
Abstract
Trialkyl-monoacetic acid derivatives of p–t-octylcalix[4]arene and calix[4]arene were prepared to investigate the effect of the alkyl branches attached to the phenoxy oxygen atoms and the p-position on the selective extraction of Li+ over Na+. Alkyl branches on the phenoxy oxygen atoms remarkably affected the Li+ selectivity, whereas those at the p-position had less effect. The former can contribute to excluding Na+ extraction while enabling Li+ extraction. Optimal selection of the alkyl branch improves the Li+ selectivity of calix[4]arene. However, sterically-hindered p–t-octylcalix[4]arene with three 2-ethylbutyl branches exhibited opposite selectivity.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Li is employed in lithium ion batteries for electric vehicles [1], ceramics and glass, greases, pharmaceuticals, and polymers [2]. From comprehensive analysis of global lithium resources and projection of the global lithium demand spanning the years 2010‒2100, a sudden surge in the demand is anticipated due to electric vehicles [1, 3]. The demand for LIBs is estimated to reach 2.2 million tonnes by 2030 [4]. The recycling rate of Li is currently less than 1% [5]; however, the price of lithium has continued to appreciate strongly each year; thus, methods of increasing the Li recycling rate have been reviewed [6].
Various ß-diketone reagents for the selective recovery of Li were also reported in a review article [7]. The lithium ion is monovalent, small, and highly hydrated. Thus, size-discrimination using macrocyclic compounds with small coordination sites has been employed for Li recovery. Since the discovery of crown ethers by Pedersen [8] and the correlation between the cavity size of these molecules and the ionic diameters by Frensdorff [9], various derivatives have been synthesized and their potential as representative macrocyclic host compounds for molecules and ions has been investigated. A number of books and review articles has correspondingly been published [10,11,12]. Katsuta et al. prepared arene trinuclear ruthenium complexes bridged by 2,3-pyridinediolate as a macrocyclic host compound for Li+ [13, 14], which exhibited high selectivity for Li+ over Na+.
Calixarenes are macrocyclic compounds prepared in a single step by the condensation reaction of the corresponding phenols and formaldehyde [15]. Various derivatives have been prepared and their ion-discrimination ability has been investigated; related books and review articles have been also published [16,17,18,19,20]. Various calixarene derivatives exhibit specific structural effects for metal extraction, as demonstrated in our studies [21,22,23]. Among calixarene derivatives, calix[4]arene derivatives have been the main focus of our research as they provide a narrow coordination site and rigid structure. Calix[4]arene was originally prepared by using Na+ as a templating ion. Most calix[4]arene derivatives exhibit selectivity for Na+ over other alkali metal ions owing to the size-discriminating effect. However, gradual substitution of the carboxyl groups of calix[4]arene tetraacetic acid with coordinative-inert propyl group(s) changes the selectivity for alkali metal ions from larger to smaller ions, demonstrating an allosteric effect involving co-extraction of the second ion with the first (Na+). The tripropyl-monoacetic acid derivative exhibits Li+ selectivity [24, 25]. It was found that slight change of substituents resulted in drastic change of metal selectivity; however, the effects of alkyl groups attached on phenoxyl oxygen atoms and at the p-position of calix[4]arene have not yet been investigated.
Herein, we report the synthesis of trialkyl-monoacetic acid derivatives of calix[4]arene and investigate the effects of alkyl groups attached to the phenoxyl oxygen atoms and at the p-position of calix[4]arene on the extraction of alkali metal ions.
Experimental
Reagents
All reagents were of analytical grade and were supplied by Wako Pure Chemical Industries, Ltd. (Osaka, Japan) and used without further purification. 25,26,27-Tripropoxy-28-carboxymethoxy-5,11,17,23-tetrakis(1,1,3,3-tetramethylbutyl)calix[4]arene 4Pr was obtained in a manner similar to that reported in a previous paper [25]. The extraction reagents, (trialkyl-monoacetic acid derivatives of p–t-octylcalix[4]arene and calix[4]arene with other alkyl branches) were synthesized by modifying this method, aiming to confirm the effects of the three alkyl branches at the lower rim and of the t-octyl groups at the upper rim of the calixarene on selective Li extraction.
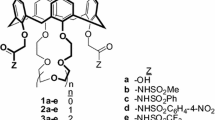
The chemical structures of the extraction reagents synthesized herein are shown in Fig. 1. The schemes for synthesizing the extraction reagents are shown in Figs. 2 and 3. 5,11,17,23-Tetrakis(1,1,3,3-tetramethylbutyl)calix[4]arene-25,26,27,28-tetrol (1) [26] and calix[4]arene-25,26,27,28-tetrol (4) [27] were synthesized by modified literature methods.

