
Abstract
A reddish orange emissive long afterglow phosphors Ca2−xSnO4:xSm3+ (x = 0.001–0.05) are prepared by solid-state reaction in air atmosphere. The synthesized phosphors are characterized and analyzed by X-ray diffraction, photoluminescence spectra, afterglow decay curves, and absorption spectra. Under excitation at 407 nm, three emission peaks locate at 565, 610 and 654 nm, respectively, which can be assigned to the 4G5/2→6HJ (J = 5/2, 7/2, 9/2) transitions of Sm3+ ion. The fluorescent intensity and the afterglow characteristic depend on the concentration of Sm3+. The optimal Sm3+ concentration is x = 0.01. The CIE 1931 chromaticity coordinates of the emission and afterglow are (x = 0.6103, y = 0.3891) and (x = 0.5668, y = 0.4325) located in the range of reddish orange light emission. The afterglow decay curves of the Ca2SnO4:Sm3+ phosphor indicate both fast and slow decay components. The striking difference of the afterglow luminescence intensities of the phosphor after irradiation under 254 and 365 nm UV is discussed in deeply with absorption spectrum.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Long-lasting phosphorescence (LLP) is a luminescence process with the decay time extending to seconds, minutes, or even hours at room temperature after removal of the excitation [1,2,3,4]. These materials are used for safety indication, emergency lighting, light guide [5], billboards, graphic arts, and interior decoration, lighting, display, radiation dosimetry [6,7,8,9], optical memory [10], erasable optical storage [11], and pressure sensors [12]. Till now, different types of inorganic lattices, including sulfides, aluminates, silicates, titanates, oxysulfides, nitrides, pyrophosphates, etc., doped with various activators, have been developed as LLP phosphors [13,14,15,16,17,18,19,20,21]. As to the activators of the LLP phosphors, almost all the lanthanides ions (excluding radioactive Pm) and transition metal ions Mn2+, Ti4+ have been reported to successfully produce afterglow emission in various host lattices [22, 23].
Among the reported long afterglow phosphors, the performance of LLP phosphors emitting in the blue [24] and green [13] spectral region almost meet the requirement for practical applications and are commercially widely available. However, orange to red LLP phosphors are still out of reach from a practical point of view. Therefore, there is still a strong need for the development of novel host materials for LLP phosphors emitting at longer, i.e. in the orange to red, wavelength range [25].
In the early time, the sulfides activated by some rare earth ions such as MS: Eu3+ (Eu2+) (M = Ca, Sr, Zn) are the main red phosphorescence materials [26, 27], but they are not very stable. Since Murazaki et al. reported a red LLP phosphor Y2O2S: Eu3+, Ti4+, Mg2+ which afterglow lasted above 3 h [26], there have been many researches on red Eu3+ activated oxysulfides LLP phosphors [28,29,30]. But such oxysulfides obtained under weak reducing atmosphere requires better equipment and complicated technique. Moreover, harm gas released in the process.
From a practical point of view, oxide phosphors are more attractive than the traditional sulfide or halide phosphors due to their resistance to moisture. Ca2SnO4, as one-dimensional chains structure, is very easy to implant other ions into the host lattice and generally exhibit special luminescence properties. During last decade, alkaline earth ortho-stannates (M2SnO4, where M = Ca, Sr and Ba) have been drawing more and more attention as host matrix for new phosphors because of their stable crystalline structure and high physical, chemical stability, environment friendly, lower cost and simple preparation technique [31,32,33,34,35]. Hence calcium stannates were chosen as the matrix in many cases, such as red Ca2SnO4:Eu3+ [3, 32, 36], Ca2SnO4:Pr3+ [37], reddish orange Ca2SnO4:Sm3+ [38], green Ca2SnO4:Tb3+ [39] and yellow Ca2SnO4:Dy3+ [40] phosphors.
In this work, an interesting reddish orange light-emitting LLP material Ca2SnO4:Sm3+ was synthesized via the conventional solid-state reaction method. Their photoluminescence and LLP properties, were systematically investigated by means of photoluminescence excitation and emission spectra, LLP emission spectra, decay curves and absorption spectra. We discussed the strongest excitation peak at 407 nm, and the enhanced afterglow by the absorption spectra.
