
Abstract
Porous CuO–CdO nanofibers with mean diameter of 463 nm and high surface-to-volume ratio were prepared by impregnating electrospun carboxylic-functionalized poly(arylene ether ketone) (PCA-PAEK) nanofibrous webs with metal acetate solution and subsequent calcination. The special morphology was demonstrated to be decided by ion exchange reaction between metal ions and functional groups on polymer. TGA and EDX analysis confirmed that Cd2+ was more readily to react with PCA-PAEK than Cu2+, and the amount of Cu2+ can be decreased by overloading of Cd2+, leading to the degradation of sensing performance. FT-IR proved the existence of the ion exchange reaction and demonstrated the products were highly pure CuO–CdO compounds with EDX and XRD spectra. The products detailedly investigated for direct electrocatalytic oxidation of glucose evaluated cyclic voltammetry and chronoamperometry. CuO–CdO nanofibers modified electrodes exhibited superiorities of good anti-interference, low detection limit and fast response of glucose, attributing to the enhanced conductivity brought by CdO, good electrocatalytic activity brought by CuO and large surface area brought by porous structure. The simple strategy of electrospinning porous nanofibers using PCA-PAEK as templates and combined CuO and CdO with good electrooxidation glucose functions opens a new route to generate novel conductive metal oxide hybrid nanofibers applied for glucose enzymeless sensors.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The emergence of nanotechnology has opened new horizons for the application of nanomaterials in bioanalytical chemistry. Nanomaterials contain a variety of structures such as nanowires, nanofibers, nanorods, nanoparticles, nanotubes and so on [1,2,3,4,5]. Among these structures, nanofibers that own high specific surface area and length-to-diameter ratios, unique and appealing properties have gained special focus in biosensor applications [6, 7]. Glucose biosensor is one of the most popular biosensors. It has been widely used in clinical diagnosis, pharmaceutical analysis and food industry [8, 9]. Amperometric glucose biosensors with and without glucose oxidase [10, 11] can achieve low detection limit easily. Especially, the enzymeless glucose biosensor possesses the advantages of good stability, simplicity, sensitivity, selectivity and so on [12, 13]. Thus, considerable attention has been given to enzyme-free electrodes [14]. Noble metals, transition metal and alloys [15,16,17,18] have been extensively investigated as non-enzymatic glucose sensors. However, these metallic nanomaterials have the drawbacks of high costs and very limited sensing utility, which hinders their practical applications. Therefore, there are considerable demands for the development of novel, cost-effective, sensitive and selective non-enzymatic glucose sensors.
CuO is a very important p-type semiconductor that owns a narrow band gap of 1.2 eV. It has been extensively utilized in fields of gas sensors, photochemical cells, catalysts and other electronic devices [19]. For its virtue of natural abundance, low production cost, good electrochemical and catalytic properties, CuO is studied for application of non-enzymatic glucose sensors as well [20]. However, lack of good conductivity may limit its application. CdO is an n-type semiconductor with direct band gap of 2.5 eV, an indirect band gap of 1.98 eV [21]. Nanoscale CdO has a very low ohmic resistivity [22], due to the native defects of oxygen vacancies. The creation of vacancies can increase the conductivity of oxides [23]. Thus, if CdO is incorporated into CuO to produce hybrid metal oxides, CdO can act as an effective dopant that enhances sensor application. Moreover, preparing metal oxide into nanofibers would combine their unique crystalline structures with large specific surface areas, leading to excellent properties [24].
It is well known that electrospinning is a straightforward, versatile and cost-effective top-down technique [24,25,26] to fabricate one-dimensional nanofibers [27]. It can produce polymer fibers in diameter ranging from micrometers to nanometers. Generally, the surface area of nanofibers prepared through electrospinning was approximately double of continuous thin films [28]. To prepare metal oxide nanofibers through electrospinning, polymer templates were essential. Carboxylic-functionalized poly(arylene ether ketone) (PCA-PAEK) (Structure 1) is a modified high-performance polymer that originates from PAEK [29]. Different from other polymer templates such as poly(l-lysine), poly(vinyl pyrrolidone) (PVP), poly(vinyl acetate) (PVAc), polylactic acid (PLA), glycerin, pentaerythritol, etc. [30,31,32,33,34], PCA-PAEK has carboxyl groups on its backbone. Introduction of –COOH can improve the solubility and chemical activity of PAEK. The functional group (–COOH) on the polymer immobilizes the inorganic ions on polymer fibers, which enables the uniformity and thinness of the metal oxides fibers. Moreover, the morphology of the final metal oxide nanofibers can be controlled by the amount of the metal ions absorbed on the polymer. Thus, PCA-PAEK can be utilized as dispersant to prepare CuO–CdO composite nanofibers with special morphology relying on the limited reaction between the functional group and metal ions. The functional group made the PCA-PAEK possible to act as templates to prepare metal oxide nanofibers via electrospinning. However, reports on this topic are really rare.

