
Abstract
A series of novel push–pull-based D–π–A-containing organic dyes have been synthesized using triphenylamine and carbazole act as π-conjugated donor and cyanoacrylic acid acceptor as well as anchoring group for application of dye-sensitized solar cells. These organic dyes D1, D2 and D3 exhibited excellent photophysical, electrochemical properties, superior stoke shift, high thermal stabilities and sufficient HOMO–LUMO energy levels, which is facilitated to dye regeneration and effective injection of electron from the excited state of the dyes into the conduction band (TiO2). Our results suggested that carbazole π-bridge has an important role in the photovoltaic performance. The D1 and D2 dyes are planar linear structure, longer π-conjugated bridging units, when compared to the starburst structure of D3 organic dye. Moreover, the carbazole π-bridged conjugated D1 dye showed promising photovoltaic conversion efficiency (η) of 1.4%, Voc = 735 mV, Jsc = 2.7 mA/cm2, ff = 0.73 (fill factor), when compared with the conventional N719 and D5 organic dyes.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Energy demands and environmental pollution result in global warming, depletion of fossil fuels and large amount of energy consumption within world population, so we need an alternative energy source. Renewable energies are one of the most important economic and never-ending sources to fulfil the world needs. In 1991, Gratzel and co-workers [1, 2] demonstrated dye-sensitized solar cells (DSSCs) to achieve 7.0% conversion efficiency, which is still considered as a milestone in the emerging photovoltaic technology. Nowadays, dye-sensitized solar cells (DSSCs) have received great attention and significant interest to solve the worldwide energy demands. As an affordable renewable energy resource, DSSCs have low-cost substitutes with easily synthesized material, have low hazard risk, are environmental-friendly and have the most efficient devices to convert sunlight to electricity compared with the conventional solid p–n junction photovoltaic devices. The power conversion efficiency (PCE) of DSSCs has reached up to 7–14.3% in the past three and a half decades under standard conditions [3,4,5,6,7,8]. In general, nanocrystalline TiO2 semiconductor photoanodes [9, 10], adsorbed by Ru(II) bipyridyldicarboxylate complex as a sensitizer, iodide/triiodide (I−/I3−) redox couple [11,12,13,14] and platinum (Pt) as the counter electrode [15,16,17]. In DSSC, dye molecules are chemisorbed on the TiO2 photoanodes and the dye molecules generate charge carriers by absorbing the visible light. The excited charge carriers will transfer electron to the conduction band of TiO2, followed by regeneration of dye by an iodide/triiodide redox couple at TiO2/dye/electrolyte interface [18]. In the past few decades, polypyridyl ruthenium complex- and metal porphyrin-based sensitizers were considered for high-performance DSSC applications [19], due to the high light-harvesting properties, high light to electric power conversion efficiency (PCE) and long-term stability, although these Ru dyes are facing the problem of costs and low abundance of ruthenium modest extinction coefficients, tricky preparation techniques, challenging synthesis, environmental issues [20, 21]. Many efforts have been made to overcome the above drawbacks associated with Ru dye. Hence, metal-free organic dyes which contain donor–π–acceptor (D–π–A) are extensively developed to study the sensitizers in DSSCs, which can be easily designed for molecular structure and convenient photochemical and photophysical properties, inexpensive, environmentally friendly and so many advantages [22]. DSSCs based on metal-free organic dyes reported by T. Takahashi et al., D–π–A-containing organic dye to achieve high PCE [23,24,25]. Encouraged by these results, many research groups, including our group, have demonstrated various metal-free organic push–pull configured dyes derived from electron-rich donors such as merocyanine-based [26], carbazole-based [27], fluorene-based [28,29,30], phenothiazine-based [31], hemicyanine-based [32, 33], coumarin-based [34, 35], triphenylamine-based [36, 37], porphyrin-based [38], squaraine-based [39, 40] and indoline-based [41] organic dyes. Recently, Berlinguette et al. have reported the thiophene-bridged triphenylamine- and phenothiazine-based organic sensitizers to achieve 7.1% PCE [42], and several research groups reported triphenylamine-based [43] and carbazole-based [44] organic dyes that have been investigated for DSSC applications [45,46,47,48]. In this regard, Zhen Li et al. reported different pyrrole- and thiophene-linked alkyl chain-containing photosensitizers used for DSSC application to achieve high conversion efficiency up to 9.7% [49]. Zhang et al. fabricated DSSCs with a novel benzothiadiazole- and ethynylthiophenezoic acid-based photosensitizer for efficient organic dye-sensitized solar cells to obtain 12.0% efficiency [50]. Subramanian et al. reported the influence of different strengths of donor units such as phenothiazine-, fluorene- and anthracene-based sensitizers with new anchoring mode to obtain 4.12% conversion efficiency [51].
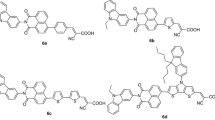
In this work, we have successfully synthesized metal-free organic dyes D1, D2 and D3, which contain carbazole and triphenylamine π-bridged (D–π–A) systems shown in Fig. 1, where triphenylamine acts as a donor and cyanoacrylic acid as an anchoring group; these organic dyes were used for fabrication of DSSCs with TiO2 (P25) as photoanode. All the prepared dyes have good thermal stability, surface morphology and required photophysical, photochemical, photovoltaic properties. Generally, carbazole and triphenylamine was included in the π-bridged system which is beneficial to reduce the band gap and cramped the dye aggregations. Hence, it has improved the light-harvesting properties. Finally, these dyes have been successfully utilized for sensitizing and high power conversion efficiency (η) 1.4% compared with traditional ruthenium dye (N719) as well as previously reported D5 organic dye [52, 53].

Molecular structure of D1, D2, D3 and D5 metal-free organic dyes
Experimental section
Materials and methods
All the starting materials and reagents were purchased from Sigma-Aldrich and Alfa Aesar and used as received. Carbazole, triphenylamine, phosphorous oxychloride, cyanoacrylic acid, methyl iodide and piperidine, all these chemicals were purchased as such used in the reaction. Other commercially available chemicals and solvents were used as received without further purification. Further, C–C cross-coupling reaction was carried out under the protection of nitrogen atmospheric conditions. Silica gel G plates (Merck) were used for TLC analysis with a mixture of petroleum ether (60–80 °C) and ethyl acetate as an eluent, and various solvents are used in this photophysical study which were purchased from commercial suppliers.