2 Experiment
The powder samples of Ca2−xSnO4:xSm3+ were prepared via conventional high temperature solid state reaction method. The starting materials used in the preparation were CaCO3 (99.99%), SnO2 (A.R.) and Sm2O3 (99.9%). The powders were mixed thoroughly for 1 h in an agate mortar and the mixtures were then transferred into corundum crucibles and calcined at 1350 °C for 4 h inside a furnace in air atmosphere. After cooling down to room temperature, the samples were ground again to fine powders in an agate mortar for measurements.
The sample structure was identified by Smart Lab X-ray powder diffraction (XRD) with Ni-filtered CuKa radiation in 2θ range from 10° to 80°. Photoluminescence spectra and afterglow characteristic were recorded on a HORIBA Fluoromax-4 Spectrofluorometer. Absorption spectra was measured on a HITACHI U-4100 Spectrophotometer. All measurements were recorded at room temperature.
3 Results and discussion
3.1 XRD pattern of Ca2SnO4:Sm3+
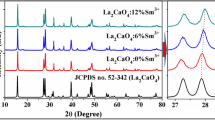
Figure 1 shows the X-ray diffraction patterns of Ca2−xSnO4:xSm3+ (x = 0.001–0.05). As a result, a typical Ca2SnO4 pattern is observed for all investigated samples which agree with the phase of Ca2SnO4 (JCPDS Card No. 46-0112), which indicates that Sm3+ ions are well doped into the lattice. The positions of the peaks in the XRD profiles shift to lower angles with the increasing Sm3+ concentration (Fig. 1b). It is known that Ca2+ is seven coordinate and Sn4+ is six coordinate in the Ca2SnO4 [11, 41]. Considering the radius of Ca2+ (1.06 Å, CN = 7), Sm3+ (1.02 Å, CN = 7) and Sn4+ (0.71 Å, CN = 6), it can be expected that the Sm3+ ions occupy the Ca2+ sites. The crystal structure of Ca2SnO4 consists of chains of edge-linking SnO6 octahedra [1, 32]. SnO6 octahedra are connected in low-dimensional form, and SnO6 octahedra are linked sharing edges with each other and forming one-dimensional chains. In this kind of low-dimensional structure, it is easy to implant other ions into the host lattice and create traps located at suitable depths that can store the excitation energy and emit light upon re-activation at room temperature [42, 43]. The extension of the lattice parameter may be due to the Sm3+ ions entering interstitial sites [3]. Since the effect of the coordination number on the radius of the ions with unsaturated electron configuration is more than that of the saturated electron configuration ones, the radius of Sm3+ ion is larger than that of Ca2+ [32]. Thus, the substitution of Ca2+ ions by Sm3+ increases the cell parameter.

XRD patterns of Ca2−xSnO4:xSm3+ (x = 0.001–0.05) and JCPDS Card No. 46-0112 (b) partially enlargement of (a)
3.2 Fluorescence properties of Ca2SnO4:Sm3+
The excitation and emission spectra of Sm3+-doped Ca2SnO4 phosphor are shown in Fig. 2. The excitation spectra consist of a series of peaks in the range of 350–550 nm. The strongest band at 407 nm (6H5/2→4L13/2 + 6P3/2 + 4F7/2) and some peaks at 362 (5D3/2 + 4F9/2 + 6P5/2), 381 (6P7/2 + 4K13/2), 416 (6P5/2) and 477 nm (4I9/2 + 4M15/2) are ascribed to the transitions from the ground state to the high energy excited levels of Sm3+ [1, 42, 44, 45]. Under excitation at 407 nm, there are three peaks situate at 565, 610 and 654 nm, respectively, which can be assigned to the 4G5/2→6HJ (J = 5/2, 7/2, 9/2) transitions of Sm3+ ion [11, 46,47,48]. Among them, the strongest peak is located at 610 nm. The most prominent emission peak situates at 610 nm combined with other two peaks make this phosphor emit reddish-orange color light which shown in Fig. 7.