Chemical structures of the extraction reagents

Schemtic of synthesis of trialkyl-monoacetic acid derivatives of p–t-octylcalix[4]arene

Schemtic of synthesis of trialkyl-monoacetic acid derivatives of calix[4]arene
Synthesis of trialkyl-monohydroxyl derivatives of t-octylcalix[4]arene (2)
Under a nitrogen stream, 1 and alkali earth hydroxide octahydrate were added to dry dimethylformamide (DMF) and the mixture was stirred. Thereafter, an alkyl halogenide was added to the mixture and stirred at 30 °C for the desired period. After filtration and removal of the solvent in vacuo, the desired compound was extracted from the residue using chloroform. The solution was washed 3 times with 1 M (M = mol dm−3) hydrochloric acid and thrice with distilled water. After drying over anhydrous magnesium sulfate, the solution was filtered, and the solvent then removed in vacuo; the desired compound was recrystallized from acetonitrile to yield a white powder. The detailed conditions of the synthesis are listed in Table 1. The 1H-NMR and FT-IR spectra of 2Oct, 2Bz, and 22EtBu are shown in Fig. S1a–c, respectively. Based on the 1H-NMR spectra, all derivatives adopted the cone conformation, in which three O-alkyl and a carboxy groups were oriented in the same direction.
25,26,27-Trioctoxy-28-hydroxy-5,11,17,23-tetrakis(1,1,3,3-tetramethylbutyl)calix[4]arene (2Oct)
TLC (SiO2, chloroform, Rf = 0.91), 1H-NMR (300 MHz, δ, CDCl3, TMS, 25 °C), 0.51 (18H, s, 2C(CH3)3), 0.76 (18H, s, 2C(CH3)3), 0.88 (12H, s, 2C(CH3)2), 0.90 (9H, t, 3CH2(CH2)5CH3), 1.2–1.4 (30H, t, 3CH2(CH2)5CH3), 1.35 (12H, s, C(CH3)2), 1.36 (4H, s, 2CCH2C), 1.74 (4H, s, 2CCH2C), 1.89 (4H, quint, 2CH2CH2(CH2)5), 2.27 (2H, quint, 2CH2CH2(CH2)5), 3.15 (2H, d, 2exo-CH2), 3.21 (2H, d, 2exo-CH2), 3.75 (4H, t, 2CH2CH2(CH2)5), 3.91 (2H, t, CH2CH2(CH2)5), 4.34 (2H, d, 2endo-CH2), 4.39 (2H, d, 2endo-CH2), 5.74 (1H, s, OH), 6.56 (4H, s, 4ArH), 7.00 (2H, s, 2ArH), 7.06 (2H, s, 2ArH). FT-IR (KBr, cm−1): 3543 (sharp peak; νFreeO–H), 3570‒3260 (low-intensity peak; νO–H), 2955 (νC–H), 1470 (νC=C), 1207 (νC–O).
25,26,27-Tribenzyloxy-28-hydroxy-5,11,17,23-tetrakis(1,1,3,3-tetramethylbutyl)calix[4]arene (2Bz)
TLC (SiO2, chloroform, Rf = 0.91), 1H-NMR (300 MHz, δ, CDCl3, TMS, 25 °C), 0.39 (18H, s, 2C(CH3)3), 0.75 (18H, s, 2C(CH3)3), 0.96 (12H, s, 2C(CH3)2), 1.31 (12H, s, 2C(CH3)2), 1.36 (4H, s, 2CCH2C), 1.68 (4H, s, 2CCH2C), 2.89 (2H, d, 2exo-CH2), 3.01 (2H, d, 2exo-CH2), 4.06 (2H, d, 2endo-CH2), 4.17 (2H, d, 2endo-CH2), 4.64 (4H, s, 2CH2Ph), 4.93 (2H, s, CH2Ph), 6.60 (2H, s, 2ArH), 6.66 (2H, s, 2ArH), 6.74 (1H, s, OH), 6.94 (2H, s, 2ArH), 7.01 (2H, s, 2ArH), 7.24 (15H, m, 3Bz-ArH). FT-IR (KBr, cm−1): 3404 (broad peak; νFreeO–H), 3570‒3260 (low-intensity peak; νO–H), 2955 (νC–H), 1476 (νC=C), 1209 (νC–O).