The melting points were measured in open capillary tubes and are uncorrected. The 1H and 13C NMR spectra were recorded on Bruker (Avance) 300 MHz and 400 MHz NMR instrument using TMS as an internal standard, and CDCl3 and DMSO-d6 were used as solvents. Electronic absorption spectra were recorded on JASCO V-630 UV–Vis spectrophotometer. The quartz cuvettes were used for all analysis by 1 cm path length. Photoluminescence (PL) was recorded on JASCO F-8500 fluorescence spectrophotometer. Furthermore, the PL spectra were recorded with corresponding absolute absorption maximum as the excitation wavelength in a 1 cm width cell, and excitation and emission slits were set to 2.5 nm for all this measurement. Electrochemical measurements were recorded on cyclic voltammeter, CH instruments—model 680 Amp Booster—at room temperature with the use of a standard three-electrode electrochemical cell in acetonitrile-containing 0.1 M tetrabutylammonium perchlorate (Bu4N+ClO ‒4 ) as a supporting electrolyte with glassy carbon working electrode, platinum wire as a counter electrode and saturated calomel as a reference electrode at the scan rate of 100 mVs−1. In addition, the resistance (RCT) value was measured on electrochemical impedance spectroscopy (EIS), bare GC, platinum wire and Ag/AgCl three-electrode system with 1 mM of K3[Fe(CN)6] containing 0.1 M KCl at scanning frequencies from 0.01 to 100,000 Hz. A quantum chemical calculation was performed by using Gaussian 09 program, and the ground-state geometries were optimized by using the restricted B3LYP/6-31G (d, p) functions at density functional theory (DFT) level.
9-Methyl-carbazole (1)
9-Methyl-carbazole was synthesized from following the procedure reported in the literature [54]. 1H NMR (300 MHz, CDCl3) δ: 8.08 (d, J = 7.8, 2H, ar CH), 7.49–7.41 (m, 2H, ar CH), 7.35 (d, J = 8.2 Hz, 2H, ar CH), 7.28–7.17 (m, 2H, ar CH), 3.77 (s, 3H, N–CH3); 13C NMR (75 MHz, CDCl3)δ: 139.61, 129.49, 124.18, 124.08, 113.95, 111.82, 28.78 (–CH3).
9-Methyl-carbazole-3-carbaldehyde (2)
9-Methyl-carbazole-3-carbaldehyde and 6-bromo-9-methyl-9H-carbazole-3-carbaldehyde were synthesized from following the reported procedure [55], 1H NMR (300 MHz, CDCl3) δ: 9.98 (s, 1H, CHO), 8.39 (s, 1H, ar CH), 8.01 (d, J = 7.6 Hz, 1H, ar CH), 7.88 (d, J = 8.5 Hz, 1H), 7.50–7.43 (d, 1H), 7.28 (dd, J = 15.8, 8.3 Hz, 3H), 3.67 (s, 3H, –CH3); 13C NMR (75 MHz, CDCl3) δ: 192.08 (–CHO), 144.40, 141.71, 128.54, 127.02, 126.96, 123.84, 122.90, 120.64, 120.53, 109.38, 108.76, 29.27 (–CH3).
6-Bromo-9-methyl-9H-carbazole-3-carbaldehyde (3)
1H NMR (300 MHz, CDCl3) δ: 10.03 (s, 1H, CHO), 8.37 (s, 1H, ar CH), 8.10 (s, 1H, ar CH), 7.97 (d, J = 8.6 Hz, 1H), 7.55 (d, J = 8.7 Hz, 1H), 7.37 (d, J = 8.6 Hz, 1H), 7.22 (d, J = 8.7 Hz, 1H), 3.77 (s, 3H, –CH3);13C NMR (75 MHz, CDCl3) δ: 191.92 (–CHO), 144.90, 140.57, 129.76, 129.19, 127.88, 124.79, 124.44, 123.66, 122.15, 113.57, 110.91, 109.37, 29.86 (–CH3).
(E)-6-(4-(diphenylamino)styryl-9-methyl-9H-carbazole-3-carbaldehyde (4)
6-Bromo-9-methyl-9H-carbazole-3-carbaldehyde 3 (0.8 g, 2.84 mmol), N,N-diphenyl-4-vinylaniline (0.85 g, 3.12 mmol), o-tolylphosphine (4 mol %) and PdCl2(PPh3)2(4 mol %) were dissolved in dry DMF/toluene (1:1) mixture. The resulting reaction mixture was purged under nitrogen atmosphere for 30 min, after that, K2CO3 (1.1 g, 8.53 mmol) was added, and then, reaction mixture was heated at 110 °C for overnight. After the completion of reaction, the resulting mixture was passed through the Celite bed and washed with ethyl acetate. Finally, organic layer was washed with saturated brine solution and dried over anhydrous Na2SO4, and the crude product was purified by column chromatography using petroleum ether/ethyl acetate (92:8) as an eluent, to afford 4 orange solid in 70% yield. (1H NMR (300 MHz, CDCl3) δ: 10.10 (s, 1H, CHO), 8.61 (s, 1H, ar CH), 8.24 (s, 1H, ar CH), 8.04–7.99 (m, 1H), 7.71 (d, J = 8.7 Hz, 1H), 7.46 (d, J = 3.7 Hz, 2H), 7.43 (d, J = 4.0 Hz, 2H), 7.31–7.26 (m, 4H), 7.25–7.30 (m, 2H), 7.18 (s, 2H), 7.11 (d, J = 5.0 Hz, 4H), 7.03 (d, J = 7.3 Hz, 1H), 3.89 (s, 3H, CH3); 13C NMR (75 MHz, CDCl3) δ: 192.13 (–CHO), 171.44, 148.0, 145.18, 141.18, 141.55, 129.81,129.69, 127.78, 127.64, 127.56, 127.04,125.82, 124.84, 124.18, 123.38, 118.81, 109.71, 109.29, 106.45, 30.11 (–CH3). ESI–MS m/z: [M + H]+ calcd. for C34H26N2O 478.2035, found 478.2038.