Excitation (λem = 610 nm) spectra of Ca2−xSnO4:xSm3+ phosphor. The inset shows the dependence of the intensity on Sm3+ concentration
The luminescence intensity of phosphor materials is always dependent on doping concentration. The dependence of the photoluminescence intensities on Sm3+ concentrations is depicted in Figs. 2 and 3. It is quite clear that the fluorescent intensity increased with the increasing concentration of Sm3+ ion up to x = 0.01, then decreased beyond the concentration due to concentration quenching. The reason lies in that odds of complex of non-radiative transition is increasing as the concentration of Sm3+ is increasing, thus the fluorescent intensity of the phosphors become lower.

Emission (λex = 407 nm) spectra of Ca2−xSnO4:xSm3+ phosphor. The inset shows the dependence of the intensity on Sm3+ concentration
3.3 Phosphorescence properties of Ca2SnO4:Sm3+
Figure 4 shows the afterglow spectra of Ca2−xSnO4:xSm3+ (x = 0.001–0.05) phosphors at 30 s after irradiation. All the samples are irradiated under 254 and 365 nm UV with a same power for 30 min, respectively. There are four peaks located at 572, 584, 612 and 626 nm, respectively. The afterglow emission spectra of Ca2−xSnO4:xSm3+ with the dopant levels ranging x = 0.001–0.05, are recorded at the same time after stoppage of irradiation. As the dopant levels ranging x = 0.001–0.05, it is noted that the optimal concentration occurred when the increasing concentration of Sm3+ ion up to x = 0.01. It is a common viewpoint that the occurrence of LLP is dependent on the presence of abundant defect traps with suitable depth used to store the incident energy. The duration time of the afterglow phosphor is markedly affected by the depth and density of the defect traps. If the trap levels are too shallow, the traps would be emptied too quickly and the LLP can only last for a too short time; on the contrary, if the trap levels are too deep, the trapped carriers cannot acquire enough energy to escape from the trap at room temperature, and then consequently cannot transfer the stored energy to the luminescent center to produce LLP emission [25, 38].

Afterglow emission spectra of Ca2−xSnO4:xSm3+ with the dopant levels ranging x = 0.001–0.05, at 30 s after irradiation under 254 and 365 nm UV respectively
It is also noted from Fig. 4 that the afterglow emission intensity of the phosphor after irradiation under 365 nm UV is relatively low. However, the initial intensity of the phosphor after irradiation under 254 nm UV is stronger about 100 times than that under 365 nm UV. The result indicates that the Sm3+–Ca2SnO4 phosphor can be more effectively activated under 254 nm UV. And so, the afterglow luminance has been greatly improved. This phenomenon will be discussed with the absorption spectrum of Ca2SnO4:Sm3+ phosphor in deeply later.
Figure 5 shows the persistent luminescence decay curves of the Ca2−xSnO4:xSm3+ (x = 0.001–0.05) phosphors after irradiation under 254 nm UV for 30 min. The afterglow intensity decreases quickly at first several minutes and then decays very slowly. The multi-exponential afterglow delay model of Ca2−xSnO4:xSm3+ phosphor is fully in agree with the behavior of a wide variety of rare-earth-ion-doped LLP phosphors [14,15,16,17,18,19, 42, 45].

Afterglow decay curves of Ca2−xSnO4:xSm3+ (x = 0.001–0.05) phosphor monitoring after irradiation
3.4 Absorption spectrum of Ca2SnO4:Sm3+
To understand the identical shape and peak positions, but remarkable different intensities (at 572, 584, 612 and 626 nm) of the afterglow emission spectra after irradiation under 254 and 365 nm UV respectively, the absorption spectra are measured. Figure 6 shows the absorption spectra of Ca2−xSnO4:xSm3+ (x = 0–0.05) phosphors measured by a HITACHI U-4100 Spectrophotometer in the spectral region from 500 to 200 nm at room temperature. The compounds exhibit a broad absorption band which enhance from blue to UV range. It can be found that the non-doped Ca2SnO4 host showed a status of lower absorption from 500 to 350 nm, a certain ascent (350–300 nm), a certain descent (300–270 nm), and then arose a notable ascent from 270 to 240 nm which peak band centered at 254 nm.