Structure of PCA-PAEK. PCA-PAEK: carboxylic-functionalized poly(arylene ether ketone)
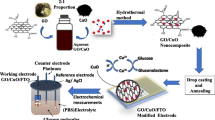
In this report, CdO is incorporated into CuO as an effective dopant to prepare hybrid metal oxide nanofibers with improved conductivity and then employed as the non-enzymatic sensing materials to detect glucose. Porous CuO–CdO composite nanofibers were prepared by impregnating electrospun PCA-PAEK nanofibrous webs with metal acetate solution and subsequent calcination. The mechanism for the formation of special porous morphology was discussed. The morphologies and structures of the as-prepared CuO–CdO composite nanofibers were characterized by SEM, TGA, XRD, FT-IR and EDX. The activity of the hybrid nanofibers electrode toward glucose electrooxidation and its sensing performances were evaluated. CuO–CdO composite nanofibers modified glassy carbon electrode showed good anti-interference, low detection limit and fast response of glucose under an applied potential of + 0.40 V. The ion exchange reaction that occurred in impregnating process between metal ions and functional group of polymer template decided the morphology and performance of hybrid nanofibers. Present study provides a simple strategy for design of electrospun CuO–CdO composite nanofibers bioelectrochemical nanodevices for glucose determination.
Experimental
Reagents
Carboxylic-functionalized poly(arylene ether ketone) (PCA-PAEK, Mn = 31,016 g mol−1) was purchased from Engineering Research Center of High Performance Plastics, Jilin University. Cupric acetate monohydrate (Cu(CH3COO)2·H2O, A.R.) and cadmium acetate (Cd(CH3COO)2·2H2O, A.R.) were obtained from Sinopharm Chemical Reagent Co., Ltd, China. Dichloromethane (DCM, Tianjin Tiantai Fine Chemical Co., Ltd., China) and N, N-dimethylformamide (DMF, Xilong Chemical Co., Ltd., China) were used as solvents directly. Nafion perfluorinated resin solution (20 wt% in lower aliphatic alcohols and water, contains 34% water) was provided by Sigma-Aldrich. d-glucose, ascorbic acid (AA), uric acid (UA) and ethanol were purchased from Beijing Chemical Plant (Beijing, China). Glucose solutions with various concentrations were prepared with redistilled water. Solutions for anti-interference testing were prepared by dissolving interfering species in glucose solution at normal physiological levels. All of the above chemicals were of analytical grade and directly used without any other treatment.
Preparation of CuO–CdO nanofibers
PCA-PAEK granules were dissolved in DMF/DCM mixture (the volume ratio was 5/5) to prepare a 20 wt% PCA-PAEK solution, which was vigorously stirred for at least 6 h at room temperature. Then, the transparent solution was put into a syringe, which was connected to a high-voltage supply (DW-P303-5AC High Voltage (0–30 kV), Dongwen High-voltage-Power-supply Company, China) subsequently. A collector placed 20 cm away from the orifice was connected to a grounded counter electrode. Then, the applied voltage was adjusted at 17 kV, and at same time, a dense web of fibers can be collected on the aluminum foil. All electrospinning processes were carried out at ambient temperature. The fibers were exposed to the air overnight to promote further evaporation of the solvent.
For the preparation of CuO–CdO nanofibers, a solution which contained 6 wt% cupric acetate monohydrate and 6 wt% cadmium acetate was prepared beforehand. The completely dried PCA-PAEK pure polymer nanofibers were then impregnated with the composite solution for 10 h at 30 °C under continuously shocking. Then, the fiber membranes were washed with water three times and dried in vacuum oven for 48 h at 45 °C to fully remove the unreacted compounds and solvent. The obtained precursor was then calcined at 600 °C for 4 h in air in order to remove matrix polymer and generate CuO–CdO nanofibers. Pure CuO nanofibers and CdO nanofibers were synthesized following a similar procedure; the only difference was to use 6 wt% cupric acetate monohydrate or 6 wt% cadmium acetate as the sole metal oxide precursor, respectively.
Preparation of CuO–CdO nanofibers modified electrodes
Prior to surface modification, a glassy carbon electrode (GCE, Ø3 mm) was polished with 1 mm and 0.05 mm alumina slurries sequentially, and then rinsed with deionized water. After that, the electrode was sonicated in deionized water, acetone, deionized water successively, dried at room temperature, respectively, and was ready for modification. To prepare 5 mg mL−1 CuO–CdO nanofibers suspension, 5 mg CuO–CdO nanofibers were first suspended in 1 mL ethanol and sonicated for 1 h to thoroughly disperse the CuO–CdO nanofibers. Then, a 10-μL suspension was dropped onto the surface of GCE. The solvent were allowed to evaporate at infrared lamp leaving the CuO–CdO nanofibers immobilized onto the electrode surface. A 5-μL 1 wt% Nafion solution was finally applied to serve both as permselective membrane and entrapment matrix for the immobilization of CuO–CdO nanofibers. The modified electrode was abbreviated as CuO–CdO NFs/GCE. A similar procedure was also applied to prepare CuO nanofibers or CdO nanofibers modified GCEs, which was labeled as CuO NFs/GCE and CdO NFs/GCE, respectively.