25,26,27-Tris(2-ethylbutyloxy)-28-hydroxy-5,11,17,23-tetrakis(1,1,3,3-tetramethylbutyl)calix[4]arene (22EtBu)
TLC (SiO2, chloroform, Rf = 0.91), 1H-NMR (300 MHz, δ, CDCl3, TMS, 25 °C), 0.50 (18H, s, 2C(CH3)3), 0.73 (9H, s, C(CH3)3), 0.79 (9H, s, C(CH3)3), 0.89 (12H, s, C(CH3)2), 0.91 (18H, t, 6CH2CH3), 1.35 (12H + 4H, s + s, 2C(CH3)2 + 2CCH2C)), 1.46 (8H, quint, 4CH2CH3), 1.63 (4H, quint, 2CH2CH3), 1.70 (2H, s, CCH2C), 1.74 (2H, s, CCH2C), 1.79 (2H, sep, 2CH2CH(Et)CH2), 2.10 (1H, sep, CH2CH(Et)CH2), 3.21 (4H, d, 4exo-CH2), 3.54 (2H, d, OCH2CH), 3.72 (2H, d, OCH2CH), 4.01 (2H, d, OCH2CH), 4.34 (2H, d, 2endo-CH2), 4.47 (2H, d, 2endo-CH2), 5.87 (1H, s, OH), 6.52 (2H, s, 2ArH), 6.62 (2H, s, 2ArH), 7.01 (2H, s, 2ArH), 7.06 (2H, s, 2ArH). FT-IR (KBr, cm−1): 3543 (sharp peak; νFreeO–H), 3570‒3260 (low-intensity peak; νO–H), 2955 (νC–H), 1475 (νC=C), 1205 (νC–O).
Synthesis of trialkyl-monoacetic acid derivatives of t-octylcalix[4]arene (3)
Under a nitrogen stream, trialkyl-monohydroxy derivatives of p–t-octylcalix[4]arene (2) were dissolved by immersion in THF for 15 min, after which sodium hydride was added to the mixture and stirred for 15 min. Ethyl bromoacetate was added and the mixture was stirred at room temperature for 24 h. After monitoring by TLC, the excess sodium hydride was deactivated with ethanol in an ice-bath; the ethanol was then removed in vacuo. The desired compound was extracted with chloroform. The organic layer was washed 3 times with 1 M hydrochloric acid and thrice with distilled water. After drying over anhydrous magnesium sulfate, the solution was filtered, and the solvent was removed in vacuo. Intermittently, sodium hydroxide, THF, and distilled water were added to the residue and the mixture was stirred at the desired temperature for a certain period. After monitoring by TLC, the solvent was removed in vacuo and chloroform was added to extract the desired compound from the residue. The solution was washed once with 6 M, twice with 1 M hydrochloric acid, and 3 times with distilled water. After drying over anhydrous magnesium sulfate, the solution was filtered, and the solvent was removed in vacuo. The desired compound was recrystallized from acetonitrile to yield a white powder. The detailed conditions of the synthesis are listed in Tables 2 and 3. The 1H-NMR and FT-IR spectra of 3Oct, 3Bz, and 32EtBu are shown in Fig. S2a–c, respectively. All derivatives adopted the cone conformation, as indicated by the 1H-NMR spectra.
25,26,27-Trioctoxyoxy-28-carboxymethoxy-5,11,17,23-tetrakis(1,1,3,3-tetramethylbutyl)calix[4]arene (3Oct)
TLC (SiO2, chloroform:methanol = 10:1, Rf = 0.77), 1H-NMR (300 MHz, δ, CDCl3, TMS, 25 °C), 0.43 (18H, s, 2C(CH3)3), 0.76 (18H, s, 2C(CH3)3), 0.86 (9H, t, (CH2)5CH3), 0.93 (12H, s, 2C(CH3)2), 1.28 (30H, m, 3(CH2)5CH3), 1.33 (4H, s, 2CCH2C), 1.37 (12H, s, 2C(CH3)2), 1.70 (4H, s, 2CCH2C)), 1.83 (6H, quint, 3CH2(CH2)5CH3), 3.16 (2H, d, 2exo-CH2), 3.21 (2H, d, 2exo-CH2), 3.75 (4H, t, OCH2CH2), 4.11 (2H, t, OCH2CH2), 4.22 (2H, d, 2endo-CH2), 4.47 (2H, d, 2endo-CH2), 4.64 (2H, s, CH2-COOH), 6.55 (2H, s, 2ArH), 6.66 (2H, s, 2ArH), 7.08 (2H, s, 2ArH), 7.10 (2H, s, 2ArH), 11.35 (1H, s, COOH). FT-IR (KBr, cm−1): peaks at 3543 (νFreeO–H) and 3570‒3260 (νO–H) disappeared, 3181 (low-intensity, broad peak; νFreeCOO–H), 2955 (νC–H), 1759 (νC=O), (1477 (νC=C), 1204 (νC–O).