Absorption spectra of Ca2−xSnO4:xSm3+ (x = 0–0.05). The inset is the partial enlarged detail
However, the Sm3+-doped Ca2SnO4 samples act out obvious differences in absorption profiles. All Sm3+-doped Ca2−xSnO4:xSm3+ (x = 0.001–0.05) phosphors contain an absorption peak at about 407 nm which is ascribed to the absorption of the transitions of Sm3+ (6H5/2→4L13/2 + 6P3/2 +4F7/2). It is quite clear that the absorptivity increased with the increasing concentration of Sm3+ ion from x = 0.001 up to x = 0.05. So, the strongest excitation peak located at 407 nm (Fig. 2). In addition, the absorption intensity of Sm3+-doped samples keep on a rise from 350 to 254 nm without a drop from 300 to 270 nm.
The Ca2−xSnO4:xSm3+ (x = 0.001–0.05) samples can be effectively activated and rapidly charged by the 254 nm UV. This results in remarkable persistent luminescence. The afterglow intensity after irradiation under 365 nm UV is much weaker than that under 254 nm UV (Fig. 4). It may be involved in the electronic trapping and detrapping recombination process. Under high-energy light (about 254 nm) excitation, the ground-state electrons of Sm3+ ions are promoted to the conduction band. The electrons trapping and detrapping processes through the conduction band contribute significantly to the persistent luminescence. Whereas the persistent luminescence after irradiated under low-energy light (about 365 nm), occurs through the quantum tunneling process. This tunneling process needs not go through the conduction band and proceeds at a slow rate. So, it produces much weaker long-persistent luminesce [49].
3.5 Luminous colors of the Ca2SnO4:Sm3+
The emission (λex = 407 nm) and afterglow (after irradiation under 254 nm UV) luminous colors of the sample with x = 0.01 are reddish orange as result of complex spectra shown in Figs. 7 and 8. In general, luminous color is represented by color coordinates and color ratios. The chromaticity coordinates of the samples have been calculated from the spectra using the CIE1931 Chromaticity Coordinate Calculation system. The chromaticity coordinates of the emission and afterglow are (x = 0.6103, y = 0.3891) and (x = 0.5668, y = 0.4325) located in the range of reddish orange light emission.

CIE of emission (λex = 407 nm) of Ca1.99SnO4:0.01Sm3+

CIE of afterglow (after irradiation under 254 nm UV) emission of Ca1.99SnO4:0.01Sm3+
4 Conclusions
A reddish orange long afterglow phosphor Ca2SnO4:Sm3+ was prepared by solid-state reaction in air atmosphere. The CIE 1931 chromaticity coordinates of the emission and afterglow are (x = 0.6103, y = 0.3891) and (x = 0.5668, y = 0.4325) located in the range of reddish orange light emission. The afterglow decay curves of the Ca2SnO4:Sm3+ phosphor indicate both fast and slow decay components. The afterglow intensity after irradiation under 254 nm UV is about 100 times than that under 365 nm UV. The striking difference of the afterglow intensity of the phosphor excited by different wavelength radiation is discussed in deeply with absorption spectrum, suggest that it may be involved in the electronic trapping and detrapping recombination process through the conduction band and the quantum tunneling process.