Characterization
A Japan SHIMDZU SSX-550 scanning electron microscope (SEM) equipped with an energy-dispersive X-ray spectrometer (EDX) was used to examine the morphology and composition of the as-prepared samples. Image visualization software ImageJ was used to analyze the mean diameters of the nanofibers. A total of 100 measurements per field were chosen based on the SEM images [35]. Thermal gravimetric analysis (TGA) was carried out by using Perkin Elmer Pyris 1 TGA (USA) from room temperature to 800 °C under a flowing air atmosphere. The vibration in functional groups of nanofibers was analyzed by Japan SHIMDZU, 1.50SU1 Fourier transform-infrared radiation (FT-IR) spectrometer. X-ray powder diffraction (XRD) patterns of the calcined nanofibers were conducted on a Siemens X-ray diffractometer (D5005XRD) with Cu Kα radiation (γ = 1.5418 Å) operating at 40 kV and 30 mA. Scans were made from 30° to 70° (2θ). All electrochemical experiments were performed on a CHI 660E Electrochemical Workstation (CH Instrument, USA), using a traditional three-electrode electrochemical cell (a working volume of 5 mL) with a working electrode, an saturated calomel electrode and a platinum wire counter electrode.
Results and discussion
Characterization of the nanofibers before and after calcination
Figure 1 illustrates SEM images of the as-spun nanofibers. The as-prepared PCA-PAEK nanofibers exhibited uniform distribution and smooth surface. Differently, the morphology of the immersed nanofibers was quite coarse and distorted. In some places, fibers stuck together, leading to the increase in fiber diameter. The phenomenon was ascribed to the longtime immersion, which resulted in swelling of the PCA-PAEK fibers. Inset images showed the difference more clearly. After the PCA-PAEK nanofibers immersed in copper acetate solution, many dots appeared on the surface (Fig. 1b). This was related to the reaction between Cu2+ and the carboxyl on PCA-PAEK. When Cu(CH3COO)2 was replaced by Cd(CH3COO)2 (Fig. 1c), the morphology was more different. Many rice-like rods appeared. The shape and size of these particles were uniform. The production of rice-like rods was on the one hand because of the reaction between Cd2+ and the carboxyl on PCA-PAEK, on the other hand due to its preferential orientation. The similar formation process of the obtained nanorods was reported by Yang Jiang et al. [36]. Figure 1d shows the morphology of PCA-PAEK/Cu(CH3COO)2/Cd(CH3COO)2 nanofibers, which was mostly similar with that of PCA-PAEK/Cu(CH3COO)2 nanofibers. In the higher magnification, many particles but not the rice-like rods irregularly distributed on the fibers. This may be because of the inhibition from Cu2+ to Cd2+ when –COOH reacted with Cu2+ and Cd2+ at the same time. From the SEM photograph, it is easy to find out that all of the particles dispersed throughout the fibers. This demonstrated that PCA-PAEK polymer chain can act as a stabilizing agent to inhibit the agglomeration of the nanoparticles [37]. The probable equation in the immersion process is shown in Eq (1).

SEM images of a pure PCA-PAEK nanofibers, b PCA-PAEK/Cu(CH3COO)2 nanofibers, c PCA-PAEK/Cd(CH3COO)2 nanofibers, d PCA-PAEK/Cu(CH3COO)2/Cd(CH3COO)2 nanofibers. Inset images are the corresponding higher magnifications
Metal acetate and PCA-PAEK took place ion exchange reaction, due to the stronger metal coordination ability of –COOH on PCA-PAEK.
In order to assess the appropriate calcination temperature and prove the speculation in SEM analysis, thermal behavior of every fiber was investigated by thermal gravimetric analyses (TGA). As seen from curve a in Fig. 2, there was only one weight loss stage for pure PCA-PAEK fibers during 400–630 °C, which corresponded to the decomposition of polymer main chain. The curve was so flat before 400 °C that demonstrated the excellent thermo-oxidative stability of the polymers. After 630 °C, no more weight loss can be seen, indicating PCA-PAEK decomposed completely. The TGA curve of PCA-PAEK/Cu(CH3COO)2 nanofibers blend is shown in Fig. 2, the curve b. The slight weight loss (~ 10%) from room temperature to 239 °C corresponded to the loss of moisture. The first dramatic weight loss (~ 45%) occurred at 315–354 °C was attributed to decomposition of Cu(PAEK-COO)2 and PCA-PAEK side chain. The second dramatic weight loss (~ 35%) occurred at 418–443 °C was owed to decomposition of PCA-PAEK main chain. The shift of the decomposition temperature of PCA-PAEK should be due to the reaction between –COOH on polymer chains and metal ions, as well as the existence of inorganic salt which facilitates the decomposition of the polymer. When the temperature reached at about 500 °C, the curve became flat, demonstrating the complete transformation of composite nanofibers to inorganic oxide. Moreover, percentage of the residual was about 11%, indicating the weight ratio of product. The weight loss procedure of PCA-PAEK/Cd(CH3COO)2 nanofibers (curve c) was similar with PCA-PAEK/Cu(CH3COO)2 nanofibers. It also had three stages corresponding to the loss of moisture and decomposition of inorganic salt and polymer. The different was the percentage of residual. After 600 °C, the weight was remained at about 30%, which was more than that of PCA-PAEK/Cu(CH3COO)2 nanofibers. This demonstrated Cd(CH3COO)2 was more likely to react with –COOH on polymer chains than Cu(CH3COO)2 at the same circumstance. The TGA curve of PCA-PAEK/Cu(CH3COO)2/Cd(CH3COO)2 nanofibers is shown in curve d. It has four stages. The first one appeared from 65 to 200 °C was related with the loss of crystal water and solvent. The second stage from 206 to 420 °C can be divided into two parts. This was because that both of Cu(PAEK-COO)2 and Cd(PAEK-COO)2 decomposed in this stage at the same time. Certainly, some of the polymer main chain also decomposed in this range. The third stage from 445 to 463 °C was due to the complete decomposition of PCA-PAEK. The fourth stage was 464 to 513 °C, corresponding to the formation of inorganic oxide [38]. After 500 °C, the curve was flat. The residual was about 17%, which was higher than PCA-PAEK/Cu(CH3COO)2 nanofibers but lower than PCA-PAEK/Cd(CH3COO)2 nanofibers. This proved the speculation in SEM analysis that the presence of Cu2+ restrained the reaction between Cd2+ and PCA-PAEK. Simultaneously, it was easily to find out that the calcinations temperature should be greater than 550 °C because all the reactions have been carried out and there was no change in weight, representing the formation of inorganic oxide.

