotonous increment in JSC, VOC and overall efficiency of devices is noticed on changing the linker from phenyl to DTP. This suggests the need to incorporate a strong donor segment in between naphthalimide and cyanoacrylic acid in the present dye design. Further studies in this direction to improve the optical and photovoltaic properties of the dyes are underway.
4 Experimental section
4.1 Characterization and instrumentation
1H and 13C NMR spectral data were collected using JEOL 400 MHz spectrometer. Mass spectra were recorded in positive-ion mode on a Bruker ESI TOF high-resolution mass spectrometer. UV–Vis absorption spectra were obtained on a Cary 100 UV–Visible spectrophotometer using freshly prepared solutions in spectroscopic grade solvents. Cyclic voltammetric experiments were carried out at room temperature in dichloromethane using 0.1 M tetrabutylammonium perchlorate as supporting electrolyte. The three-electrode configuration comprising a glassy carbon working electrode, a platinum wire counter electrode, and a non-aqueous Ag/AgNO3 reference electrode was used. The E1/2 values were determined as 1/2(Epa + Epc), where Epa and Epc are the anodic and cathodic peak potentials, respectively for reversible waves. The values of irreversible redox potentials reported are observed from DPV. All the potentials are quoted against the ferrocene internal standard.
4.2 Device fabrication and characterization of DSSC
The method and instrumentation utilized for device fabrication and characterization was same as our earlier report [44]. The active area of the DSSC was restricted to 0.16 cm2 by using a mask. The IPCE (λ) is defined by IPCE (λ) = 1240 (JSC/λφ), where λ is the wavelength, JSC is short-circuit photocurrent density (mA cm−2) and ϕ is the incident radiative flux (W m−2). The dye-loading data were obtained by estimating the concentration of the unabsorbed dyes in the bath solution by measuring its molar extinction coefficient and utilizing the calibration curve.
4.3 Computational methods
All the computational calculations were performed using Gaussian 09 program package [74]. The ground-state geometries were fully optimized without any symmetry constraints using DFT [75] employing M062x [76] functional and 6-31G(d,p) basis set. The vibrational frequency analysis on the optimized structures was performed to confirm the structure of the compounds belong to minima. TD-DFT calculations under the same level was used to compute the excitation energies and oscillator strengths for the lowest 10 singlet–singlet transitions at the optimized geometry in the ground state.
4.4 Synthetic details
All the reactions were performed under nitrogen atmosphere unless otherwise mentioned by following standard inert atmosphere procedures. Solvents were dried by following standard procedures and distilled freshly from the suitable drying agent prior to use. All column chromatography purifications were performed using 100–200 mesh silica gel or neutral alumina as the stationary phase in a column measuring 30 cm long and 2.0 cm diameter.
4.4.1 6-(4-(9,9-Dibutyl-9H-fluoren-2-yl)-4H-dithieno[3,2-b:2′,3′-d]pyrrol-2-yl)-2-(9-ethyl-9H-carbazol-3-yl)-1H-benzo[de]isoquinoline-1,3(2H)-dione (4)
A mixture of 6-bromo-2-(9-ethyl-9H-carbazol-3-yl)-1H-benzo[de]isoquinoline-1,3(2H)-dione (3) (240 mg, 0.5 mmol); 4-(9,9-dibutyl-9H-fluoren-2-yl)-2-(tributylstannyl)-4H-dithieno[3,2-b:2′,3′-d]pyrrole (500 mg, 0.5 mmol), Pd(PPh3)2Cl2 (7 mg, 1 mol%) and DMF (5 mL) was heated at 80 °C for 12 h. After the completion of reaction, the reaction mixture was poured into cold water and extracted with CHCl3, washed thoroughly with brine solution and dried over Na2SO4. Volatiles were removed by rotary vacuum evaporation to yield a dark orange residue, which was purified by column chromatography using alumina as stationary phase. Eluent: 40% CHCl3 in hexanes. Red solid; 200 mg (47%), mp: 322 °C; IR (KBr, cm−1) 2958, 2927, 2858 (νCHstretch), 1706, 1667, 1586 (νC=O), 1586 (νC=C), 1490 (νC–N); 1H-NMR (400 MHz, CDCl3) δ 8.89 (dd, J = 8.5, 1.1 Hz, 1H), 8.74–8.67 (m, 2H), 8.60 (d, J = 7.3 Hz, 1H), 8.48 (dd, J = 8.5, 1.1 Hz, 1H), 8.09–8.02 (m, 2H), 7.96 (d, J = 7.3 Hz, 1H), 7.87 (d, J = 8.2 Hz, 1H), 7.82 (dd, J = 8.7, 7.3 Hz, 1H), 7.78–7.72 (m, 1H), 7.66–7.61 (m, 2H), 7.57 (dd, J = 8.5, 7.1 Hz, 2H), 7.53 (s, 1H), 7.50–7.35 (m, 2H), 7.34 (d, J = 5.5 Hz, 1H), 7.24 (s, 1H), 7.23–7.18 (m, 1H), 4.49–4.37 (m, 4H), 3.26–3.21 (m, 2H), 2.10–1.94 (m, 4H), 1.52–1.47 (4H), 1.02 (t, J = 7.3 Hz, 3H), 0.66 (t, J = 7.3 Hz, 6H); 13C-NMR (100 MHz, CDCl3) δ 165.0, 164.8, 152.7, 150.6, 147.5, 144.4, 140.4, 140.0, 139.6, 138.2, 137.2, 132.6, 131.7, 131.6, 131.2, 131.1, 130.5, 129.9, 129.4, 129.0, 128.6, 127.4, 127.3, 127.1, 126.5, 125.9, 125.6, 125.2, 123.6, 123.3, 122.9, 122.8, 121.8, 121.5, 120.8, 120.6, 119.7, 118.9, 118.6, 117.4, 116.8, 114.3, 112.2, 109.1, 108.5, 55.3, 40.1, 37.7, 33.2, 26.0, 23.0, 13.9, 13.85; HRMS calcd for C55H45N3O2S2 [M + Na] m/z 894.2794, found 894.2795.