References
B. Lei, H. Zhang, W. Mai, S. Yue, Y. Liu, S. Man, Luminescent properties of orange-emitting long-lasting phosphorescence phosphor Ca2SnO4:Sm3+. Solid State Sci. 13, 525–528 (2011)
P. Chau, K. Ryu, C. Yo, Influence of the technological conditions on the luminescence of Eu3 + ions in Sr2SnO4. J. Mater. Sci. 33, 1299–1302 (1998)
Y.-C. Chen, Y.-H. Chang, B.-S. Tsai, Synthesis and the luminescent properties of europium-activated Ca2SnO4 phosphors. Opt. Mater. 27, 1874–1878 (2005)
F. Clabau, X. Rocquefelte, S. Jobic, P. Deniard, M.H. Whangbo, A. Garcia, T. Le Mercier, Mechanism of phosphorescence appropriate for the long-lasting phosphors Eu2+-Doped SrAl2O4 with Codopants Dy3+ and B3+. Chem. Mater. 17, 3904–3912 (2005)
U. Caldiño, A. Speghini, S. Berneschi, M. Bettinelli, M. Brenci, S. Pelli, G.C. Righini, Optical spectroscopy and waveguide fabrication in Sm3+/Tb3+ doped zinc–sodium–aluminosilicate glasses. Opt. Mater. 34, 1067–1071 (2012)
G. Okada, B. Morrell, C. Koughia, A. Edgar, Spatially resolved measurement of high doses in microbeam radiation therapy using samarium doped fluorophosphate glasses. Appl. Phys. Lett. 99, 1395 (2011)
G. Belev, G. Okada, D. Tonchev, C. Koughia, C. Varoy, A. Edgar, T. Wysokinski, D. Chapman, S. Kasap, Valency conversion of samarium ions under high dose synchrotron generated X-ray radiation. Physica Status Solidi 8, 2822–2825 (2011)
G. Okada, S. Vahedi, B. Morrell, C. Koughia, G. Belev, T. Wysokinski, D. Chapman, C. Varoy, A. Edgar, S. Kasap, Examination of the dynamic range of Sm-doped glasses for high-dose and high-resolution dosimetric applications in microbeam radiation therapy at the Canadian synchrotron. Opt. Mater. 35, 1976–1980 (2013)
G. Okada, J. Ueda, S. Tanabe, G. Belev, T. Wysokinski, D. Chapman, D. Tonchev, S. Kasap, Samarium-doped oxyfluoride glass-ceramic as a new fast erasable dosimetric detector material for microbeam radiation cancer therapy applications at the canadian synchrotron. J. Am. Ceram. Soc. 97, 2147–2153 (2014)
K. Hirao, S. Todoroki, D.H. Cho, N. Soga, Room-temperature persistent hole burning of Sm2+ in oxide glasses. Opt. Lett. 18, 1586–1587 (1993)
Z.-H. Ju, S.-H. Zhang, X.-P. Gao, X.-L. Tang, W.-S. Liu, Reddish orange long afterglow phosphor Ca2SnO4:Sm3+ prepared by sol–gel method. J. Alloy. Compd. 509, 8082–8087 (2011)
Q. Jing, Q. Wu, L. Liu, J.A. Xu, Y. Bi, Y. Liu, H. Chen, S. Liu, Y. Zhang, L. Xiong, An experimental study on SrB4O7:Sm2+ as a pressure sensor. J. Appl. Phys. 113, 284 (2013)
T. Matsuzawa, Y. Aoki, N. Takeuchi, Y. Murayama, A new long phosphorescent phosphor with high brightness, SrAl2O4: Eu2+, Dy3+. J. Electrochem. Soc. 143, 2670–2673 (1996)
J. Trojan-Piegza, J. Niittykoski, J. Hölsä, E. Zych, Thermoluminescence and kinetics of persistent luminescence of vacuum-sintered Tb3+-doped and Tb3+, Ca2+-codoped Lu2O3 materials. Chem. Mater. 20, 2252–2261 (2008)
J. Wang, S. Wang, Q. Su, The role of excess Zn2+ ions in improvement of red long lasting phosphorescence (LLP) performance of β-Zn3(PO4)2:Mn phosphor. J. Solid State Chem. 177, 895–900 (2004)