TGA curves of (a) pure PCA-PAEK nanofibers, (b) PCA-PAEK/Cu(CH3COO)2 nanofibers, (c) PCA-PAEK/Cd(CH3COO)2 nanofibers, (d) PCA-PAEK/Cu(CH3COO)2/Cd(CH3COO)2 nanofibers
Thus, the calcined temperature was determined at 600 °C. The morphology of calcined samples at 600 °C for 3 h is presented in Fig. 3. Rope-like CuO fibers with diameter of 348 nm are shown in Fig. 3a. They were not continuous fibers with length of centimeter any more. The fibers were slim and short. This may be related with the little amount of Cu2+ reacted with –COOH on the polymer, which had been analyzed in TGA curves. The mean diameter of CdO fibers was about 766 nm, which was twice bigger than CuO fibers. The result was coincidence with the residual amount of PCA-PAEK/Cd(CH3COO)2 nanofibers in TGA curves, demonstrating that Cd(CH3COO)2 was more likely to react with –COOH on polymer chains than Cu(CH3COO)2 at the same circumstance once again. In addition, many pores can be seen on CdO fibers (Fig. 3b), which were conducive to increase the specific surface area. The formation of these pores was because of the large amount of Cd2+ reacted with polymer. In general, the following reaction can take place during calcinations process and lead to the formation of metal oxide:

SEM images of a PCA-PAEK/Cu(CH3COO)2 nanofibers, b PCA-PAEK/Cd(CH3COO)2 nanofibers, c PCA-PAEK/Cu(CH3COO)2/Cd(CH3COO)2 nanofibers after calcination at 600 °C and the corresponding fiber diameter distribution. Inset images are the higher magnifications
With the increase in temperature, main chain of PAEK was decomposed into CO2 and H2O. Due to the thick absorbed layer outside the polymer, the generated gas did not have another exit but only to crack the surface. As a result, pores were appeared. Figure 3c gives the diameter and morphology of calcined PCA-PAEK/Cu(CH3COO)2/Cd(CH3COO)2 nanofibers. The average diameter was 463 nm, between that of PCA-PAEK/Cu(CH3COO)2 nanofibers and PCA-PAEK/Cd(CH3COO)2 nanofibers. Interestingly, the morphology was also in the middle. It was slim and had many pores on the fiber, presented a hollow out fiber. This indicated that the product was probable a compound of CuO and CdO.
Based on the analysis above, the possible formation mechanism of the special morphology of CuO–CdO nanofibers can be speculated and is shown in Fig. 4. When pure PCA-PAEK with –COOH on side chains was immersed in Cu2+ or Cd2+ solution, the metal ions reacted with –COO− and absorbed on the fibers. In the immobilization process, due to different activity of metal ions, Cd ions were more readily to react with –COOH on polymer, whereas Cu ions were somewhat inert. At the same time, Cu2+ acts as an obstruction to restrain the reaction between Cd2+ and –COOH. As a result, there are many Cd ions and little Cu ions absorbed on the polymer. With the increase in temperature, polymer decomposed and metal oxide generated gradually. Due to the good stability of PAEK, fiber morphology was retained. The formation of pores on the one hand due to the large amount of Cd2+ absorbed on the polymer which formed thick layer like that in pure CdO nanofibers, on the other hand due to the little amount of Cu2+ absorbed on the polymer which formed break like that in pure CuO nanofibers. It was easy to find out that in the formation of metal oxide nanofibers, ion exchange reaction between metal acetate and PCA-PAEK was the key point. The porous morphology may enhance its application ascribed to the enlarged specific surface area.

The possible formation mechanism of CuO–CdO nanofibers
FT-IR was investigated in order to demonstrate the presence of the intermediate products and identify the structure of the final products preliminarily. As shown in Fig. 5a, the weak peak at 1642 cm−1 belonged to the stretching vibration of C=O on the main chain of PCA-PAEK. After reacted with metal salt, the peak did not shift (Fig. 5b–d). The peak at 1701 cm−1 in Fig. 5a corresponded to the asymmetric stretching vibration of C=O on the side chain of pure PCA-PAEK [39]. However, it had shifted to 1712 cm−1 in Fig. 5b–d. This demonstrated that –COOH on the side chain of PCA-PAEK had reacted with metal ions already. After treating at 600 °C, organic groups disappeared and new peaks came out. In Fig. 5e, the peak existed at about 535 cm−1 was the stretching vibration of Cu–O [40], demonstrating the production of pure CuO. In Fig. 5f, the new peak appeared at 473 cm−1, corresponding to the stretching vibration of Cd–O [41]. This demonstrated that PCA-PAEK/Cd(CH3COO)2 nanofibers converted into CdO thoroughly under the function of heat treatment. As to PCA-PAEK/Cu(CH3COO)2/Cd(CH3COO)2 nanofibers, it can be seen from Fig. 5g that there were two characteristic peaks: one was at 474 cm−1, the other was at 535 cm−1. The two peaks corresponded to the stretching vibration of Cd–O and Cu–O, respectively. This indicated that the product for calcinating PCA-PAEK/Cu(CH3COO)2/Cd(CH3COO)2 nanofibers was indeed the compound of CdO and CuO.