(E)-6-(4-(bis(4-methoxyphenyl)amino)styryl)-9-methyl-9H-carbazole-3-carbaldehyde (5)
Compound 5 was synthesized according to procedure 4 using compound 3 (0.8 g, 2.84 mmol); 4-methoxy-N-(4-methoxyphenyl)-N-(4-vinylphenyl)aniline (0.94 g, 2.84 mmol), o-tolylphosphine (4 mol %) and PdCl2(PPh3)2 (5 mol %) were dissolved in dry DMF/toluene (1:1) mixture. After the completion of the reaction, the crude product was purified by column chromatography using petroleum ether/ethyl acetate (90:10) as an eluent, to afford 5 as yellow solid in 68% yield; 1H NMR (300 MHz, CDCl3) δ: 10.09 (s, 1H, CHO), 8.60 (d, J = 1.0 Hz, 1H, ar CH), 8.22 (s, 1H, ar CH), 8.03–7.99 (m, 1H, ar CH), 7.71–7.67 (m, 1H, ar CH), 7.44 (d, J = 8.5 Hz, 2H, ar CH), 7.39 (d, J = 2.3 Hz, 1H, ar CH), 7.37 (d, J = 2.3 Hz, 1H, ar CH), 7.12 (dd, J = 3.5 Hz, 2H, ar CH), 7.08 (d, J = 8.9 Hz, 4H, ar CH), 6.94 (d, J = 8.6 Hz, 2H, ar CH), 6.84 (d, J = 8.9 Hz, 4H), 3.87 (s, 3H, N–CH3), 3.81 (s, 6H, OCH3); 13C NMR (75 MHz, CDCl3) δ: 192.15 (–CHO), 156.30, 145.22, 141.43, 141.22,131.08, 129.01, 127.59, 127.45, 127.41, 127.26, 127.06, 126.77, 126.98, 125.98, 125.73, 123.66, 121.20, 121.09, 121.06, 118.63, 115.12, 109.68, 107.03, 55.91, 29.88 (–CH3). ESI–MS m/z: [M + H]+ calcd for C36H30N2O3 538.2325, found 538.3347.
(E)-2-cyano-3-(6-((E)-4-(diphenylamino)styryl)-9-methyl-9H-carbazol-3-yl)acrylic acid (D1)
A mixture of compound 4 (0.2 g, 0.44 mmol) and cyanoacetic acid (45 mg, 0.53 mmol) was dissolved in 10 mL of acetonitrile; subsequently, piperidine (56 mg, 0.66 mmol) was added into reaction mixture at refluxed for about 6 h. After the completion of the reaction, the reaction mixture was quenched with dilute HCl to remove the unreacted piperidine and then extracted with ethyl acetate two times, and the collective organic solvent was washed with brine solution and dried over anhydrous sodium sulphate. The solvent was evaporated under reduced pressure, and the crude product was purified by column chromatography using petroleum ether/chloroform (70:30); red-coloured solid was obtained. Yield is 85%; FT-IR (powder, cm−1) 3430, 2924, 2214, 1666, 1547, 1493, 1428, 1363, 1285, 1118, 749, 695. 1H NMR (400 MHz, DMSO-d6) δ: 12.35 (b s, 1H, COOH) 8.80 (s, 1H, ar CH), 8.38 (s, 1H, ar CH), 8.27 (s, 1H, ar CH), 8.22 (d, J = 8.7 Hz, 1H, ar CH), 7.79 (d, J = 8.8 Hz, 1H, ar CH), 7.75 (d, J = 8.8 Hz, 1H, ar CH), 7.64 (d, J = 6.5 Hz, 1H, ar CH), 7.51 (d, J = 8.8 Hz, 2H, ar CH), 7.30–7.26 (m, 5H, ar CH), 7.24 (d, J = 7.2 Hz, 1H, ar CH), 7.02–6.98 (m, 6H, ar CH), 6.93 (d, J = 8.4 Hz, 2H, ar CH), 3.90 (s, 3H, N-CH3); 13C NMR (75 MHz, DMSO-d6) δ: 164.65 (–COOH), 155.15, 147.55, 143.98, 141.41, 132.41, 131.23, 130.12, 126.02, 128.48, 127.90, 126.56, 125.82, 125.67, 124.57, 123.77, 123.70, 123.17, 122.99, 128.82, 118.10, 119.03, 110.94, 110.86, 30.04 (–CH3). ESI–MS m/z: [M + H]+ calcd for C37H27N3O2 545.210, found 545.2097.
(E)-3-(6-((E)-4-(bis(4-methoxyphenyl)amino)styryl)-9-methyl-9H-carbazol-3-yl)-2-cyanoacrylic acid (D2)
A mixture of compound 5 (0.5 g, 0.82 mmol) and cyanoacetic acid (84 mg, 0.9 mmol) was dissolved in 10 mL of acetonitrile solvent; subsequently, piperidine (0.1 g, 1.23 mmol) was added into the reaction mixture and refluxed for about 6 h. After the completion of the reaction, the reaction mixture was quenched with dilute HCl to remove the unreacted piperidine and then extracted with ethyl acetate two times; after that, collective organic solvent was washed with brine solution and dried over anhydrous sodium sulphate. Finally, the solvent was evaporated under reduced pressure and the crude product was purified by column chromatography using petroleum ether/chloroform (70:30) to get red solid. Yield is 85%; FT-IR (powder, cm−1) 3428, 2921, 2206, 1575, 1491, 1334, 1256, 1114, 970, 821, 697, 619. 1H NMR (400 MHz, DMSO-d6) δ: 12.31 (b s, 1H, COOH) 8.76 (s, 1H), 8.34 (s, 1H, ar CH), 8.23 (s, 1H, ar CH), 8.20 (d, J = 6.8 Hz, 1H, ar CH), 7.72 (d, J = 9.2 Hz, 1H, ar CH), 7.75 (d, J = 8.8 Hz, 1H, ar CH), 7.65 (d, J = 8.5 Hz, 1H, ar CH), 6.99 (d, J = 8.3 Hz, 4H, ar CH), 6.88 (d, J = 8.4 Hz 4H, ar CH), 6.73 (d, J = 8.4 Hz, 2H, ar CH), 3.89 (s, 3H, –CH3), 3.70 (s, 6H, OCH3);13C NMR (75 MHz, DMSO-d6) δ: 164.73, 156.28, 154.43, 154.40, 148.14, 143.80, 141.23, 141.20, 140.52, 130.70, 129.97, 128.30, 127.66, 127.19, 126.79, 123.32, 122.97, 128.82,119.92, 118.76, 115.48, 110.85, 110.77, 101.35, 55.76 (–OCH3), 29.99 (–CH3). ESI–MS m/z: [M + H]+ calcd. for C39H31N3O4 605.2317, found 605.2328.