B. Lei, Y. Liu, G. Tang, Z. Ye, C. Shi, Spectra and long-lasting properties of Sm3+-doped yttrium oxysulfide phosphor. Mater. Chem. Phys. 87, 227–232 (2004)
M. Fang, H. Wang, X. Tan, B. Cheng, L. Zhang, Z. Xiao, One-dimensional hollow SrS nanostructure with red long-lasting phosphorescence. J. Alloy. Compd. 457, 413–416 (2008)
X. Zhang, J. Zhang, X. Zhang, L. Chen, S. Lu, X.-J. Wang, Enhancement of red fluorescence and afterglow in CaTiO3: Pr3+ by addition of Lu2O3. J. Lumin. 122–123, 958–960 (2007)
Y. Liu, B. Lei, C. Shi, Luminescent properties of a white afterglow phosphor CdSiO3:Dy3+. Chem. Mater. 17 (2005) 2108–2113
B. Lei, K. Machida, T. Horikawa, H. Hanzawa, N. Kijima, Y. Shimomura, H. Yamamoto, Reddish-orange long-lasting phosphorescence of Ca2Si5N8:Eu2+, Tm3+ phosphor. J. Electrochem. Soc. 157, J196-J201 (2010)
B. Lei, Y. Liu, J. Zhang, J. Meng, S. Man, S. Tan, Persistent luminescence in rare earth ion-doped gadolinium oxysulfide phosphors. J. Alloy. Compd. 495, 247–253 (2010)
C.-C. Kang, R.-S. Liu, J.-C. Chang, B.-J. Lee, Synthesis and luminescent properties of a new yellowish-orange afterglow phosphor Y2O2S:Ti, Mg. Chem. Mater. 15, 3966–3968 (2003)
Y. Lin, Z. Tang, Z. Zhang, C.W. Nan, Luminescence of Eu2+ and Dy3+ activated R3MgSi2O8-based (R = Ca, Sr, Ba) phosphors. J. Alloy. Compd. 348, 76–79 (2003)
Y. Lin, Z. Tang, Z. Zhang, Preparation of long-afterglow Sr4Al14O25-based luminescent material and its optical properties. Mater. Lett. 51, 14–18 (2001)
B.-F. Lei, S. Yue, Y.-Z. Zhang, Y.-L. Liu, Luminescence properties of Sr2Sn4:Sm3+ afterglow phosphor. Chin. Phys. Lett. 27, 037201 (2010)
X. Gao, Z. Zhang, C. Wang, J. Xu, Z. Ju, Y. An, W. Liu, The persistent energy transfer and effect of oxygen vacancies on red long-persistent phosphorescence phosphors Ca2SnO4: Gd3+, Eu3+. J. Electrochem. Soc. 158, J405 (2011)
D. jia, Enhancement of long-persistence by Ce Co-doping in CaS: Eu2+, Tm3+ red phosphor. J. Electrochem. Soc. 153, H198–H201 (2006)
W. Li, Y. Liu, P. Ai, Synthesis and luminescence properties of red long-lasting phosphor Y2O2S:Eu3+, Mg2+, Ti4+ nanoparticles. Mater. Chem. Phys. 119, 52–56 (2010)
S. Yuan, Y. Yang, B. Fang, G. Chen, Effects of doping ions on afterglow properties of 2S:Eu phosphors. Opt. Mater. 30, 535–538 (2007)
X. Wang, Z. Zhang, Z. Tang, Y. Lin, Characterization and properties of a red and orange Y2O2S-based long afterglow phosphor. Mater. Chem. Phys. 80, 1–5 (2003)
R.C. Ropp, Luminescence and the solid state, Elsevier 2004
H.M. Yang, J.X. Shi, M.L. Gong, A novel red emitting phosphor Ca2SnO4:Eu3+. J. Solid State Chem. 178, 917–920 (2005)
F. Shi-Liu, Y. Tao, C. Fei, Synthesis and characterization of Ca2Sn1–xCexO4 with blue luminescence originating from Ce4+ charge transfer transition. Chin. Phys. 16, 3129–3133 (2007)
T. Yamashita, K. Ueda, Blue photoluminescence in Ti-doped alkaline-earth stannates. J. Solid State Chem. 180, 1410–1413 (2007)
Y. Suzuki, M. Kakihana, Parallel solution-based synthesis approach for search of lanthanoid-activated Ca2SnO4 phosphor materials, J. Am. Ceram. Soc. 92, S168–S171 (2009)