4.4.2 4-(2-(9-Ethyl-9H-carbazol-3-yl)-1,3-dioxo-2,3-dihydro-1H-benzo[de]isoquinolin-6-yl)benzaldehyde (5a)
A mixture of 3 (469 mg, 1 mmol); 4-formylphen-1-ylboronic acid (245 mg, 1.5 mmol), K2CO3 (414 mg, 3 mmol), Pd(PPh3)4 (23 mg, 3 mol%) and 20 mL DMF/H2O (3:1) was heated at 80 °C. The progress of the reaction was monitored by TLC. After 12 h, the starting material disappeared indicating the completion of the reaction. The reaction was quenched by the addition of ice-water. The organic product was extracted with chloroform. The combined organic layer was thoroughly washed with brine solution and dried over anhydrous Na2SO4. Solvent was evaporated under reduced pressure to yield a residue. Desired product was obtained by silica column chromatography. Eluent: hexanes: CHCl3 (1:8); Pale yellow; 350 mg (71%), mp: 342 °C; IR (KBr, cm−1) 3053, 2975, 2929, 2719 (νCHstretch), 1703 (νCHO), 1663, 1594 (νC=O), 1489 (νC–N); 1H NMR (400 MHz, CDCl3) δ 10.17 (s, 1H), 8.78–8.71 (m, 2H), 8.26 (dd, J = 8.5, 0.9 Hz, 1H), 8.11 (d, J = 8.1 Hz, 2H), 8.08–8.03 (m, 2H), 7.81–7.72 (m, 4H), 7.58 (d, J = 8.7 Hz, 1H), 7.53–7.38 (m, 3H), 7.25–7.18 (m, 1H), 4.44 (q, J = 7.2 Hz, 2H), 1.50 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 191.7, 165.0, 164.8, 145.4, 144.9, 140.4, 139.7, 136.1, 132.2, 131.8, 131.1, 130.7, 130.0, 129.9, 129.0, 127.9, 127.4, 126.2, 126.0, 125.5, 123.6, 123.4, 122.9, 122.8, 120.8, 120.6, 119.0, 108.2, 108.6, 37.7, 13.9; HRMS calcd for C33H22N2O3 [M + Na] m/z 517.1522, found 517.1524.
4.4.3 General method for the synthesis of aldehydes 5b and 5c
A mixture of 3 (469 mg, 1 mmol); respective tin reagent, (5-(1,3-dioxolan-2-yl)thiophen-2-yl)tributylstannane (1.2 g, 1.5 mmol) or (5′-(1,3-dioxolan-2-yl)-[2,2′-bithiophen]-5-yl)tributylstannane (1.01 g, 1.5 mmol), Pd(PPh3)2Cl2 (7 mg, 2 mol%) and DMF (10 mL) was heated at 80 °C for 24 h. After the completion of reaction, it was poured into cold water and extracted with CHCl3, washed thoroughly with brine solution and dried over Na2SO4. Solvent was removed using rotary evaporator to yield a solid residue. It was mixed with glacial acetic acid (5 mL) and heated at 60 °C for 1 h. After addition of water (15 mL) it was heated further for 4 h. The cooled reaction mixture was extracted with CHCl3. The extract was washed thoroughly with brine solution and dried over anhydrous sodium sulfate. The solid obtained on evaporation of the solvent was purified by silica column chromatography.
4.4.4 5-(2-(9-Ethyl-9H-carbazol-3-yl)-1,3-dioxo-2,3-dihydro-1H-benzo[de]isoquinolin-6-yl)thiophene-2-carbaldehyde (5b)
Purified using silica gel by using hexanes/CHCl3 (1:8) as eluent; Pale yellow; Yield: 460 mg (92%), mp: 301 °C; IR (KBr, cm−1) 3061, 2972, 2920, 2868 (νCHstretch), 1705 (νCHO), 1667, 1589 (νC=O), 1483 (νC–N); 1H NMR (400 MHz, CDCl3) δ 10.04 (s, 1H), 8.76 (dd, J = 7.3, 1.0 Hz, 1H), 8.72 (d, J = 7.5 Hz, 1H), 8.60 (dd, J = 8.5, 1.1 Hz, 1H), 8.06 (d, J = 2.0 Hz, 1H), 8.04 (s, 1H), 7.93 (d, J = 3.7 Hz, 1H), 7.91–7.82 (m, 2H), 7.58 (d, J = 8.5 Hz, 1H), 7.53–7.42 (m, 3H), 7.40 (dd, J = 8.5, 2.0 Hz, 1H), 7.25–7.17 (m, 1H), 4.44 (q, J = 7.2 Hz, 2H), 1.50 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 182.8, 164.8, 164.5, 149.3, 145.1, 140.5, 139.7, 137.6, 136.5, 132.1, 131.8, 130.9, 129.8, 129.1, 129.0, 127.9, 127.3, 126.1, 125.5, 123.7, 123.5, 122.8, 120.8, 120.6, 119.1, 109.2, 108.6, 37.8, 13.9; HRMS calcd for C31H20N2O3S [M + H] m/z 501.1267, found 501.1277.