25,26,27-Tribenzyloxy-28-carboxymethoxy-5,11,17,23-tetrakis(1,1,3,3-tetramethylbutyl)calix[4]arene (3Bz)
TLC (SiO2, chloroform:methanol = 10:1, Rf = 0.63), 1H-NMR (300 MHz, δ, CDCl3, TMS, 25 °C), 0.41 (18H, s, 2C(CH3)3), 0.72 (9H, s, C(CH3)3), 0.81 (9H, s, C(CH3)3), 0.90 (12H, s, 2C(CH3)2), 1.26 (12H, s, 2C(CH3)2), 1.33 (4H, s, 2CCH2C), 1.62 (2H, s, CCH2C)), 1.73 (2H, s, CCH2C)), 2.62 (2H, d, 2exo-CH2), 2.78 (2H, d, 2exo-CH2), 3.68 (2H, d, 2endo-CH2), 3.90 (2H, d, 2endo-CH2), 4.43 (2H, s, CH2-COOH), 4.53 (2H, s, CH2Ph), 5.04 (2H, s, CH2Ph), 5.07 (2H, s, CH2Ph), 6.47 (2H, s, 2ArH), 6.67 (2H, s, 2ArH), 6.92 (2H, s, 2ArH), 7.03 (2H, s, 2ArH), 7.1–7.4 (15H, m, CH2Ph), 11.65 (1H, s, COOH). FT-IR (KBr, cm−1): peaks at 3543 (νFreeO–H) and 3570‒3260 (νO–H) disappeared, 3379 (low-intensity, broad peak; νFreeCOO–H), 2955 (νC–H), 1759 (νC=O), 1477 (νC=C), 1204 (νC–O).
25,26,27-Tris(2-ethylbutyloxy)-28-carboxymethoxy-5,11,17,23-tetrakis(1,1,3,3-tetramethylbutyl)calix[4]arene (32EtBu)
TLC (SiO2, chloroform:methanol = 10:1, Rf = 0.77), 1H-NMR (300 MHz, δ, CDCl3, TMS, 25 °C), 0.52 (18H, s, 2C(CH3)3), 0.73 (9H, s, C(CH3)3), 0.80 (9H, s, C(CH3)3), 0.86 (12H, s, 2C(CH3)2), 0.91 (18H, t, 3CH(CH2CH3)2), 1.33 (4H, s, 2CCH2C), 1.38 (12H, s, 2C(CH3)2), 1.3–1.6 (12H, m, 3CH(CH2CH3)2), 1.72 (3H, sept, 3 CH2CH(CH2CH3)2), 1.74 (4H, s, CCH2C), 3.21 (4H, d, 2exo-CH2), 3.52 (2H, d, CH2CH), 3.71 (2H, d, CH2CH), 4.01 (2H, d, CH2CH), 4.25 (2H, d, 2endo-CH2), 4.44 (2H, d, 2endo-CH2), 4.46 (2H, s, CH2-COOH), 6.47 (2H, s, 2ArH), 6.63 (2H, s, 2ArH), 7.08 (2H, s, 2ArH), 7.11 (2H, s, 2ArH), 10.82 (1H, s, COOH). FT-IR (KBr, cm−1): peaks at 3543 (νFreeO–H) and 3570‒3260 (νO–H) disappeared, 3426 (νCOO–H), 3424 (low-intensity, broad peak; νFreeCOO–H), 2955 (νC–H), 1764 (νC=O), 1477 (νC=C), 1204 (νC–O).
Synthesis of trialkyl-monohydroxyl derivatives of calix[4]arene (5)
Only propyl, octyl, and benzyl groups were selected as the introduced alkyl branches, because 2-ethylbutyl-branched p–t-octylcalixarene exhibited selectivity for sodium ion over the lithium ion.
Under a nitrogen stream, 4 and strontium hydroxide octahydrate were added to dry DMF, and the mixture was stirred. Thereafter, alkyl halogenide was added to the mixture and stirred at 30 °C for the desired period. The remaining procedures were the same as those for 2. The detailed synthesis conditions are listed in Table 4. The 1H-NMR and FT-IR spectra of 5Oct, 5Bz, and 52EtBu are shown in Fig. S3a–c, respectively. All derivatives adopted the cone conformation, as confirmed by the 1H-NMR spectra.