FT-IR spectra of (a) pure PCA-PAEK nanofibers, (b) PCA-PAEK/Cu(CH3COO)2 nanofibers, (c) PCA-PAEK/Cd(CH3COO)2 nanofibers, (d) PCA-PAEK/Cu(CH3COO)2/Cd(CH3COO)2 nanofibers. (e–g) are the corresponding spectra of (b–d) after calcination at high temperature
EDX and XRD were used to identify the structure of the final products further more. From Fig. 6a, it can be seen clearly that there were O and Cu in calcined PCA-PAEK/Cu(CH3COO)2 nanofibers, demonstrating the existence of CuO products. Figure 6b and c intuitionisticly indicated that the products were CdO and CuO–CdO composite, respectively. What is more, it can be seen from Table 1 that in CuO–CdO composite, atomic percent of Cd element (~ 25%) was more than Cu element (~ 11%), indicating the amount of CdO was more than that of CuO. This demonstrated that –COOH was more likely to react with Cd2+ when Cu2+ and Cd2+ existed at the same time. The result proved the speculation in SEM and TGA analysis further more.

EDX spectra of a PCA-PAEK/Cu(CH3COO)2 nanofibers, b PCA-PAEK/Cd(CH3COO)2 nanofibers, c PCA-PAEK/Cu(CH3COO)2/Cd(CH3COO)2 nanofibers after calcination at high temperature
XRD spectra of the calcined samples are shown in Fig. 6. After PCA-PAEK/Cu(CH3COO)2 nanofibers calcined at high temperature (shown in Fig. 7a), well-defined diffraction peaks were observed. All of the diffraction peaks can be assigned to the monoclinic structured CuO (Joint Committee for Powder Diffraction Studies (JCPDS) File No. 05-0661) [42]. The sharp peaks indicated that the products are perfectly crystallized. The major peaks located at 2θ = 35.5° and 38.7° indexed as (111)–(002) and (111)–(200) planes, respectively, were the characteristics for the pure phase monoclinic CuO crystallites. As shown in Fig. 7b, it can be seen that the calcined PCA-PAEK/Cd(CH3COO)2 nanofibers presented diffraction peaks at 2θ = 33.02°, 38.28°, 55.31°, 65.93°, 69.29°, corresponding to (111), (200), (220), (311), (222) planes of cubic crystalline CdO (JCPDS-05-0640), respectively [23]. After PCA-PAEK/Cu(CH3COO)2/Cd(CH3COO)2 nanofibers calcined at high temperature (shown in Fig. 7c), it can be seen that both of monoclinic structured CuO and cubic crystalline CdO appeared in the XRD spectra. This demonstrated that the calcined product was compound of CuO and CdO.

XRD spectra of the calcined (a) PCA-PAEK/Cu(CH3COO)2 nanofibers, (b) PCA-PAEK/Cd(CH3COO)2 nanofibers and (c) PCA-PAEK/Cu(CH3COO)2/Cd(CH3COO)2 nanofibers
Sensitive and selective glucose sensing at CuO–CdO nanofibers modified electrode
To examine the electrocatalytic properties after the incorporation of CuO into CdO, pure CuO, CdO and CuO–CdO nanofibers were deposited on a GCE, respectively. The three different modified electrodes toward oxidation of glucose were tested through CV, which was carried out in the potential range from 0 to + 0.70 V in 0.1 M NaOH. As shown in Fig. 8A, contrasted with CdO NFs/GCE, CuO–CdO NFs/GCE presented a pair of quasi-reversible redox peaks, which can be assigned to the redox couple of Cu (II) and Cu (III) (CuO + OH− → CuOOH +e−) [43]. Moreover, CuO–CdO NFs/GCE exhibited a more dramatic increase in current signal toward the positive end of the potential range than that obtained at CdO NFs/GCE, which can be attributed to the enhanced electrocatalytic activity by the incorporation of CuO into the hybrid nanofibers. The background current of CdO NFs/GCE is low, possibly because of the limited capacity produced by the unchanged oxidation state during the electrochemical scan. Thus, the background current of CuO–CdO NFs/GCE which is lower than CuO NFs/GCE can be explained. Pure CuO possesses a good electrocatalytic performance; however, adding CdO into hybrid nanofibers could reduce the amount of CuO, just like the results showed in EDX and TGA analysis. Meanwhile, the morphology of the nanofibers influenced the performance. Although the surface area of porous continuous CuO–CdO nanofibers was improved from CdO nanofibers, it was still smaller than that of slim short CuO nanofibers. The increased surface area further enhanced the performance. However, the total catalytic sites reduced by the reduction of CuO and thus led to a decreased response. It proved that the incorporation of CuO can enhance the electrocatalytic performance of CdO.