(Bis(4-(bis(4-methoxyphenyl)amino)phenyl)amino)benzaldehyde (6)
4-(Bis(4-iodophenyl)amino)benzaldehyde (0.5 g, 0.95 mmol), bis(4-methoxyphenyl)amine (0.43 g, 1.90 mmol) and bis(tri-tert-butylphosphine)palladium(0) (4 mol %) were added into the Schlenk tube and dissolved in dry toluene under the protection of nitrogen atmosphere for about 30 min. After that, NatOBu (0.22 g, 2.3 mmol) was added into the reaction mixture and heated at 95 °C for about 12 h. After the completion of the reaction to monitor TLC, the resulting mixture was passed through the Celite bed and washed with ethyl acetate. The organic layer was further washed with a saturated brine solution and dried over anhydrous Na2SO4. The crude product was purified by column chromatography silica gel, using petroleum ether/ethyl acetate (85:15) as an eluent to afford yellow solid. Yield is 72%; 1H NMR (300 MHz, CDCl3) δ: 9.75 (s, 1H, –CHO), 7.64 (d, J = 8.8 Hz, 2H, ar CH), 7.08 (d, J = 9.0 Hz, 8H, ar CH), 6.99 (d, J = 8.9 Hz, 4H), 6.93 (d, 2H, ar CH), 6.89 (d, J = 9.1 Hz, 4H, ar CH), 6.84 (d, J = 9.0 Hz, 8H, CH), 3.80 (s, 12H, OCH3); 13C NMR (75 MHz, CDCl3) δ: 192.55 (–CHO), 155.99, 147.54, 130.79, 129.54, 129.21,128.79, 127.72, 127.27, 127.15, 126.52, 126.43, 124.45, 123.56, 122.97, 120.47, 114.77, 55.49 (–OCH3). ESI–MS m/z: [M + H]+ calcd. for C47H41N3O5, 727.3045 found 727.3016.
(E)-3-(4-(bis(4-(bis(4-methoxyphenyl)amino)phenyl)amino)phenyl)-2-cyanoacrylicacid (D3)
A mixture of compound 6 (0.5 g, 0.69 mmol) and cyanoacetic acid (64 mg, 0.76 mmol) was dissolved in 10 mL of acetonitrile solvent, and piperidine (85 mg, 1.03 mmol) was added to reaction mixture at refluxed for about 6 h. After the completion of reaction, the reaction mixture was quenched with dilute HCl to remove unreacted piperidine base and finally extracted with ethyl acetate two times; the collective organic solvent was washed with brine solution and dried over anhydrous sodium sulphate. Finally, the collective solvent was evaporated under reduced pressure; the crude product was purified by column chromatography using petroleum ether/chloroform (65:35) to obtain the dark red-coloured solid. Yield is 80%; FT-IR (powder, cm−1) 3432, 2919, 2218, 1585, 1504, 1238, 1175, 1030, 827, 713, 574. 1H NMR (400 MHz, DMSO-d6) δ: 12.01 (b s, 1H, COOH), 7.98 (s, 1H), 7.88 (d, J = 7.2 Hz, 2H, ar CH), 7.02 (d, J = 9.0 Hz, 10H, ar CH), 6.88 (d, J = 7.8 Hz, 10H), 6.72 (d, J = 7.8 Hz, 4H), 6.71 (d, J = 9.1 Hz, 2H), 3.11 (s, 12H, OCH3); 13C NMR (75 MHz, CDCl3) δ:159.94 (–COOH), 157.94, 149.50, 146.65, 139.69, 130.28, 129.46, 128.24, 127.47, 126.93, 125.50, 124.14, 124.08, 118.80, 114.31, 55.36 (–OCH3). ESI–MS m/z: [M + H]+ calcd. for C50H42N4O6 794.3107, found 794.3109.
(E)-2-cyano-3-(5-((E)-4-(diphenylamino)styryl)thiophen-2-yl)acrylic acid (D5)
The D5 (E)-2-cyano-3-(5-((E)-4-(diphenylamino)styryl)thiophen-2-yl)acrylic acid dye was synthesized from following the reported procedure [52, 53].
Results and discussion
Synthesis and characterization of organic dyes
Synthesis of (E)-2-cyano-3-(6-((E)-4-(diphenylamino)styryl)-9-methyl-9H-carbazol-3-yl) acrylic acid (D1), (E)-3-(6-((E)-4-(bis(4-methoxyphenyl)amino)styryl)-9-methyl-9H-carbazol-3-yl)-2-cyanoacrylic acid (D2) and (E)-3-(4-(bis(4-(bis(4-methoxyphenyl)amino) phenyl) amino) phenyl)-2-cyanoacrylic acid (D3) is shown in Scheme 1 and Fig. 1. A new series of D1, D2 and D3 dyes are synthesized from commercially available starting materials such as carbazole and triphenylamine. The formylation reaction of aldehyde derivatives like carbazole and triphenylamine 2 was prepared by Vilsmeier–Haack reaction; then, bromination was carried out using N-bromosuccinimide in DMF solvent; further, Mizoroki–Heck and Buchwald–Hartwig C–C and C–N coupling reactions were carried out, respectively. Finally, the desired products of D1–D3 were obtained by condensation of the respective aldehydes with cyanoacetic acid by Knoevenagel condensation reaction in the presence of piperidine. Further, the synthesized materials were confirmed by 1H NMR, 13C NMR, ESI–MS, FT-IR and their photophysical and optical properties such as UV–visible, fluorescence, thermogravimetry, electrochemical impedance spectroscopy and photovoltaic performance were studied.

Synthetic routes of target organic dyes D1, D2 and D3. Reagent and conditions: (a) DMF, POCl3, rt; (b) N-bromosuccinimide, DMF; (c) N,N-diphenyl-4-vinylaniline, PdCl2(PPh3)2, tri(o-tolyl)phosphine, K2CO3, 110 °C, overnight; (d) cyanoacrylic acid, piperidine, acetonitrile, rt; (e) bis(4-methoxyphenyl)amine, bis(tri-tert-butylphosphine)palladium(0), NatOBu, toluene, 110 °C, overnight
Optical properties
The electronic absorption spectra of compounds D1, D2 and D3 were recorded in dry THF solution and are shown in Fig. 2a, and corresponding data are listed in Table 1. In this case, compound shows a major absorption band appeared at 360, 370 and 434 nm, respectively, which are attributed to intramolecular charge transfer (ICT) through the whole molecule such as triphenylamine to cyanoacrylic acid group, providing efficient charge separation at the excited state. We observed such charge transfer transition is dependent on the solvent polarity, which is confirmed by solvatochromism. Moreover, in the solvatochromism studies, similar absorption bands were observed at fixed wavelength in different solvents, such as toluene, dichloromethane, THF and ethanol, but slightly positive shift in DMF solvent was observed (Fig. S1), which suggested that DMF has slightly basic character in nitrogen lone pair and interaction with cyanoacrylic acid. In addition to that, the absorption spectra were strongly affected by the acidity or alkalinity of the solutions (Fig. S2). In addition, molar extinction coefficient of maximum absorption band for D1 is ε = 33,650 M−1 cm−1, D2ε = 31,100 M−1 cm−1 and D3ε = 13,350 M−1 cm−1. Moreover, the D3 dyes show lower molar extinction coefficient; it can be speculated that there is an increase in co-planarity between electron donor part triphenylamine to electron acceptor part cyanoacrylic acid in the ground state. In addition to that, the longer wavelength has high molar extinction coefficient and broader absorption region is beneficial to improve the light-harvesting properties which leads to the improved photovoltaic performance and is more useful for the photocurrent generation in DSSC applications [56]. The D3 dye is redshifted due to an increase in the conjugation length and the number of triarylamine units, when compared with D1 and D2 dyes [57]. The dye D2 represents redshift for 10 nm compared with D1 dye; it may be attributed to electron-donating group (–OMe) attached to the triarylamine moieties. Furthermore, the Stokes shift decreases from D1 to D3, probably due to donor group equally shared the electron transportation with the acceptor group in D3 [58].