4.4.5 5′-(2-(9-Ethyl-9H-carbazol-3-yl)-1,3-dioxo-2,3-dihydro-1H-benzo[de]isoquinolin-6-yl)-[2,2′-bithiophene]-5-carbaldehyde (5c)
Purified on silica gel using hexanes/EtOAc (4:1) as eluent; Pale orange solid. Yield: 410 mg (70%), mp: 310 °C; IR (KBr, cm−1) 2967, 2923, 2846, (νCHstretch), 1704 (νCHO), 1664, 1585 (νC=O), 1462 (νC–N); 1H-NMR (400 MHz, CDCl3) δ 9.92 (s, 1H), 8.81–8.64 (m, 3H), 8.13–7.97 (m, 2H), 7.95–7.80 (m, 2H), 7.74 (d, J = 3.9 Hz, 1H), 7.60–7.55 (m, 1H), 7.52 (d, J = 3.7 Hz, 1H), 7.50–7.42 (m, 2H), 7.42–7.36 (m, 2H), 7.31 (d, J = 4.1 Hz, 1H), 7.25–7.19 (m, 1H), 4.44 (q, J = 7.2 Hz, 2H), 1.50 (t, J = 7.1 Hz, 3H); 13C-NMR (100 MHz, CDCl3) δ 182.6, 164.9, 164.6, 146.0, 142.3, 141.2, 140.4, 139.7, 138.1, 138.1, 137.3, 132.1, 132.0, 131.0, 129.9, 129.3, 128.6, 127.6, 126.8, 126.2, 126.0, 125.5, 124.8, 123.6, 123.4, 122.8, 120.8, 120.6, 119.0, 109.2, 108.6, 37.7, 13.9; HRMS calcd for C35H22N2O3S2 [M + Na] m/z 605.0964, found 605.0970.
4.4.6 4-(9,9-Dibutyl-9H-fluoren-2-yl)-6-(2-(9-ethyl-9H-carbazol-3-yl)-1,3-dioxo-2,3-dihydro-1H-benzo[de]isoquinolin-6-yl)-4H-dithieno[3,2-b:2′,3′-d]pyrrole-2-carbaldehyde (5d)
A solution of 4 (175 mg, 0.2 mmol) in 4 mL DMF maintained at 0 °C in ice bath was slowly added POCl3 (0.1 mL). After stirring the mixture for 30 min at this temperature, brought to room temperature and heated at 45 °C for 2 h. The reaction was monitored by TLC. After the completion of reaction, ice cold water was added followed by the aq. NaOH solution to neutralize excess of HCl. Compound was extracted with CHCl3 and washed with brine solution. The organic layer was separated and dried over Na2SO4. Removal of solvent yielded a crude product which was purified by alumina column chromatography. Eluent: 80% CHCl3 in hexanes. Dark orange solid; 90 mg (51%), mp: 334 °C; IR (KBr, cm−1) 2952, 2924, 2855 (νCHstretch), 1707 (νCHO), 1662, 1583, 1491 (νC=O), 1458 (νC–N); 1H-NMR (400 MHz, CDCl3) δ 9.94 (s, 1H), 8.82 (dd, J = 8.7, 0.9 Hz, 1H), 8.78–8.69 (m, 2H), 8.10–8.01 (m, 2H), 7.97 (d, J = 7.3 Hz, 1H), 7.91 (d, J = 7.8 Hz, 1H), 7.86 (s, 1H), 7.84 (dd, J = 8.2, 7.3 Hz, 1H), 7.79–7.74 (m, 1H), 7.66–7.55 (m, 3H), 7.53–7.47 (m, 2H), 7.46 (s, 1H), 7.44–7.37 (m, 4H), 7.25–7.19 (m, 1H), 4.43 (q, J = 7.2 Hz, 2H), 2.03 (q, J = 5.6 Hz, 4H), 1.49 (t, J = 7.3 Hz, 3H), 1.18–1.00 (m, 4H), 0.91–0.78 (m, 2H), 0.73–0.68 (m, 2H), 0.67 (t, J = 7.3 Hz, 6H); 13C-NMR (100 MHz, CDCl3) δ 183.0, 164.9, 164.6, 153.1, 150.7, 147.8, 143.6, 142.4, 141.8, 140.5, 140.4, 139.7, 139.7, 139.2, 137.2, 132.2, 131.9, 130.9, 129.9, 129.3, 128.9, 127.7, 127.6, 127.2, 126.2, 126.0, 125.5, 124.3, 123.6, 123.4, 123.0, 122.8, 122.7, 121.8, 121.1, 120.7, 120.6, 119.9, 119.0, 118.1, 117.7, 113.8, 109.1, 108.6, 55.3, 40.0, 37.7, 26.1, 23.0, 13.9, 13.9; HRMS calcd for C56H45N3O3S2 [M + Na] m/z 894.2794, found 894.2795.
4.4.7 General method for the synthesis of dyes 6a–6d
A mixture of respective aldehyde (5a–5d), cyanoacetic acid (68 mg, 0.8 mmol), acetic acid (10 mL) and ammonium acetate (5 mg) was heated at 120 °C for 8 h. The resulting solution was poured into ice-cold water and filtered. The solid residue was washed thoroughly with water and dried to obtain the desired compound. Analytically pure compound was obtained by recrystallization from chloroform/hexanes mixture.
4.4.8 (E)-2-cyano-3-(4-(2-(9-ethyl-9H-carbazol-3-yl)-1,3-dioxo-2,3-dihydro-1H-benzo[de]isoquinolin-6-yl)phenyl)acrylic acid (6a)
It was synthesized as described above using 5a (0.25 g, 0.5 mmol). Yellow solid; Yield: 240 mg (86%), mp: 318 °C; IR (KBr, cm−1) 3048, 2969, 2926 (νCHstretch), 2225 (νC≡N), 1696, 1669 (νC=O), 1590 (νC=C), 1488 (νC–N); 1H NMR (400 MHz, DMSO-d6) δ 8.60 (m, 2H), 8.48 (s, 1H), 8.35–8.21 (m, 3H), 8.20–8.15 (m, 1H), 8.13 (d, J = 7.3 Hz, 1H), 7.96–7.87 (m, 2H), 7.83 (dd, J = 8.2, 2.7 Hz, 2H), 7.74 (dd, J = 8.5, 3.0 Hz, 1H), 7.70–7.62 (m, 1H), 7.53–7.38 (m, 2H), 7.21 (d, J = 7.3 Hz, 1H), 4.52 (m, 2H), 1.38 (t, J = 6.9 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 164.2, 164.0, 163.3, 153.6, 144.7, 142.6, 140.0, 139.0, 132.0, 131.7, 131.0, 130.8, 130.4, 129.2, 128.4, 128.3, 127.9, 127.0, 126.5, 126.1, 123.2, 122.6, 122.4, 122.0, 120.9, 120.5, 119.0, 116.2, 109.4, 109.3, 104.7, 37.2, 13.8; HRMS calcd for C36H23N3O4 [M + Na] m/z 584.1581, found 584.1589.