A CVs of (a) CuO NFs/GCE, (b) CdO NFs/GCE, (c) CuO–CdO NFs/GCE in 0.1 M NaOH. Scan rate: 50 mV s−1. B CVs of CuO–CdO NFs/GCE in the absence (a) and presence of 4 mM glucose (b) and 8 mM glucose (c) in 0.1 M NaOH. C Amperometric response of (a) CuO NFs/GCE, (b) CdO NFs/GCE, (c) CuO–CdO NFs/GCE with successive additions of 0.1 mM glucose to 0.1 M NaOH at + 0.40 V. Inset shows the corresponding calibration curve. D Amperometric responses of (a) CuO NFs/GCE, (b) CdO NFs/GCE, (c) CuO–CdO NFs/GCE with successive additions of 3 mM glucose, 0.1 mM AA, 0.1 mM UA, 10 mM ethanol to 0.1 M NaOH at +0.40 V
Then, potential application in glucose detection of CuO–CdO NFs/GCE was investigated. Figure 8B presents CVs of CuO–CdO NFs/GCE in the absence and presence of glucose in 0.1 M NaOH. With the concentration of glucose increased, the background current and the anodic peak increased, which can be attributed to the catalytic effect of the redox couple for oxidation of glucose to glucose acid (CuOOH + e− + glucose → CuO + OH− + glucose acid). The results indicated that the electrodes may have potential application in the glucose detection.
Amperometric analysis was carried out at + 0.40 V at the three different modified electrodes, respectively, by successive additions of glucose to 0.1 M NaOH. From Fig. 8C, it can be seen that both of the CuO NFs/GCE and CuO–CdO NFs/GCEs responded rapidly to the change in glucose concentration, whereas the CdO NFs/GCEs have no obvious response, indicating a poor electrocatalytic activity to glucose. Time for achieving 95% of the steady-state current at CuO NFs/GCE and CuO–CdO NFs/GCEs was within 3 s and 2 s, respectively, indicating a better catalytic property of the CuO–CdO NFs/GCEs. Moreover, the current response of CuO–CdO NFs/GCEs to 2 mM glucose was approximately 0.2 μA, which was significantly higher than those of CdO NFs/GCEs. Inset image showed the corresponding calibration curve. Due to the negligible response to glucose, calibration curve for CdO NFs/GCEs was not shown. A better linear response of the electrocatalytic current of glucose can be seen at the CuO–CdO NFs/GCEs. As listed in Table 2, CuO–CdO NFs/GCEs exhibited lower detection limit and wider linear range than CuO NFs/GCEs. The better behaviors of CuO–CdO NFs/GCEs can be attributed to the high conductivity of CdO, which further enhanced the performance in the sensor application [23]. In addition, CuO–CdO NFs/GCEs showed a lower sensitivity than CuO NFs/GCEs. This was because of the CdO component, which was an ineffective material to catalyze glucose electrooxidation for single use. Although doping CdO can increase the conductivity and the porous continuous morphology can enhance the surface area, which was beneficial to enhance the performance, it cannot counteract the negative function of the overloading of CdO. Superfluous CdO excessively diluted the content of CuO, which provided catalytic sites to catalyze glucose electrooxidation, leading to significant reduction in total catalytic sites. As a result, a decreased response showed up.
Good anti-interference is a very important but challenging aspect to a non-enzymatic glucose sensor. It was because that the coexistence of electroactive compounds in real blood (such as AA and UA) interfered with the determination of glucose. It is well known that the common concentration of glucose in blood is 3–8 mM and the interfering species are about 0.1 mM. Therefore, we examined electrochemical response of the possible interfering species such as UA, AA and ethanol. The experiment was carried out by adding 3 mM glucose in 0.1 M NaOH followed by 0.1 mM AA, 0.1 mM UA and 10 mM ethanol. The results are shown in Fig. 8D. At CuO NFs/GCE, signal was evident. UA caused a significant interference in physiological level, AA and ethanol did not cause any observable interference, indicating a good anti-interference ability. At CdO NFs/GCE, the response curve was so flat, indicating the ineffective performance in glucose detection. While at CuO–CdO NFs/GCE, all of the interfering substances did not cause significant interference, implying a better anti-interference ability. A remarkable difference can be seen when the electrodes responded to UA. The difference can be explained by a repelling model that has been reported by Ding et al. [23]. In strong alkaline solution, surfaces of the nanofibers contained negative charge, which repelled the deprotonated UA. Therefore, the electrooxidation of UA on surface of the nanofibers was reduced. Due to the ion exchange reaction between PCA-PAEK and metal ions, nanofibers with different morphology formed which increases the electroactive surface area. In this process, surface area of porous continuous CuO–CdO nanofibers was smaller than that of slim short CuO nanofibers. Thus, CuO–CdO nanofibers provided limited possibilities to repel deprotonated UA, resulting in a not obvious response.
In a word, CuO–CdO NFs/GCE exhibited better performances such as anti-interference, detection limit and response time. The enhanced performance was benefited from the increased surface area, conductivity of CdO and electrocatalytic activity of CuO.