a Normalized UV–Vis absorption spectra of organic dyes D1, D2 and D3 in dry THF solutions. b Solid-state absorption spectra of the organic dyes (thin film)
The organic dyes anchored to a nanocrystalline TiO2 surface is also shown in Fig. 2b. Although the absorption wavelength of organic dyes adsorbed on TiO2 film D1, D2 and D3 is redshifted to 10, 12 and 26 nm, respectively, and broader absorption band was observed in comparison with those in solution state which is shown in Fig. 2a, this positive shift attributed to the interactions between TiO2 and organic dyes, restricted rotation of dye molecule and π–π stacking interaction between the organic dyes [59] are listed in Table 1. The fluorescence spectra of all the compound D1, D2 and D3 were also recorded in dry THF solution and are shown in Fig. 3a. The compounds shows emission band appeared at 471, 487 and 533 nm, respectively.

a Emission spectra of organic dyes in dry THF solutions. b–d FT-IR spectra of organic dyes D1, D2 and D3 coated on a TiO2 surface
FT-IR spectra
The FT-IR spectrum of compound D1, D2 and D3 powder dyes is adsorbed on TiO2 film and is shown in Fig. 3b–d; for the powder sample of three dyes, D1, D2 and D3, characteristic stretching bands for carboxylic acid functional group C=O are clearly observed at around 1670, 1653 and 1623 cm−1, respectively. In addition to that, we measured all the dye molecules adsorbed on nanocrystalline TiO2 surfaces [60, 61] and found that around 1725, 1703 and 1659 cm−1, respectively, from this observed results C=O stretching frequency shifted to 45 cm−1 higher wavenumber than the powder IR spectrum. Its suggested that restricted rotation of dye molecules, π–π stacking attraction between the organic dye and Bronsted acid site on TiO2 surfaces are strongly binding with –COO− acid group.
Electrochemical characteristics
In order to investigate the electrochemical properties of D1, D2 and D3 dyes, they are characterized by cyclic voltammetry techniques in acetonitrile-containing 0.1 M solution of tetrabutylammonium perchlorate (Bu4N+ClO ‒4 ) as a supporting electrolyte, with the scan rate 100 mv/s. The potentials were referenced to ferrocene/ferrocenium (Fe/Fe+) as an internal standard, which is shown in Figs. 4, 5. All these characteristic data are listed in Table 1. The D1, D2 and D3 showed quasi-reversible oxidation peak potential at 0.67, 0.48 and 0.34 V, respectively. It is suggested that a successive formation of the cation radical should be attributed to the removal of electrons from triphenylamine donor to acceptor carboxylic acid group. In addition, the corresponding reduction peak potential was also appeared; these results suggested that dye molecules are stable in that particular oxidized state. The HOMO levels of D1, D2 and D3 (0.67 V, 0.48 V, 0.34 V versus NHE, respectively) are adequately positive than the iodide/triiodide (0.40 V) redox potential value, which suggests that the oxidized dyes could be efficiently regenerated with enough driving force. Hence, the oxidized dyes and electron injected were increased in respective order D1 > D2 > D3. Further, the dyes accept electrons from redox couple electrolyte (I−/I3−) thermodynamically [62]. The HOMO and LUMO energy levels of all the dyes were analysed from absorption spectral analysis, and all these data provided in Table 2.

Cyclic voltammograms corresponding to oxidation of organic dyes in acetonitrile 0.1 M Bu4NClO4.a D1; b D2; and c D3

Molecular orbital energy-level diagram of all dyes, the TiO2 CB and the I−/I3− redox potential
Thermal properties
The thermal stability of D1, D2 and D3 dyes was carried out thermogravimetric analysis under nitrogen atmosphere, temperature range up to 800 °C, heating flow rate at 5 °C min−1 which is represented in Fig. 6. The thermogram of three dyes represented good thermal stability with no weight loss observed at low temperature. Moreover, the decomposition temperature starts at D1 dye, range of 247–291 °C, D2 dye the range of 285–371 °C and D3 dye range of 335–424 °C, corresponding to 10%, 14% and 15% weight loss observed respectively, the order of stability D3 > D2 > D1. Furthermore, the D3 dye represented more thermally stable when compared with D2 and D1 dyes, due to C2 symmetric nature of dye [60]. Based on these observed results, all these dyes are thermally stable, suitable for device fabrication to facilitate an enhanced lifetime for DSSC device.

Thermogravimetric analysis of D1, D2 and D3 dyes
The SEM images of bare TiO2 film and D1, D2 and D3 dyes adsorbed on TiO2 surface are presented in Fig. 7. Hence, the TiO2 film showed irregularly arranged particle-like morphological structure; this TiO2 film was prepared by doctor blade method and then immersed into a dye solution of (0.3 mM in DMF) for about 12 h; the structural morphology of dyes that adsorbed TiO2 film showed disturbed particle arrangement and porous structure, which indicates that all these dye molecules are adsorbed on TiO2 surfaces.

SEM images of a crystalline TiO2b–d D1, D2 and D3 dye molecules adsorbed on TiO2 film
Theoretical investigations
In order to investigate effect of electronic structure and electron distributions of D1, D2 and D3 on the performance of DSSCs, geometries and energy levels were optimized by density functional theory (DFT) and calculated by B3LYP/6-31G (d, p) level as shown in Fig. 8. The energy band gaps and optimized structure for D1, D2 and D3 dyes were calculated to be 3.16, 3.26 and 2.62 eV, respectively. At the HOMO level, all the dyes D1, D2 and D3 have high electron density mainly across the donor triphenylamine moieties [63]. The electron densities of the LUMO of the three dyes are located the cyanoacrylic acid group through the π-conjugated linkers; further, the excited state of all dyes responsible for strong visible absorption is potentially strongly coupled to TiO2 surface by virtue of cyanoacrylic contribution, which is favourable for efficient electron injection to anode. The calculated experimental and theoretical values are summarized in Table 2.