4.4.9 (E)-2-cyano-3-(5-(2-(9-ethyl-9H-carbazol-3-yl)-1,3-dioxo-2,3-dihydro-1H-benzo[de]isoquinolin-6-yl)thiophen-2-yl)acrylic acid (6b)
It was synthesized by following general procedure and using 5b (0.25 g, 0.5 mmol). Bright yellow solid; Yield: 230 mg (81%), mp: 323 °C; IR (KBr, cm−1) 2973, 2927, 2857 (νCHstretch), 2221 (νC≡N), 1664, 1621 (νC=O), 1586 (νC=C), 1466 (νC–N); 1H NMR (400 MHz, DMSO-d6) δ 8.68 (d, J = 8.7 Hz, 1H), 8.59 (d, J = 6.6 Hz, 1H), 8.55 (d, J = 7.5 Hz, 1H), 8.21 (s, 1H), 8.17 (d, J = 1.8 Hz, 1H), 8.08 (d, J = 7.6 Hz, 1H), 8.02 (d, J = 7.6 Hz, 1H), 7.98–7.92 (m, 1H), 7.90 (d, J = 4.0 Hz, 1H), 7.74–7.69 (m, 1H), 7.66 (d, J = 3.7 Hz, 1H), 7.65 (s, 1H), 7.52–7.41 (m, 2H), 7.18 (t, J = 7.5 Hz, 1H), 4.50 (q, J = 7.0 Hz, 2H), 1.37 (t, J = 7.1 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 164.3, 164.0, 144.0, 140.7, 140.0, 139.1, 137.2, 136.1, 131.8, 131.2, 130.4, 129.0, 128.6, 128.3, 127.0, 126.6, 126.1, 123.3, 122.8, 122.4, 122.1, 121.0, 120.5, 119.2, 119.1, 110.4, 109.4, 37.2, 13.9; HRMS calcd for C34H21N3O4S [M + Na] m/z 590.1144, found 590.1161.
4.4.10 (E)-2-cyano-3-(5′-(2-(9-ethyl-9H-carbazol-3-yl)-1,3-dioxo-2,3-dihydro-1H-benzo[de]isoquinolin-6-yl)-[2,2′-bithiophen]-5-yl)acrylic acid (6c)
Prepared by following the general procedure and using 5c (0.25 g, 0.4 mmol). Orange solid. Yield: 213 mg (82%), mp: 325 °C; IR (KBr, cm−1) 3067, 2973, 2927, 2852 (νCHstretch), 2216 (νC≡N), 1698, 1667 (νC=O), 1579 (νC=C), 1536 (νC–N);1H NMR (400 MHz, DMSO-d6) δ 8.79 (d, J = 8.2 Hz, 1H), 8.61 (d, J = 7.3 Hz, 1H), 8.56 (d, J = 7.8 Hz, 1H), 8.40 (s, 1H), 8.17 (d, J = 1.8 Hz, 1H), 8.12 (d, J = 7.8 Hz, 1H), 8.06 (d, J = 7.8 Hz, 1H), 8.00 (dd, J = 8.7, 7.3 Hz, 1H), 7.94 (d, J = 4.1 Hz, 1H), 7.83–7.78 (m, 1H), 7.73 (d, J = 8.7 Hz, 1H), 7.71–7.64 (m, 3H), 7.50 (d, J = 7.8 Hz, 1H), 7.48–7.42 (m, 1H), 7.21 (t, J = 7.3 Hz, 1H), 4.52 (q, J = 7.0 Hz, 2H), 1.38 (t, J = 7.1 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) Could not be recorded even at high scans due to poor solubility; HRMS calcd for C38H23N3O4S2 [M + H] m/z 650.1202, found 650.1201.
4.4.11 (E)-2-cyano-3-(4-(9,9-dibutyl-9H-fluoren-2-yl)-6-(2-(9-ethyl-9H-carbazol-3-yl)-1,3-dioxo-2,3-dihydro-1H-benzo[de]isoquinolin-6-yl)-4H-dithieno[3,2-b:2′,3′-d]pyrrol-2-yl)acrylic acid (6d)
It was synthesized using procedure described above and using 5d (80 mg, 0.1 mmol). Brick red solid; Yield: 70 mg (82%), mp: 338 °C; IR (KBr, cm−1) 2952, 2926, 2858 (νCHstretch), 2210 (νC≡N), 1696, 1667 (νC=O), 1579 (νC=C), 1491 (νC–N); 1H-NMR (400 MHz, DMSO-d6) δ 9.07–8.89 (m, 1H), 8.66–8.49 (m, 3H), 8.27 (s, 1H), 8.17 (d, J = 1.8 Hz, 1H), 8.12–8.01 (m, 3H), 7.98 (d, J = 1.8 Hz, 1H), 7.96–7.85 (m, 2H), 7.76 (dd, J = 8.2, 1.8 Hz, 1H), 7.73–7.62 (m, 3H), 7.53–7.40 (m, 3H), 7.40–7.31 (m, 2H), 7.20 (t, J = 7.6 Hz, 1H), 4.51 (q, J = 6.9 Hz, 2H), 2.27–2.08 (m, 2H), 2.04–1.98 (m, 2H), 1.37 (t, J = 7.1 Hz, 3H), 1.10–0.98 (m, 4H), 0.68–0.41 (m, 10H); 13C-NMR (100 MHz, DMSO-d6) δ 164.2, 163.9, 152.5, 152.2, 150.5, 147.3, 143.0, 142.5, 140.0, 139.7, 139.5, 139.0, 138.6, 137.0, 134.9, 132.1, 131.1, 130.7, 130.3, 129.1, 129.0, 128.7, 128.4, 128.1, 127.6, 127.2, 127.0, 126.5, 126.1, 125.0, 123.4, 123.2, 123.0, 122.4, 122.0, 121.6, 121.4, 120.9, 120.5, 120.3, 120.2, 119.0, 118.1, 117.6, 114.5, 109.4, 109.2, 79.2, 55.2, 37.2, 25.9, 22.4, 13.8; HRMS calcd for C59H46N4O4S2 [M + H] m/z 939.3033, found 939.3044.