Conclusion
Porous CuO–CdO composite nanofibers with mean diameter of 463 nm were prepared by a facile three-step procedure consisting of electrospinning, liquid–solid reaction and subsequent calcination. Benefiting from the –COOH functional group on PCA-PAEK template, liquid–solid reaction occurred and led to the absorbance of metal ions on the template nanofibers. Heat treatment resulted in the elimination of polymer. SEM and TGA proved that the formation of porous morphology was decided by the ion exchange reaction between template and metal ions. TGA and EDX analysis confirmed that Cd2+ was more readily to react with PCA-PAEK compared with Cu2+ and the amount of Cu2+ can be decreased by overloading of Cd2+. FT-IR spectra illustrated the reaction between metal ions and –COOH, and demonstrated the products were highly pure CuO–CdO compounds with EDX and XRD spectra. The products detailedly investigated for direct electrocatalytic oxidation of glucose evaluated cyclic voltammetry and chronoamperometry. Results revealed a good anti-interference, low detection limit and fast response of glucose at +0.40 V, attributing to the enhanced conductivity brought by CdO, good electrocatalytic activity brought by CuO and large surface area brought by porous structure, which benefited from ion exchange reaction between template and metal ions. Moreover, the CuO–CdO nanofibers modified electrodes showed a lower sensitivity than CuO nanofibers, due to the decreased amount of CuO that proved by TGA and EDX. The good features indicated that CuO–CdO composite nanofibers have a great potential in the development of sensitive and selective non-enzymatic glucose sensor.
References
Hwang B, Kim M, Cho SM, Becker S, Kim YH, Kim H (2017) Embedded silver-nanowire electrode in an acrylic polymer-silicate nanoparticle composite for highly robust flexible devices. J Appl Polym Sci 134:45203. https://doi.org/10.1002/app.45203
Liu M, Cheng Z, Yan J, Qiang L, Ru X, Liu F, Ding D, Li J (2013) Preparation and characterization of TiO2 nanofibers via using polylactic acid as template. J Appl Polym Sci 128:1095–1100
Liu Y, Zhang B, Luo L, Chen X, Wang Z, Wu E, Su DS, Huang W (2015) TiO2/Cu2O core/ultrathin shell nanorods as efficient and stable photocatalysts for water reduction. Angew Chem Int Edit 54:15260–15265
Ding H, Ma Y (2015) Theoretical and computational investigations of nanoparticle–biomembrane interactions in cellular delivery. Small 11:1055–1071
Zhang L, Wang X, Xu W, Zhang Y, Li Q, Bradford PD, Zhu Y (2015) Strong and conductive dry carbon nanotube films by microcombing. Small 11:3830–3836
Wang W, Li Z, Zheng W, Dong B, Li S, Wang C (2010) A novel non-enzymatic glucose sensor based on nickel (II) oxide electrospun nanofibers. J Nanosci Nanotechno 10:7537–7540
Islam AB, Islam SK, Rahman T (2010) A highly selective mediator less glucose detector employing vertically aligned carbon nanofiber (VACNF). In: Ieee sensors 2010 conference, pp 42–46
Wang J (2008) Electrochemical glucose biosensors. Chem Rev 108:814–825
Terry LA, White SF, Tigwell LJ (2005) The application of biosensors to fresh produce and the wider food industry. J Agric Food Chem 53:1309–1316
Lee DY, Kafi AKF, Choi WS, Park SH, Kwon YS (2008) Glucose sensor based on glucose oxidase-lipid LB film immobilized in prussian blue layer. J Nanosci Nanotechnol 8:4543–4547
Manesh KM, Kim JH, Santhosh P, Gopalan AY, Lee KP, Kang HD (2007) Fabrication of a gold nanoparticles decorated carbon nanotubes based novel modified electrode for the electrochemical detection of glucose. J Nanosci Nanotechnol 7:3365–3372
Wilson R, Turner APF (1992) Glucose oxidase: an ideal enzyme. Biosens Bioelectron 7:165–185
Senthamizhan A, Balusamy B, Uyar T (2016) Glucose sensors based on electrospun nanofibers: a review. Anal Bioanal Chem 408:1285–1306
Li J, Hu H, Li H, Yao C (2017) Recent developments in electrochemical sensors based on nanomaterials for determining glucose and its byproduct H2O2. J Mater Sci 52:10455–10469. https://doi.org/10.1007/s10853-017-1221-4
Liu H, Su X, Tian X, Huang Z, Song W, Zhao J (2006) Preparation and electrocatalytic performance of functionalized copper-based nanoparticles supported on the gold surface. Electroanalysis 18:2055–2060
Zhou L, Gan T, Zheng DY, Yan JJ, Hu CG, Hu SS (2012) High-density gold nanoparticles on multi-walled carbon nanotube films: a sensitive electrochemical nonenzymatic platform of glucose. J Exp Nanosci 7:263–273
Sun YP, Buck H, Mallouk TE (2001) Combinatorial discovery of alloy electrocatalysts for amperometric glucose sensors. Anal Chem 73:1599–1604
Liu YX, Ding Y, Zhang YC, Lei Y (2012) Pt-Au nanocorals, Pt nanofibers and Au microparticles prepared by electrospinning and calcination for nonenzymatic glucose sensing in neutral and alkaline environment. Sens Actuators B 171:954–961