Frontier molecular orbital of HOMOs and LUMOs calculated at B3LYP/6-31G (d, p) level and optimized structure of dyes D1, D2 and D3
Photovoltaic performance
The current–voltage (I–V) curves of synthesized organic and commercial dyes (D1, D2, D3, D5 and N719) were measured under AM 1.5 sun illumination (100 mW cm−2) and are shown in Fig. 9, and the corresponding photovoltaic parameters are summarized in Table 2. DSSCs made with D1 dye presented best performance for power conversion efficiency of 1.4% (compared to conventional N719 dye which has 6.1% and D5 organic dye which has 2.2% under the same condition), which has Voc of 0.735 V, Jsc of 2.7 mA cm−2 and ff of 0.73. Notably, devices made with D3 dye also showed similar photovoltaic performance (η = 1.3%) with D1 dye, but interestingly the Jsc value of D3 dye is slightly higher (3.3 mA cm−2) and Voc (0.700 V) is much lower, when compared with D1 device; this may be due to a large planar π-conjugated system extending from the electron donor moiety to the electron acceptor moiety and D3 dye molecule contains two effective (–OMe) donating groups introduced in triphenylamine core; anchoring groups also directly attached to the triphenylamine core lead to larger wavelength and lower band gap to obtain higher Jsc. Moreover, the D1 and D2 dyes have intramolecular charge transfer effect over long distance [64]; upon light illumination conditions, electrons are not effectively injected into the TiO2 conduction band, so overall efficiency is reduced; the power conversion efficiency was calculated according to the following equation.
where Jsc is the photocurrent density of short-circuit current, Pin is the incident power density, Voc is the open-circuit voltage and ff denotes the fill factor.

J − V curves of DSSCs based on D1, D2, D3, D5 and N719 dyes
On the other hand, the significantly low photovoltaic performance of DSSC based on D2 device is 0.8%, when compared with D1 and D3 devices, due to the back electron transfer from the electrons injected into TiO2 to excited dye. The Jsc and Voc for D1 and D3 devices (2.7 mA cm−2, 0.735 V) and (3.3 mA cm−2, 0.700 V) are higher than those for D2 dye (1.6 mA cm−2 and 0.67 V). In addition to that, the formation of intermolecular π–π stacking aggregations, recombination process and self-quenching of the dyes are the major drawbacks for the low power conversion efficiencies. The photovoltaic performance of all the dyes shows maximum values; each dye was constructed and fabricated for four devices.
After that, scrutinizing the obtained photovoltaic parameters of sensitizer DSSCs, the incident photon-to-current conversion efficiency (IPCE) studies were performed for D1, D2 and D3 dye-fabricated devices; images of the cells are shown in Figs. 10, 11, and their corresponding results are listed in Table 3. Generally, higher IPCE spectra correspond to better Jsc values. In addition, the light-harvesting properties and electron injection ability depend on the Jsc value. The IPCE spectra of all the organic dyes D1, D2 and D3 exhibited a similar shape with different IPCE values 24.4%, 15.2% and 18.1%, and the wavelengths 418, 400 and 540, respectively, which may be attributed to the varied electron collection efficiencies with different core units. These results indicated that the D1 dye has better efficiencies compared with other two organic dyes.

IPCE spectra of D1, D2 and D3 dye-based DSSCs

Photographs of the dyes D1, D2 and D3 a in DMF solvent and b adsorbed on TiO2 film
Electrochemical impedance spectroscopy
In order to investigate the open-circuit voltage properties and charge recombination process in DSSCs, the electrochemical impedance spectroscopy was measured under dark condition. Th obtained Nyquist plots best fit to the Randles circuit model (Rs[C1 – Rp]) for dyes D1, D2 and D3 shown in Fig. 12a, where Rs represents the solution resistance, C1 represents constant phase element and Rp represents polarization resistance. In addition to that, Nyquist plot shows that larger semicircle in higher-frequency regions corresponds to charge recombination resistance at TiO2/dye/electrolyte interface [15, 17]. In addition, the RCT values of these dyes D1, D2 and D3 are calculated from EIS shown in the following order: D2 (9.014 KΩ) > D3 (8.008 KΩ) > D1(4.979 KΩ). After that, lower value of RCT indicates high charge loss in TiO2/dye/electrolyte interface, and also faster charge recombination and larger dark current [15, 44]. The larger value of RCT indicates more effective suppression and recombination of the injected electrons with I−/I3− in electrolyte [18]. Furthermore, Bode phase plot is drawn to analyse electron lifetime at the TiO2/dye/electrolyte interface calculated using τn = 1/(2πf). Moreover, the f values of D1, D2 and D3 dyes at 32.1 Hz, 120.2 Hz and 10.5 Hz are shown in Fig. 12b, c. The electron lifetime values are represented in the following order: D3 (15 ms) > D1 (4.96 ms) > D2 (1.32 ms), respectively, because of larger lifetime that the current in dark for DSSCs. Bode phase plots show phase angles of D1, D2 and D3 dyes as 62.5, 68.1 and 72.4 theta, respectively; the D1 has less phase angle compared to other dyes. It is suggested that electron transfer reaction was facile at these interfaces [65].

a Nyquist plots of DSSCs based on D1, D2 and D3 dyes, the experimental data, fitted curves, inner parts of the equivalent circuit model used in the studies, b Bode amplitude, c Bode phase angle plots obtained in dark conditions
The results show promise for D1, D2 and D3 dyes are generally stimulate in this field of dye development, but are disappointing. After that the synthesized organic dye 5 was compared with commercially available bench mark N719 dye [52]. In future to improve the metal-free organic dye development in combination with the co-absorbent and additives or perovskite absorber materials (CH3NH3PbI3), it may be possible to achieve high power conversion efficiencies.