Z. Fu, H.K. Yang, B.K. Moon, B.C. Choi, J.H. Jeong, Synthesis and luminescent properties of europium-activated Ca2SnO4 phosphors by sol–gel method. J. Lumin. 129, 1669–1672 (2009)
Y. Jin, Y. Hu, L. Chen, X. Wang, G. Ju, Luminescent properties of a red afterglow phosphor Ca2SnO4:Pr3+. Opt. Mater. 35, 1378–1384 (2013)
Z. Ju, R. Wei, J. Zheng, X. Gao, S. Zhang, W. Liu, Synthesis and phosphorescence mechanism of a reddish orange emissive long afterglow phosphor Sm3+-doped Ca2SnO4. Appl. Phys. Lett. 98, 121906 (2011)
Y. Jin, Y. Hu, L. Chen, X. Wang, G. Ju, Z. Mu, Luminescent properties of Tb3+-doped Ca2SnO4 phosphor. J. Lumin. 138, 83–88 (2013)
M. Shi, D. Zhang, C. Chang, Dy3+:Ca2SnO4, a new yellow phosphor with afterglow behavior. J. Alloy. Compd. 639, 168–172 (2015)
M. Trömel, Die Kristallstruktur der Verbindungen vom Sr2PbO4-Typ. Zeitschrift für anorganische und allgemeine Chemie 371, 237–247
A. Stanulis, A. Katelnikovas, D. Enseling, D. Dutczak, S. Šakirzanovas, M.V. Bael, A. Hardy, A. Kareiva, T. Jüstel, Luminescence properties of Sm3+-doped alkaline earth ortho-stannates. Opt. Mater. 36, 1146–1152 (2014)
X. Xu, Y. Wang, W. Zeng, Y. Gong, Luminescence and storage properties of Sm-doped alkaline-earth atannates. J. Electrochem. Soc. 158, J305 (2011)
B. Lei, B. Li, H. Zhang, W. Li, Preparation and luminescence properties of CaSnO3:Sm3+ phosphor emitting in the reddish orange region. Opt. Mater. 29, 1491–1494 (2007)
B.-F. Lei, S. Yue, Y.-Z. Zhang, Y.-L. Liu, Luminescence properties of Sr2SnO4:Sm3+ afterglow phosphor. Chin. Phys. Lett. 27, 037201 (2010)
X. Lin, X. Qiao, X. Fan, Synthesis and luminescence properties of a novel red SrMoO4:Sm3+,R+ phosphor. Solid State Sci. 13, 579–583 (2011)
E.L. Cates, M. Cho, J.-H. Kim, Converting visible light into UVC: microbial inactivation by Pr3+-activated upconversion materials. Environ. Sci. Technol. 45, 3680–3686 (2011)
Z. Xu, X. Kang, C. Li, Z. Hou, C. Zhang, D. Yang, G. Li, J. Lin, Ln3+ (Ln = Eu, Dy, Sm, and Er) ion-doped YVO4 nano/microcrystals with multiform morphologies: hydrothermal synthesis, growing mechanism, and luminescent properties. Inorg. Chem. 49, 6706–6715 (2010)
Z. Pan, Y.-Y. Lu, F. Liu, Sunlight-activated long-persistent luminescence in the near-infrared from Cr3+-doped zinc gallogermanates. Nat. Mater. 11, 58–63 (2012)
Acknowledgements
This work is supported by Scientific and Technological Cooperation Program of Guizhou Province (2015-7644, 2015-7643), the National Natural Science Foundation of China (51462003, 51762010), the Science Research Plan Funds of Guizhou province of China (Nos. 2015-4006, 2014-001, 2014-7611) and the Education Office Research Foundation of Guizhou Province, China (Grant No. KY2013-193).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Gong, X., Cui, R., Li, X. et al. The photoluminescence and afterglow properties of Ca2SnO4:Sm3+ phosphor. J Mater Sci: Mater Electron 29, 5668–5674 (2018). https://doi.org/10.1007/s10854-018-8536-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10854-018-8536-z