References
B. O’Regan, M. Grätzel, A low-cost, high-efficiency solar cell based on dye-sensitized colloidal TiO2 films. Nature 353, 737–740 (1991)
A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo, H. Pettersson, Dye-sensitized solar cells. Chem. Rev. 110, 6595–6663 (2010)
M. Grätzel, Photoelectrochemical cells. Nature 2001, 338–344 (2001)
M.K. Nazeeruddin, F. De Angelis, S. Fantacci, A. Selloni, G. Viscardi, P. Liska, S. Ito, B. Takeru, M. Grätzel, Combined experimental and DFT-TDDFT computational study of photoelectrochemical cell ruthenium sensitizers. J. Am. Chem. Soc. 127, 16835–16847 (2005)
M.K. Nazeeruddin, A. Kay, L. Rodicio, R. Humphry-Baker, E. Müller, P. Liska, L. Cevey, E. Costa, V. Shklover, L. Spiccia, G.B. Deacon, C.A. Bignozzi, M. Grätzel, Conversion of light to electricity by cis-X2bis(2,2′-bipyridyl-4,4′-dicarboxylate)ruthenium(II) charge-transfer sensitizers (X = Cl-, Br-, I-, CN-, and SCN-) on nanocrystalline titanium dioxide electrodes. J. Am. Chem. Soc. 115, 6382–6390 (1993)
C.Y. Chen, S.J. Wu, C.G. Wu, J.G. Chen, K.C. Ho, A ruthenium complex with superhigh light-harvesting capacity for dye-sensitized solar cells. Angew. Chem. Int. Ed. 45, 5822–5825 (2006)
A. Mishra, M.K.R. Fischer, P. Bäuerle, Metal-free organic dyes for dye-sensitized solar cells, from structure, property relationships to design rules. Angew. Chem. Int. Ed. 48, 2474–2499 (2009)
Y. Ooyama, Y. Harima, Photophysical and electrochemical properties and molecular structures of organic dyes for dye-sensitized solar cells. Chem. Phys. Chem. 13, 4032–4080 (2012)
S. Kim, J.K. Lee, S.O. Kang, J. Ko, J.H. Yum, S. Fantacci, F. De Angelis, D. Di Censo, M.K. Nazeeruddin, M. Grätzel, Molecular engineering of organic sensitizers for solar cell applications. J. Am. Chem. Soc. 128, 16701–16707 (2016)
J. Yang, P. Ganesan, J. Teuscher, T. Moehl, Y.J. Kim, C. Yi, P. Comte, K. Pei, T.W. Holcombe, M.K. Nazeeruddin, Influence of the donor size in D-π-A organic dyes for dye-sensitized solar cells. J. Am. Chem. Soc. 136, 5722–5730 (2014)
G. Li, K. Jiang, Y. Li, S. Li, L. Yang, Efficient structural modification of triphenylamine-based organic dyes for dye-sensitized solar cells. J. Phys. Chem. C 15, 11591–11599 (2008)
M. Liang, J. Chen, Arylamine organic dyes for dye-sensitized solar cells. Chem. Soc. Rev. 42, 3453–3488 (2013)
Z. Ning, Q. Zhang, W. Wu, H. Pei, B. Liu, H. Tian, Starburst triarylamine based dyes for efficient dye-sensitized solar cells. J. Org. Chem. 73, 3791–3797 (2008)
A. Baheti, P. Tyagi, K.R.J. Thomas, Y. Hsu, J.T. Lin, Simple triarylamine-based dyes containing fluorene and biphenyl linkers for efficient dye-sensitized solar cells. J. Phys. Chem. C 113, 8541–8547 (2009)
L.L. Estrella, M.P. Balanay, D.H. Kim, The effect of donor group rigidification on the electronic and optical properties of arylamine-based metal-free dyes for dye-sensitized solar cells: a computational study. J. Phys. Chem. A 120, 5917–5927 (2016)
Y.Z. Wu, X. Zhang, W.Q. Li, Z.S. Wang, H. Tian, W.H. Zhu, Hexylthiophene-featured D-A-π-A structural indoline chromophores for coadsorbent-free and panchromatic dye-sensitized solar cells. Adv. Energy Mater. 2, 149–156 (2012)
S. Kajiyama, Y. Uemura, H. Miura, K. Hara, N. Koumura, Organic dyes with oligo-n-hexylthiophene for dye-sensitized solar cells: relation between chemical structure of donor and photovoltaic performance. Dyes Pigments 92, 1250–1256 (2012)
Y. Wu, M. Marszalek, S.M. Zakeeruddin, Q. Zhang, H. Tian, M. Grätzel, W. Zhu, High-conversion-efficiency organic dye-sensitized solar cells: molecular engineering on D–A–π-A featured organic indoline dyes. Energy Environ. Sci. 5, 8261–8272 (2012)
B. Liu, Q. Liu, D. You, X. Li, Y. Naruta, W. Zhu, Molecular engineering of indoline based organic sensitizers for highly efficient dye-sensitized solar cells. J. Mater. Chem. 22, 13348–13356 (2012)
A. Venkateswararao, K.R.J. Thomas, C.P. Lee, C.T. Li, K.C. Ho, Organic dyes containing carbazole as donor and π-linker: optical, electrochemical, and photovoltaic properties. ACS Appl. Mater. Interfaces 6, 2528–2539 (2014)
J. Tang, J. Hua, W. Wu, J. Li, Z. Jin, Y. Long, H. Tian, New starburst sensitizer with carbazole antennas for efficient and stable dye-sensitized solar cells. Energy Environ. Sci. 3, 1736–1745 (2010)
A. Venkateswararao, K.R.J. Thomas, C.P. Lee, K.C. Ho, Synthesis and characterization of organic dyes containing 2,7-disubstituted carbazole π-linker. Tetrahedron Lett. 54, 3985–3989 (2014)