Conclusions
In this study, we have successfully synthesized three push–pull organic dyes D1, D2 and D3 containing triphenylamine as a donor and cyanoacrylic acid as an auxiliary acceptor and employed them as photosensitizers in DSSCs. D1 and D2 composed of carbazole π-conjugated spacer unit has planar linear structure and has longer π-conjugated bridging units. We also believe that the flattened structure of D1 and D2 leads to more efficient charge transport, which is compared to triphenyl-bridged starburst structure of D3 dye; D1 dye exhibited comparatively high molar extinction coefficients, better electron collection efficiency, light-harvesting properties and slow carrier recombination than D2 and D3 dyes, which shows that all properties are beneficial to enhance the device performance. Furthermore, DSSC performance based on these dyes provides different results. Hence, D1 dye gives highest η values compared to D2 and D3 dyes, due to excellent photophysical, electrochemical properties, superior stoke shift, efficient electron injection and fast dye regenerations. A maximum power conversion efficiency value of 1.4% has been obtained under simulated AM 1.5 irradiation (100 mW/cm2) with a DSSCs based on D1 dye (Voc = 735 mV, Jsc = 2.70 mA/cm2, ff = 0.73); in addition to that, the comparison with 6.1% of N719 reported inorganic dye and that with 2.2% of D5 reported organic dyes under the same conditions. Overall dyes structure D1 appear more promising to the development of efficient organic sensitizer, which can be possible through the more sophisticated structural modification for the future material development.
Supplementary Information
All the structural characterization data are available in electronic supplementary information’s section.
References
B.O. Regan, M. Gratzel, Nature 353, 737 (1991)
P. Wang, C. Klein, T.H. Baker, S.M. Zakeeruddin, M. Gratzel, J. Am. Chem. Soc. 127, 808 (2005)
K.A. Kakiage, Y. Yano, T. Oya, K. Fujisawa, J.I. Hanaya, J.J.H.M. Bearpark, E. Brothers, K.N. Kudin, V.N. Staroverov, R. Kobayashi, Chem. Commun. 51, 15894 (2015)
R. Yan, X. Qian, Y. Jiang, Y. He, Y. Hang, L. Hou, (2017). https://doi.org/10.1016/j.dyepig.2017.02.011
J. Pan, H. Song, C. Lian, H. Liu, Y. Xie, Dyes and Pigments (2017). https://doi.org/10.1016/j.dyepig.2017.01.027
W.J. Fan, Y.Z. Chang, J.L. Zhao, Z.N. Xu, D.Z. Tan, Y.G. Chend, New J. Chem. 42, 20163 (2018)
Y. Mu, H. Wu, G. Dong, Z. Shen, S. Li, M. Zhang, J. Mater. Chem. A 6, 21493 (2018)
P. Naik, I.M. Abdellah, M.A. Shakour, M. Acharaya, N. Pilicode, A.E. Shafei, A. Vasudeva Adhikari, Chem. Sel. 3, 12297 (2018)
F. Bellaa, A. Verna, C. Gerbaldi, Materials Science Semiconductor Processing (2017). https://doi.org/10.1016/j.mssp.2017.07.030
A. Carella, R. Centore, F. Borbone, M. Toscanesi, M. Trifuoggi, F. Bella, C. Gerbaldi, S. Galliano, E. Schiavo, A. Massaro, A.B. Munoz-Garcia, M. Pavonea, Electrochimica Acta. https://doi.org/10.1016/j.electacta.2018.09.204
S. Galliano, F. Bella, G. Piana, G. Giacona, G. Viscardi, C. Gerbaldi, M. Grätzel, C. Barolo, Sol. Energy 163, 251 (2018)
A. Scalia, F. Bella, A. Lamberti, C. Gerbaldi, E. Tresso, Energy 166, 789 (2019)
F. Bella, J. Popovic, A. Lamberti, E. Tresso, C. Gerbaldi, J. Maier, A.C.S. Appl, Mater. Interfaces 9, 37797 (2017)
K.M. Manikandan, A. Yelilarasi, P. Senthamaraikannan, S.S. Saravanakumar, A. Khan, A.M. Asiri, J. Solid State Electrochem. 22, 3785 (2018)
V. Ganapathy, B. Karunagaran, S.W. Rhee, J. Power Sour. 195, 5138 (2010)
A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo, H. Pettersson, Chem. Rev. 110, 6595 (2010)
V. Karthikeyan, S. Maniarasu, V. Manjunath, E. Ramasamy, G. Veerappan, Sol. Energy 147, 202 (2017)
G. Veerappan, D.W. Jung, J. Kwon, J.M. Choi, N. Heo, G.R. Yi, J.H. Park, Langmuir 30, 3010 (2014)
R. Sivakumar, R. Recabarren, S. Ramkumar, A. Manivel, J.A. Morales, D. Contreras, P. Manidurai, New J. Chem. 41, 5605 (2017)
X. Li, J. Gui, H. Yang, W. Wu, F. Li, H. Tian, C. Huang, Inorg. Chim. Acta 361, 2835 (2008)
H. Tian, X. Yang, R. Chen, Y. Pan, L. Li, A. Hagfeldt, L. Sun, Chem. Commun. 36, 3741 (2007)
Y.S. Yen, H.H. Chou, Y.C. Chen, C.Y. Hsu, J.T. Lin, J. Mater. Chem. 22, 8734 (2012)
H. Masui, M.M. Maitani, S. Fuse, A. Yamamura, Y. Ogomi, S. Hayase, T. Kaiho, H. Tanaka, Y. Wada, T. Takahashi, Asian J. Org. Chem. 7, 458 (2018)
T.Y. Wu, M.H. Tsao, F.L. Chen, S.G. Su, C.W. Chang, H.P. Wang, Y.C. Lin, I.W. Sun, J. Iran. Chem. Soc. 7, 707 (2010)
F. Zanjanchi, J. Beheshtian, J. Iran. Chem. Soc. (2018). https://doi.org/10.1007/s13738-018-1561-2
K. Sayama, K. Hara, N. Mori, M. Satsuki, S. Suga, S. Tsukagoshi, Y. Abe, H. Sugihara, H. Arakawa, Chem. Commun. 0, 1173 (2000)