25,26,27-Tripropoxy-28-hydroxy-calix[4]arene (5Pr)
TLC (SiO2, chloroform, Rf = 0.90), 1H-NMR (300 MHz, δ, CDCl3, TMS, 25 °C), 0.92 (3H, t, CH2CH3), 1.11 (6H, t, 2CH2CH3), 1.89 (4H, sext, 2CH2CH2CH3), 2.27 (2H, sext, CH2CH2CH3), 3.21 (2H, d, 2exo-CH2), 3.29 (2H, d, 2exo-CH2), 3.74 (4H, t, 2CH2CH2CH3), 3.84 (2H, t, CH2CH2CH3), 4.38 (2H, d, 2endo-CH2), 4.42 (2H, d, 2endo-CH2), 4.68 (1H, s, OH), 6.37 (6H, m, 6ArH), 6.77 (1H, t, ArH), 6.97 (1H, t, ArH), 7.09 (2H, d, ArH), 7.17 (2H, d, ArH). FT-IR (KBr, cm−1): 3545 (sharp peak; νFreeO–H), 3655‒2955 (low-intensity peak; νO–H), 2963 (νC–H), 1459 (νC=C), 1200 (νC–O), 762 (σ∠HCC(Ar)).
25,26,27-Trioctoxy-28-hydroxy-calix[4]arene (5Oct)
TLC (SiO2, chloroform, Rf = 0.90), 1H-NMR (300 MHz, δ, CDCl3, TMS, 25 °C), 0.88 (9H, t, 3CH2(CH2)5CH3), 1.50 (30H, m, 3CH2(CH2)5CH3), 1.89 (4H, quint, 2CH2CH2(CH2)5), 2.20 (2H, quint, 2CH2CH2(CH2)5), 3.20 (2H, d, 2exo-CH2), 3.28 (2H, d, 2exo-CH2), 3.78 (4H, t, 2OCH2), 3.90 (2H, t, OCH2), 4.37 (2H, d, 2endo-CH2), 4.41 (2H, d, 2endo-CH2), 4.72 (1H, s, OH), 6.37 (6H, m, 6ArH), 6.77 (1H, t, ArH), 6.96 (1H, t, ArH), 7.09 (2H, d, ArH), 7.16 (2H, d, ArH). FT-IR (KBr, cm−1): 3545 (sharp peak; νFreeO–H), 3655‒2955 (low-intensity peak; νO–H), 2926 (νC–H), 1458 (νC=C), 1199 (νC–O), 761 (σ∠HCC(Ar)).
25,26,27-Tribenzyloxy-28-hydroxy-calix[4]arene (5Bz)
TLC (SiO2, chloroform, Rf = 0.88), 1H-NMR (300 MHz, δ, CDCl3, TMS, 25 °C), 2.96 (2H, d, 2exo-CH2), 3.14 (2H, d, 2exo-CH2), 4.12 (2H, d, 2endo-CH2), 4.34 (2H, d, 2endo-CH2), 4.73 (4H, s + s, 2CH2Ph), 5.05 (2H, s, CH2Ph), 5.29 (1H, s, OH), 6.41 (6H, m, 6ArH), 6.75 (1H, t, 1ArH), 6.89 (1H, t, ArH), 7.05 (2H, d, 2ArH), 7.11 (2H, d, 2ArH), 7.2–7.4 (15H, m, 3Bz-ArH). FT-IR (KBr, cm−1); 3545 (broad peak; νO–H), 2963 (νC–H), 1459 (νC=C), 1200 (νC–O), 762 (σ∠HCC(Ar)).
Synthesis of trialkyl-monoacetic acid derivatives of calix[4]arene (6)
Under a nitrogen stream, trialkyl-monohydroxy derivatives of calix[4]arene (5) were dissolved by immersion in THF for 15 min, after which sodium hydride was added to the mixture and stirred for 15 min. Ethyl bromoacetate was added and the mixture was stirred at room temperature for 24 h. The rest procedures were same with those for 3. The detailed synthetic condition is listed in Tables 5 and 6. The 1H-NMR and FT-IR spectra of 6Oct, 6Bz, and 62EtBu are shown in Fig. S4a–c, respectively. All derivatives were confirmed to take cone conformation by 1H-NMR spectra.
25,26,27-Tripropoxy-28-carboxymethoxy-calix[4]arene (6Pr)
TLC (SiO2, chloroform:methanol = 10:1, Rf = 0.57), 1H-NMR (300 MHz, δ, CDCl3, TMS, 25 °C), 0.86 (3H, t, (CH2)5CH3), 1.02 (6H, t, (CH2)5CH3), 1.90 (6H, sext, 3CH2CH2CH3), 3.19 (2H, d, 2exo-CH2), 3.30 (2H, d, 2exo-CH2), 3.77 (4H, t, 2OCH2), 3.98 (2H, t, 2OCH2), 4.33 (2H, d, 2endo-CH2), 4.46 (2H, d, 2endo-CH2), 4.88 (2H, s, CH2-COOH), 6.2–6.4 (6H, m, 6ArH), 6.96 (1H, t, ArH), 6.99 (1H, t, ArH), 7.15 (4H, d, 2ArH), 11.35 (1H, s, COOH). FT-IR (KBr, cm−1): peak at 3543 (νFreeO–H) disappeared, 3197 (broad peak; νFreeCOO–H), 2961 (νC–H), 1763 (νC=O), 1460 (νC=C), 1200 (νC–O), 762 (σ∠HCC(Ar)).