Z.S. Wang, N. Koumura, Y. Cui, M. Takahashi, H. Sekiguchi, A. Mori, T. Kubo, A. Furube, K. Hara, Hexylthiophene-functionalized carbazole dyes for efficient molecular photovoltaics: tuning of solar-cell performance by structural modification. Chem. Mater. 20, 3993–4003 (2008)
C.-J. Yang, Y.J. Chang, M. Watanabe, Y.-S. Hon, T.J. Chow, Phenothiazine derivatives as organic sensitizers for highly efficient dye-sensitized solar cells. J. Mater. Chem. 22, 4040–4049 (2012)
A. Baheti, K.R.J. Thomas, C.T. Li, C.P. Lee, K.C. Ho, Fluorene-based sensitizers with a phenothiazine donor: effect of mode of donor tethering on the performance of dye-sensitized solar cells. ACS Appl. Mater. Interfaces 7, 2249–2262 (2015)
Z.Q. Wan, C.Y. Jia, Y.D. Duan, L.L. Zhou, Y. Lin, Y. Shi, Phenothiazine–triphenylamine based organic dyes containing various conjugated linkers for efficient dye-sensitized solar cells. J. Mater. Chem. 22, 25140–25147 (2012)
A. Saini, K.R.J. Thomas, C.T. Li, K.C. Ho, Organic dyes containing fluorenylidene functionalized phenothiazine donors as sensitizers for dye sensitized solar cells. J. Mater. Sci. Mater. Electron. 27, 12392–12404 (2016)
L.E. Polander, A. Yella, J. Teuscher, R. Humphry-Baker, B.F.E. Curchod, N. Astani, P. Gao, J.E. Moser, I. Tavernelli, U. Rothlisberger, Unravelling the potential for dithienopyrrole sensitizers in dye-sensitized solar cells. Chem. Mater. 25, 2642–2648 (2013)
S. Kumar, K.R.J. Thomas, C.-T. Li, K.-C. Ho, Synthesis and photovoltaic properties of organic dyes containing N-fluoren-2-yl dithieno[3,2-b:2′,3′-d]pyrrole and different donors. Org. Electron. 26, 109–116 (2015)
D. Sahu, H. Padhy, D. Patra, J.F. Yin, Y.C. Hsu, J.T.S. Lin, K.L. Lu, K.H. Wei, H.C. Lin, Synthesis and applications of novel acceptor-donor-acceptor organic dyes with dithienopyrrole- and fluorene-cores for dye-sensitized solar cells. Tetrahedron 67, 303–311 (2011)
B. Liu, B. Wang, R. Wang, L. Gao, S. Huo, Q. Liu, X. Li, W. Zhu, Influence of conjugated π-linker in D–D–π–A indoline dyes, towards long-term stable and efficient dye-sensitized solar cells with high photovoltage. J. Mater. Chem. A 2, 804–812 (2014)
A. Venkateswararao, K.R.J. Thomas, A. Tiwari, R. Boukherroub, M. Sharon, Solar Cell Nanotechnology, vol. 2 (Wiley-Scrivener, Beverly, 2014), pp. 41–96
P. Naik, M.R. Elmorsy, R. Su, D.D. Babu, A. El-Shafei, A.V. Adhikari, New carbazole based metal-free organic dyes with D-π-A-π-A architecture for DSSCs: synthesis, theoretical and cell performance studies. Sol. Energy 153, 600–610 (2017)
R. Samae, P. Surawatanawong, U. Eiamprasert, S. Pramjit, L. Saengdee, P. Tangboriboonrat, S. Kiatisevi, Effect of thiophene spacer position in carbazole-based dye-sensitized solar cells on photophysical, electrochemical and photovoltaic properties. Eur. J. Org. Chem. 21, 3536–3549 (2016)
T.N. Murakami, N. Koumura, T. Uchiyama, Y. Uemura, K. Obuchi, N. Masaki, M. Kimura, S. Mori, Recombination inhibitive structure of organic dyes for cobalt complex redox electrolytes in dye-sensitised solar cells. J. Mater. Chem. A 1, 792–798 (2013)
S. Haid, M. Marszalek, A. Mishra, M. Wielopolski, J. Teuscher, J.E. Moser, R. Humphry-Baker, S.M. Zakeeruddin, M. Grätzel, P. Bäuerle, Significant improvement of dye-sensitized solar cell performance by small structural modification in π-conjugated donor-acceptor dyes. Adv. Funct. Mater. 22, 1291–1302 (2012)
W.Q. Li, Y.Z.; Li, X.Y.S. Wu, W.H. Zhu, Absorption and photovoltaic properties of organic solar cell sensitizers containing fluorene unit as conjunction bridge. Energy Environ. Sci. 4, 1830–1837 (2011)
R. Li, X. Lv, D. Shi, D. Zhou, Y. Cheng, G. Zhang, P. Wang, Dye-sensitized solar cells based on organic sensitizers with different conjugated linkers: furan, bifuran, thiophene, bithiophene, selenophene, and biselenophene. J. Phys. Chem. C 113, 7469–7479 (2009)
X. Wang, L. Guo, P.F. Xia, F. Zheng, M.S. Wong, Z. Zhu, Dye-sensitized solar cells based on organic dyes with naphtho[2,1-b,3,4-b′]dithiophene as the conjugated linker. J. Mater. Chem. A 1, 13328–13336 (2013)
M. Katono, M. Wielopolski, M. Marszalek, T. Bessho, J.E. Moser, R. Humphry-Baker, S.M. Zakeeruddin, M. Grätzel, Effect of extended π-conjugation of the donor structure of organic D-A-π-A dyes on the photovoltaic performance of dye-sensitized solar cells. J. Phys. Chem. C 118, 16486–16493 (2014)
S. Namuangruk, R. Fukuda, M. Ehara, J. Meeprasert, T. Khanasa, S. Morada, T. Kaewin, S. Jungsuttiwong, T. Sudyoadsuk, V. Promarak, D-D-π-A-type organic dyes for dye-sensitized solar cells with a potential for direct electron injection and a high extinction coefficient, synthesis, characterization, and theoretical investigation. J. Phys. Chem. C 116, 25653–26110 (2012)