Sahay R, Kumar PS, Aravindan V, Sundaramurthy J, Ling WC, Mhaisalkar SG, Ramakrishna S, Madhavi S (2012) High aspect ratio electrospun CuO nanofibers as anode material for lithium-ion batteries with superior cycleability. J Phys Chem C 116:18087–18092
Zhang Y, Liu Y, Su L, Zhang Z, Huo D, Hou C, Lei Y (2014) CuO nanowires based sensitive and selective non-enzymatic glucose detection. Sens Actuators B 191:86–93
Kanjwal MA, Barakat NAM, Sheikh FA, Kim HY (2010) Electronic characterization and photocatalytic properties of TiO2/CdO electrospun nanofibers. J Mater Sci 45:1272–1279. https://doi.org/10.1007/s10853-009-4078-3
Wang SH, Yang SH (2001) Spectroscopic characterization of the copper sulphide core/shell nanowires. Mater Sci Eng C 16:37–40
Ding Y, Wang Y, Zhang LC, Zhang H, Lei Y (2012) Preparation, characterization and application of novel conductive NiO–CdO nanofibers with dislocation feature. J Mater Chem 22:980–986
Savva I, Kalogirou AS, Chatzinicolaou A, Papaphilippou P, Pantelidou A, Vasile E, Vasile E, Koutentis PA, Christoforou TK (2014) PVP-crosslinked electrospun membranes with embedded Pd and Cu2O nanoparticles as effective heterogeneous catalytic supports. RSC Adv 4:44911–44921
Deshawar D, Chokshi P (2017) Stability analysis of an electrospinning jet of a polymeric fluid. Polymer 131:34–49
Wittmer CR, Hebraud A, Nedjari S, Schlatter G (2014) Well-organized 3D nanofibrous composite constructs using cooperative effects between electrospinning and electrospraying. Polymer 55:5781–5787
Dai Y, Liu W, Formo E, Sun Y, Xia Y (2011) Ceramic nanofibers fabricated by electrospinning and their applications in catalysis, environmental science, and energy technology. Polym Adv Technol 22:326–338
Dersch R, Steinhart M, Boudriot U, Greiner A, Wendorff JH (2005) Nanoprocessing of polymers: applications in medicine, sensors, catalysis, photonics. Polym Adv Technol 16:276–282
Liu M, Wang Y, Cheng Z, Zhang M, Hu M, Li J (2015) Electrospun carboxylic-functionalized poly(arylene ether ketone) ultrafine fibers: optimization, characterization, and water absorption behavior. High Perform Polym 27:939–949
Sarah LS, David WW (2006) Biomimetic synthesis of titanium dioxide utilizing the R5 peptide derived from cylindrotheca fusiformis. Chem Mater 18:3108–3113
Nuansing WW, Ninmuang S, Jarernboon W, Maensiria S, Seraphin S (2006) Structural characterization and morphology of electrospun TiO2 nanofibers. Mater Sci Eng, B 131:147–155
Ding B, Kim CK, Kim HY, Seo MK, Park SJ (2004) Titanium dioxide nanofibers prepared by using electrospinning method. Fiber Polym 5:105–109
Zheng JY, Pang JB, Qiu KY, Wei Y (2001) Synthesis of mesoporous titanium dioxide materials by using a mixture of organic compounds as a non-surfactant template. J Mater Chem 11:3367–3372
Zheng JY, Qiu KY, Feng QW, Xu JG, Wei Y (2000) Sol–Gel synthesis of mesoporous titania using nonsurfactant organic compounds as templates. Mol Cryst Liq Cryst 354:183–194
Liu M, Wang Y, Cheng Z, Zhang M, Hu M, Li J (2014) Electrospun Mn2O3 nanowrinkles and Mn3O4 nanorods: morphology and catalytic application. Appl Surf Sci 313:360–367
Yang J, Zeng JH, Yu SH, Yang L, Zhou G, Qian Y (2000) Formation process of CdS nanorods via solvothermal route. Chem Mater 12:3259–3263
Liu M, Song L, Wang Y, Cheng Z, Li J (2014) High-effective preparation of ultrafine poly-(L-lactide-co-ε-caprolactone-diOH) fibers containing silver nanoparticles. High Perform Polym 26:483–487
Liu M, Wang Y, Cheng Z, Song L, Zhang M, Hu M, Li J (2014) Function of NaOH hydrolysis in electrospinning ZnO nanofibers via using polylactide as templates. Mater Sci Eng B 187:89–95
Zhang Y (2016) Synthesis and photoluminescence study of zinc sulfide–poly(arylene ether ketone) nanocomposite materials. High Perform Polym 28:198–205
Guan H, Shao C, Chen B, Gong J, Yang X (2003) A novel method for making CuO superfine fibres via an electrospinning technique. Inorg Chem Commun 6:1409–1411
Bazargan AM, Fateminia SMA, Ganji ME, Bahrevar MA (2009) Electrospinning preparation and characterization of cadmium oxide nanofibers. Chem Eng J 155:523–527
Wang W, Zhang L, Tong S, Li X, Song W (2009) Three-dimensional network films of electrospun copper oxide nanofibers for glucose determination. Biosens Bioelectron 25:708–714
Tian K, Prestgard M, Tiwari A (2014) A review of recent advances in nonenzymatic glucose sensors. Mat Sci Eng C 41:100–118
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interests regarding the publication of this article.
Rights and permissions
About this article

Cite this article
Liu, M., Wang, Y. & Lu, D. Sensitive and selective non-enzymatic glucose detection using electrospun porous CuO–CdO composite nanofibers. J Mater Sci 54, 3354–3367 (2019). https://doi.org/10.1007/s10853-018-3034-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-018-3034-5