25,26,27-Trioctoxy-28-carboxymethoxy-calix[4]arene (6Oct)
TLC (SiO2, chloroform:methanol = 10:1, Rf = 0.92), 1H-NMR (300 MHz, δ, CDCl3, TMS, 25 °C), 0.86 (9H, t, 3(CH2)5CH3), 1.29 (30H, m, 3(CH2)5CH3), 1.87 (6H, quint, 3CH2(CH2)5CH3), 3.18 (2H, d, 2exo-CH2), 3.30 (2H, d, 2exo-CH2), 3.79 (4H, t, OCH2), 4.01 (2H, t, OCH2), 4.32 (2H, d, 2endo-CH2), 4.44 (2H, d, 2endo-CH2), 4.88 (2H, s, CH2-COOH), 6.20 (2H, t, 2ArH), 6.28 (4H, d, 4ArH), 6.97 (2H, t, 2ArH), 7.15 (4H, d, 4ArH), 11.63 (1H, s, COOH). FT-IR (KBr, cm−1): peak at 3543 (νFreeO–H) disappeared, 3237 (νFreeCOO–H), 2961 (νC–H), 1762 (νC=O), 1459 (νC=C), 1198 (νC–O), 760 (σ∠HCC(Ar)).
25,26,27-Tribenzyloxy-28-carboxymethox-calix[4]arene (6Bz)
TLC (SiO2, chloroform:methanol = 10:1, Rf = 0.75), 1H-NMR (300 MHz, δ, CDCl3, TMS, 25 °C), 2.82 (2H, d, 2exo-CH2), 2.83 (2H, d, 2exo-CH2), 3.93 (2H, d, 2endo-CH2), 3.97 (2H, d, 2endo-CH2), 4.65 (2H, s, CH2Ph), 4.67 (2H, s, CH2-COOH), 4.96 (2H, s, CH2Ph), 5.01(2H, s, CH2Ph), 6.26 (2H, t, 2ArH), 6.36 (4H, d, 4ArH), 6.90 (2H, t, 2ArH), 7.01 (4H, d, 4ArH), 7.1–7.4 (15H, m, 3CH2Ph), 11.80 (1H, s, COOH). FT-IR (KBr, cm−1): peak at 3543 (νFreeO–H) disappeared, 3160 (broad peak; νFreeCOO–H), 2961 (νC–H), 1755 (νC=O), 1460 (νC=C), 1192 (νC–O), 754 (σ∠HCC(Ar)).
Distribution study
The extraction study was performed using a previously described procedure [28]. The organic solution was prepared by dissolving each trialkyl-monoacetic acid reagent, 4, in chloroform to a concentration of 5 mM. The aqueous solution was prepared by dissolving each alkali metal chloride in 0.1 M HEPES buffer solution (N-(2-hydroxyethyl)piperazin-N′-(2-ethanesulfonic acid)). The pH of this solution was adjusted by adding 1 M ammonia solution. Equal volumes (5 cm3) of both the organic and aqueous phases, were mixed and shaken (150 rpm) for an adequate time at 30 °C. After phase separation, the pH after reaching equilibrium and the metal concentrations before and after reaching equilibrium were measured using a pH meter (HM-30R, TOA-DKK, Tokyo) and atomic absorption spectrophotometer (abbreviated as AAS, Shimadzu Instruments AA-6800). The pH meter was pre-calibrated by using pH 4.01, 6.86, and 9.18 standard solutions.
The extraction percentage (%E) and distribution ratio (D) were respectively calculated using Eqs. (1 and 2).
where Ci and Ce represent the initial and equilibrium metal concentrations in the aqueous phase, respectively.
Results and discussion
Synthesis of trialkyl-monoacetic acid derivatives
Tri-substitution of the alkyl branches on the phenol groups was successfully carried out, and was facilitated by using strontium salts instead of barium salts as the templating ion. All spectral data support the successful preparation of the desired compounds, including the intermediate compounds. The hydroxy peaks in the 1H-NMR spectra of the trialkyl-monohydroxy derivatives appeared at 4.68–6.74 ppm, where the chemical shifts are much lower than those of the tetrahydroxy derivative (9.95 ppm for 1 [26], 10.20 ppm for 4 [29]). This means that the strong hydrogen bonds were broken by the introduction of the three alkyl groups. This was supported by the decreased intensity of the broad νO–H peaks in the IR spectra. The carboxy peaks in the 1H-NMR spectra of the trialkyl-monoacetic acid derivatives appeared at 11.35–11.80 ppm, corresponding to a higher magnetic field compared with that of normal carboxylic acid derivatives, consistent with the appearance of the IR peaks of the carboxylic acid at a higher (more than 3100 cm−1) wavenumber. These data also indicate that the strong hydrogen bonds were broken by introducing the three alkyl groups.