A. Baheti, P. Singh, C.-P. Lee, K.R.J. Thomas, K.-C. Ho, 2,7-Diaminofluorene-based organic dyes for dye-sensitized solar cells, effect of auxiliary donor on optical and electrochemical properties. J. Org. Chem. 76, 4910–4920 (2011)
S.Y. Qu, C.J. Qin, A. Islam, Y.Z. Wu, W.H. Zhu, J.L. Hua, H. Tian, L.Y. Han, A novel D–A-π-A organic sensitizer containing a diketopyrrolopyrrole unit with a branched alkyl chain for highly efficient and stable dye-sensitized solar cells. Chem. Commun. 48, 6972–6974 (2012)
S. Kumar, K.R.J. Thomas, C.T. Li, K.C. Ho, Effect of electron-deficient linkers on the physical and photovoltaic properties of dithienopyrrole-based organic dyes. J. Mater. Sci. Mater. Electron. 28, 1–14 (2017)
W. Zhu, Y. Wu, S. Wang, W. Li, X. Li, J. Chen, Z.S. Wang, H. Tian, Organic D-A-π-A solar cell sensitizers with improved stability and spectral response. Adv. Funct. Mater. 21, 756–763 (2011)
Y. Wu, W. Zhu, Organic sensitizers from D–π–A to D–A–π–A: effect of the internal electron-withdrawing units on molecular absorption, energy levels and photovoltaic performances. Chem. Soc. Rev. 42, 2039–2058 (2013)
X. Zhang, L. Chen, X. Li, J. Mao, W. Wu, H. Ågren, J. Hua, Photovoltaic properties of bis(octyloxy)benzo-[c][1,2,5]thiadiazole sensitizers based on an N,N-diphenylthiophen-2-amine donor. J. Mater. Chem. C 2, 4063–4072 (2014)
M. Velusamy, K.R.J. Thomas, J.T. Lin, Y.C. Hsu, K.C. Ho, Organic dyes incorporating low-band-gap chromophores for dye-sensitized solar cells. Org. Lett. 7, 1899–1902 (2005)
T.W. Holcombe, J.-H. Yum, Y. Kim, K. Rakstysac, M. Grätzel, Diketopyrrolopyrrole-based sensitizers for dye-sensitized solar cell applications, anchor engineering. J. Mater. Chem. A 1, 13978–13983 (2013)
S.Y. Qu, B. Wang, F.L. Guo, J. Li, W.J. Wu, C. Kong, Y.T. Long, J.L. Hua, New diketo-pyrrolo-pyrrole (DPP) sensitizer containing a furan moiety for efficient and stable dye-sensitized solar cells. Dyes Pigments 92, 1384–1393 (2012)
K. Pei, Y. Wu, W. Wu, Q. Zhang, B. Chen, H. Tian, W. Zhu, Constructing organic D–A–π-A-featured sensitizers with a quinoxaline unit for high-efficiency solar cells: the effect of an auxiliary acceptor on the absorption and the energy level alignment. Chem. A Eur. J. 18, 8190–8200 (2012)
X. Lu, Q. Feng, T. Lan, G. Zhou, Z.S. Wang, Molecular engineering of quinoxaline-based organic sensitizers for highly efficient and stable dye-sensitized solar cells. Chem. Mater. 24, 3179–3187 (2012)
Y. Cui, Y. Wu, X. Lu, X. Zhang, G. Zhou, F.B. Miapeh, W. Zhu, Z.S. Wang, Incorporating benzotriazole moiety to construct D-A-π-A organic sensitizers for solar cells: significant enhancement of open-circuit photovoltage with long alkyl group. Chem. Mater. 23, 4394–4401 (2011)
W. Li, Y. Wu, Q. Zhang, H. Tian, W. Zhu, D-A-π-A featured sensitizers bearing phthalimide and benzotriazole as auxiliary acceptor: effect on absorption and charge recombination dynamics in dye-sensitized solar cells. ACS Appl. Mater. Interfaces 4, 1822–1830 (2012)
J.Y. Mao, F.L. Guo, W.J. Ying, W.J. Wu, J. Li, J.L. Hua, Benzotriazole-bridged sensitizers containing a furan moiety for dye-sensitized solar cells with high open-circuit voltage performance. Chem. Asian J. 7, 982–991 (2012)
S.R. Bobe, A. Gupta, A. Rananaware, A. Bilic, W. Xiang, J.L. Li, S.V. Bhosale, S.V. Bhosale, R.A. Evans, Insertion of a naphthalenediimide unit in a metal-free donor–acceptor organic sensitizer for efficiency enhancement of a dye-sensitized solar cell. Dyes Pigments 134, 83–90 (2016)
Q. Zhang, J. He, H. Li, N. Li, Q. Xu, D. Chen, J. Lu, A novel ternary memory property achieved through rational introduction of end-capping naphthalimide acceptors. J. Mater. Chem. C 5, 7961–7968 (2017)
A. Saini, K.R.J. Thomas, Bis-naphthalimides bridged by electron acceptors: optical and self-assembly characteristics. RSC Adv. 6, 71638–71651 (2016)
D. Dang, Y. Zhi, X. Wang, B. Zhao, C. Gao, L. Meng, A1-A-A1 type small molecules terminated with naphthalimide building blocks for efficient non-fullerene organic solar cells. Dyes Pigments 137, 43–49 (2017)
X. Huang, Y. Fang, X. Li, Y. Xie, W. Zhu, Novel dyes based on naphthalimide moiety as electron acceptor for efficient dye-sensitized solar cells. Dyes Pigments 90, 297–303 (2011)
A. Margalias, K. Seintis, M.Z. Yigit, M. Can, D. Sygkridou, V. Giannetas, M. Fakis, E. Stathatos, The effect of additional electron donating group on the photophysics and photovoltaic performance of two new metal free D-π-A sensitizers. Dyes Pigments 121, 316–327 (2015)