C. Chen, J.Y. Liao, Z. Chi, B. Xu, X. Zhang, D.B. Kuang, Y. Zhang, S. Liu, J. Xu, J. Mater. Chem. 22, 8994 (2012)
W. Li, Y. Wu, X. Li, Y. Xe, W. Zhu, Energy Environ. Sci. 4, 1830 (2011)
D.Y. Chen, Y.Y. Hsu, H.C. Hsu, B.S. Chen, Y.T. Lee, H. Fu, M.W. Chung, S.H. Liu, H.C. Chen, Y. Chi, P.T. Chou, Chem. Commun. 46, 5256 (2010)
H. Zhou, P. Xue, Y. Zhang, X. Zhao, J. Jia, X. Zhang, X. Lu, R. Lu, Tetrahedron 67, 8477 (2011)
B. Nagarajan, S. Kushwaha, R. Elumalai, S. Mandal, K. Ramanujam, D. Raghavachari, J. Mater. Chem. A. 5, 10289 (2017)
K.B. Fadadu, S.S. Soni, Electrochim. Acta 88, 270 (2013)
X. Ma, J. Hua, W. Wu, Y. Jin, F. Meng, W. Zhan, H. Tian, Tetrahedron 64, 345 (2008)
K.D. Seo, I.T. Choi, Y.G. Park, S. Kang, J.Y. Lee, H.K. Kim, Dyes Pigm. 94, 469 (2012)
K. Hara, Z.S. Wang, T. Sato, A. Furube, R. Katoh, H. Sugihara, Y.D. Oh, C. Kasada, A. Shinpo, S. Suga, J. Phys. Chem. B. 109, 15476 (2005)
Z. Shen, J. Chen, X. Li, X. Li, Y. Zhou, Y. Yu, H. Ding, J. Li, L. Zhu, J. Hua, ACS Sustinable Chem. Eng. 4, 3518 (2016)
X. Liu, Z. Cao, H. Huang, X. Liu, Y. Tan, H. Chen, Y. Pei, S. Tan, J. Power Sour. 248, 400 (2014)
X.F. Wang, H. Tamiaki, O. Kitao, T. Ikeuchi, S.I. Sasaki, J. Power, Sources 242, 860 (2013)
J. Chung, C.P. Lee, D. Kumar, P.W. Chen, L.Y. Lin, K.R.J. Thomas, K.C. Ho, J. Power Sour. 240, 779 (2013)
D. Pugliese, A. Lamberti, F. Bella, A. Sacco, S. Bianco, E. Tresso, Org. Electron. 15, 3715 (2014)
T. Horiuchi, H. Miura, K. Sumioka, S. Uchida, J. Am. Chem. Soc. 126, 12218 (2004)
V.A. Chiykowski, B. Lam, C. Du, C.P. Berlinguette, Chem. Commun. 53, 2367 (2017)
G. Zhang, H. Bala, Y. Cheng, D. Shi, X. Lv, Q. Yu, P. Wang, Chem. Commun. 16, 2198 (2009)
T.B. Raju, J.V. Vaghasiya, M.A. Afroz, S.S. Soni, P. Krishnan Iyer, Phys. Chem. Chem. Phys. 18, 28485 (2016)
A. Karuppasamy, K. Stalindurai, J. Peng, K.C. Ho, C. Ramalingan, Phys. Chem. Chem. Phys. 18, 30105 (2016)
D. Kumar, K.T. Wong, Mater. Today Energy 5, 243 (2017)
Y. Ooyama, N. Yamaguchi, I. Imae, K. Komaguchi, J. Ohshita, Y. Harima, Chem. Commun. 49, 2548 (2013)
A. Abbotto, N. Manfredi, C. Marinzi, F.D. Angelis, E. Mosconi, J. Yum, Z. Xianxi, M.K. Nazeeruddin, M. Gratzel, Energy Environ. Sci. 2, 1094 (2009)
J. Wang, S. Liu, Z. Chai, K. Chang, M. Fang, M. Han, Y. Wang, S. Li, H. Han, Q. Li, Z. Li, J. Mater. Chem. A 6, 22256 (2018)
G. Prakash, K. Subramanian, New J. Chem. 42, 17939 (2018)
D.P. Hagberg, T. Edvinsson, T. Marinado, G. Boschloo, A. Hagfeldtb, L. Sun, Chem. Commun. 0, 2245 (2006)
M.A. Mamun, Q. Qiao, B.A. Logue, RSC Adv. 8, 31943 (2018)
A. Karuppasamy, K. Stalindurai, J.D. Peng, K.C. Ho, C. Ramalingan, Phys. Chem. Chem. Phys. 18, 30105 (2016)
F. Meng, Y. Liu, X. Yu, W. Lin, New J. Chem. 40, 7399 (2016)
G. Li, Y.F. Zhou, X.B. Cao, P. Bao, K.J. Jiang, Y. Lin, L.M. Yang, Chem. Commun. 0, 2201 (2009)
Z. Ning, Q. Zhang, W. Wu, H. Pei, B. Liu, H. Tian, J. Org. Chem. 73, 3791 (2008)
R. Balasaravanan, V. Sadhasivam, G. Sivaraman, A. Siva, Asian J. Org. Chem. 5, 399 (2016)
R.Y.Y. Lin, H.W. Lin, Y.S. Yen, C.H. Chang, H.H. Chou, P.W. Chen, C.Y. Hsu, Y.C. Chen, J.T. Lin, K.C. Ho, Energy Environ. Sci. 6, 2477 (2013)
Y. Ooyama, K. Furue, T. Enoki, M. Kanda, Y. Adachi, J. Ohshita, Phys. Chem. Chem. Phys. 18, 30662 (2016)
A. Jankovic, Z.V. Saponjic, M.I. Comor, J.M. Nedeljkovic, J. Phys. Chem. C 113, 12645 (2009)
J.V. Vaghasiya, K.K. Sonigara, J. Prasad, T. Beuvier, A. Gibaud, S.S. Soni, J. Mater. Chem. A. 5, 5373 (2017)
B.G. Kim, K.M. Chung, J.S. Kim, Chem. Eur. J. 19, 5220 (2013)
Y.A. Sadiki, S.M. Bouzzine, L. Bejjit, G.S. Moran, M. Hamidi, M. Bouachrine, F. Serein-Spirau, J.P. Lere Porte, J. Marc Sotiropoulos, L.G. Candia, D.G. Mitnik, J. Iran. Chem. Soc. 13, 37 (2016)
A.N. Krol, B. Filmmel, M. Son, D. Kim, F. Warthner, Faraday Discuss. 185, 507 (2015)
P. Arul, S.A. John, Electrochim. Acta 235, 680 (2017)
Acknowledgements
The authors acknowledge the financial support from the Department of Science and Technology, SERB, Extramural Major Research Project (Grant No. EMR/2015/000969), the Department of Science and Technology, CERI, New Delhi, India (Grant No. DST/TM/CERI/C130(G), and the University Grants Commission, New Delhi, India (Grant No. UGC No.41-215/2012 (SR). We acknowledge the DST-FIST, DST-PURSE, UPE programme for providing instrumental support.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Velu, S., Muniyasamy, H., Ayyanar, S. et al. Design, synthesis of organic sensitizers containing carbazole and triphenylamine π-bridged moiety for dye-sensitized solar cells. J IRAN CHEM SOC 16, 1923–1937 (2019). https://doi.org/10.1007/s13738-019-01663-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13738-019-01663-w