The peak position of the aryl protons in the 1H-NMR spectra of 3 and 6 can provide an indicator of the coordination site, because these protons are far from the coordination site for alkali metal ions and are thus not electrostatically affected by the alkyl branches introduced on the phenoxy oxygen atoms. The appearance of the aryl proton peaks at relatively higher magnetic field indicates that the four benzene rings are positioned upright, whereas their appearance at lower magnetic field indicates that the calixarene changed to a flattened orientation. The former structure would provide a broader coordination site, whereas the latter structure would provide a narrower site, which would consequently contribute to efficient separation among the alkali metal ions with different ionic sizes. However, the difference in the positions were insignificant and the introduced alkyl branches may not have a significant impact on the extraction behavior.
Extraction time to reach equilibrium
The extraction time required to reach equilibration was first investigated. The effect of the shaking time on the percentage extraction of lithium and sodium ions with 3 and 6 is shown in Figs. 4a and b, and 5 a and b, respectively. The extraction times to reach equilibration for Li+ and Na+ with 3 were 6‒24 h, and 6‒12 h, respectively. The extraction times to reach equilibration for Li+ and Na+ with 6 were 4 h to longer than 12 h, and 3 h, respectively. Li+ extraction with 3 and 6 required a longer time than Na+ extraction. Thus, the shaking times were set for longer than the obtained periods.

Effect of shaking time on the percentage extraction of a Li+ and b Na+ with 3Pr (open triangle), 3Oct (closed diamond), 3Bz (closed circle), and 32EtBu (open square) in competitive system. [M+]i = 0.1 mM, [Extractant] = 5.0 mM, [HEPES] = 0.1 M + [NH3] = 1.0 M

Effect of shaking time on the percentage extraction of a Li+ and b Na+ with 6Pr (open triangle), 6Oct (closed diamond), and 6Bz (closed circle) in competitive system. [M+]i = 0.1 mM, [Extractant] = 5.0 mM, [HEPES] = 0.1 M + [NH3] = 1.0 M
Extraction of various alkali metal ions in competitive systems
In the previous paper [25], the extraction of Li+ with 3Pr was expressed as the loading percentage, which is the extent to which certain amounts of alkali metal ions in high concentrations (0.10 M) are loaded on a lower concentration of 3Pr (5 mM). In this study, the data were reconstructed by changing the metal concentration for comparison with the data for 3 with other alkyl branches.
The effects of the equilibrium pH on the percentage extractions (%Extraction) of Li+, Na+, K+, Rb+, and Cs+ using 3Pr, 3Oct, 3Bz, and 32EtBu, in a competitive system are shown in Fig. 6a–d, respectively. As reported in the previous paper using 3Pr in the individual system [25], trialkyl-monoacetic acid derivatives were ineffective for extracting larger K+, Rb+, and Cs+, but extracted smaller Li+ and Na+. In the previous section, it was expected that little difference in the extraction behavior would be observed by using 3 with different alkyl branches; the slight difference in the peak position of the aryl protons was expected to have a minimal effect on the extraction behavior. However, the alkyl branches obviously affected the extraction behavior. In particular, the separation efficiency was higher for Li+ than for Na+, with an obvious difference in the pH of the extraction region. Careful observation shows that sodium extraction was affected with 3Pr, 3Oct, and 3Bz. Because the original calix[4]arene was synthesized by using sodium base as the templating ion, most of the calix[4]arene derivatives exhibited selectivity for Na+ over the other alkali metal ions. Nevertheless, 3Pr, 3Oct, and 3Bz providing metal coordination sites closely similar to the size of Li+ and thus exhibited selectivity for Li+ extraction over Na+ extraction, consequently affecting the separation efficiency. Therefore, the introduced alkyl branches effectively exclude the sodium ion by lipophilic-hydrophobic repulsion and steric hindrance. A significant effect was also observed for more sterically-hindered 32EtBu. In contrast, selectivity for Na+ over Li+ was observed with 32EtBu, which suppressed Li+ extraction, although a definite reason was not clarified.