A. Saini, K.R.J. Thomas, A. Sachdev, P. Gopinath, Photophysics, electrochemistry, morphology, and bioimaging applications of new 1,8-naphthalimide derivatives containing different chromophores. Chem. Asian J. 12, 2612–2622 (2017)
E. Knoevenagel, Condensation von Malonsäure mit aromatischen Aldehyden durch Ammoniak und Amine. Eur. J. Inorg. Chem. 31, 2596–2619 (1898)
R.A. Irgashev, G.A. Kim, G.L. Rusinov, 5-(Methylidene) barbituric acid as a new anchor unit for dye-sensitized solar cells (DSSC). Arkivoc 2014, 123–131 (2014)
K. Sayama, S. Tsukagoshi, K. Hara, Y. Ohga, A. Shinpou, Y. Abe, S. Suga, H. Arakawa, Photoelectrochemical properties of J aggregates of benzothiazole merocyanine dyes on a nanostructured TiO2 film. J. Phys. Chem. B 106, 1363–1371 (2002)
D.P. Hagberg, T. Edvinsson, T. Marinado, G. Boschloo, A. Hagfeldt, L.A. Sun, Novel organic chromophore for dye-sensitized nanostructured solar cells. Chem. Commun. 21, 2245 (2006)
J.R. Mann, M.K. Gannon, T.C. Fitzgibbons, M.R. Detty, D.F. Watson, Optimizing the photocurrent efficiency of dye-sensitized solar cells through the controlled aggregation of chalcogenoxanthylium dyes on nanocrystalline titania films. J. Phys. Chem. C 112, 13057–13061 (2008)
M.K. Nazeeruddin, S.M. Zakeeruddin, R. Humphry-Baker, M. Jirousek, P. Liska, N. Vlachopoulos, V. Shklover, C.-H. Fischer, M. Grätzel, Acid–base equilibria of (2,2′-bipyridyl-4,4′-dicarboxylic acid)ruthenium(II) complexes and the effect of protonation on charge-transfer sensitization of nanocrystalline titania. Inorg. Chem. 38, 6298–6305 (1999)
C. Reichardt, Solvatochromic dyes as solvent polarity indicators. Chem. Rev. 94, 2319–2358 (1994)
X.-H. Zhang, Y. Cui, R. Katoh, N. Koumura, K. Hara, Organic dyes containing thieno[3,2-B]indole donor for efficient dye-sensitized solar cells. J. Phys. Chem. C 114, 18283–18290 (2010)
J. Preat, C. Michaux, J.-M. André, E.A. Perpète, Pyrrolidine-based dye-sensitized solar cells: a time-dependent density functional theory investigation of the excited state electronic properties. Int. J. Quantum Chem. 112, 2072–2084 (2012)
A.C. Khazraji, S. Hotchandani, S. Das, P.V. Kamat, Controlling dye (merocyanine-540) aggregation on nanostructured TiO2 films. An organized assembly approach for enhancing the efficiency of photosensitization. J. Phys. Chem. B 103, 4693–4700 (1999)
J. Lagemaat, N.-G. Park, A.J. Frank, Influence of electrical potential distribution, charge transport, and recombination on the photopotential and photocurrent conversion efficiency of dye-sensitized nanocrystalline TiO2 solar cells: a study by electrical impedance and optical modulation techniques. J. Phys. Chem. B 104, 2044–2052 (2000)
M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G.A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H.P. Hratchian, A.F. Izmaylov, J. Bloino, G. Zheng, J.L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J.A. Montgomery, J.E. Peralta, F. Ogliaro, M. Bearpark, J.J. Heyd, E. Brothers, K.N. Kudin, V.N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J.C. Burant, S.S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N.J. Millam, M. Klene, J.E. Knox, J.B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R.E. Stratmann, O. Yazyev, A.J. Austin, R. Cammi, C. Pomelli, J.W. Ochterski, R.L. Martin, K. Morokuma, V.G. Zakrzewski, G.A. Voth, P. Salvador, J.J. Dannenberg, S. Dapprich, A.D. Daniels, O. Farkas, J.B. Foresman, J.V. Ortiz, J. Cioslowski, D.J. Fox, Gaussian 09 (Revision-A. 02) (Gaussian, Inc., Wallingford, 2009)
C. Lee, W. Yang, R.G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785–789 (1988)
Y. Zhao, D.G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 120, 215–241 (2008)
Acknowledgements
KRJT is thankful to DST, New Delhi (DST/TSG/PT/2013/09) and CSIR (Ref. No. 02/(0230)/15/EMR-II dated 05-06-2015), New Delhi for generous financial support.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Absorption spectra of precursors of dyes in DCM, Absorption spectra and data of dyes in different solvents, CV and DPV of the dyes, Frontier molecular orbitals of dyes, Optical and electrochemical data of precursors of dyes, copies of 1H and 13C-NMR spectra, Cartesian coordinates for the optimized geometry.
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Saini, A., Thomas, K.R.J., Huang, YJ. et al. Synthesis and characterization of naphthalimide-based dyes for dye sensitized solar cells. J Mater Sci: Mater Electron 29, 16565–16580 (2018). https://doi.org/10.1007/s10854-018-9750-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10854-018-9750-4