Effect of equilibrium pH on percentage extraction of alkali metal ions with a 3Pr, b 3Oct, c 3Bz, and d 32EtBu in competitive system. Li+ (closed triangle), Na+ (open circle), K+ (open diamond), Rb+ (closed square), and Cs+ (open up triangle), [M(I)]i = 0.1 mM, [Extractant] = 5.0 mM
The effect of the alkyl branches at the p-position on the extraction was also investigated. The effects of the equilibrium pH on the percentage extractions (%Extraction) of Li+, Na+, K+, Rb+, and Cs+ using 6Pr, 6Oct, and 6Bz, in a competitive system are shown in Fig. 7a–d, respectively. 62EtBu was not prepared because 32EtBu exhibited Na+ selectivity. Dealkylated reagents, 6, exhibited less extraction ability compared with 3, plausibly because the extraction shifted with the pH region. This is plausible attributed to the lower lipophilicity in the absence of alkyl branches at the p-position. The separation efficiency of 6 was similar to that of 3, which means that the alkyl branches at the p-position have less effect on the separation efficiency.

Effect of equilibrium pH on percentage extraction of alkali metal ions with a 6Pr, b 6Oct, and c 6Bz in competitive system. Li+ (closed triangle), Na+ (open circle), K+ (open diamond), Rb+ (closed square), and Cs+ (open up triangle), [M(I)]i = 0.1 mM, [Extractant] = 5.0 mM
Extraction reactions, and extraction equilibrium constants and separation factors
The extraction of monovalent alkali metal ions using trialkyl-monoacetic acid derivatives 3 and 6 was investigated because trialkyl-monoacetic acid derivatives are mono-ionizable and release a single proton during the extraction. The reaction proposed in the previous paper [25] is represented by Eq. (3):
where RH and M represent 3 or 6, and the alkali metal ion, respectively. The extraction equilibrium constant was obtained using Eq. (4),
where the distribution ratio, D, is the ratio of the metal concentrations in the organic and aqueous phases, and defined in Eq. (2). After taking logarithms, Eq. (5) was obtained.
The effects of the equilibrium pH on the distribution ratio of Li+ and Na+ with 3Pr, 3Oct, 3Bz, and 32EtBu are shown in Fig. 8a–d, respectively. The effects of the equilibrium pH on the distribution ratio of Li+ and Na+ with 6Pr, 6Oct, and 6Bz are shown in Fig. 9a–c, respectively. All plots lie on straight lines, each with a slope of unity. The slope corresponds to the monovalent charge of both metal ions. The data confirm that the extraction takes place by a simple ion-exchange mechanism. The concentration of the extraction reagent was sufficiently higher than the total concentration of the metal ions. The extraction equilibrium constants for both ions with all reagents were obtained from the intercepts of log D vs. pH. The extraction equilibrium constants, Kex, of Li+ and Na+ with 3 and 6, as well as the separation factors, ß, for Li+ and Na+ are listed in Table 7. The separation factors, ß, is defined by Eq. (6).

Effect of equilibrium pH on distribution ratio of Li+ (closed triangle) and Na+ (open circle) with a 3Pr, b 3Oct, c 3Bz, and d 32EtBu in competitive system. [M(I)]i = 0.1 mM, [Extractant] = 5.0 mM

Effect of equilibrium pH on distribution ratio of Li+ (close triangle) and Na+ (open circle) with a 6Pr, b 6Oct, and c 6Bz in competitive system. [M(I)]i = 0.1 mM, [Extractant] = 5.0 mM
The theoretical curve and straight lines drawn in Figs. 6a–d, 7a–c, 8a–d, and 9a–c were obtained by using the extraction equilibrium constants.
Conclusions
Four and three trialkyl-monoacetic acid derivatives of p–t-octylcalix[4]arene and calix[4]arene were respectively prepared to investigate the effect of the alkyl branches attached to the phenoxy oxygen atoms and to the p-position on the selective extraction of Li+ over Na+. Alkyl branches on the phenoxy oxygen atoms remarkably affected the selectivity for Li+ over Na+ by excluding Na+ extraction while maintaining Li+ extraction. However, the introduction of a more-hindered alkyl branch depressed the Li+ extraction. The data can contribute to elucidating the origin of the high Li+ selectivity and the Li+ extraction mechanism. Optimization of the alkyl branch improves the Li+ selectivity.
Data availability
No datasets were generated or analysed during the current study.
References
Gruber, P.W., Medina, P.A., Keoleian, G.A., Kesler, S.E., Everson, M.P., Wallington, T.J.: Global lithium availability, a constraint for electric vehicles? J. Ind. Ecol. 15(5), 760–775 (2011)
Liu, G., Zhao, Z., Ghahreman, A.: Novel approaches for lithium extraction from salt-lake brines: a review. Hydrometallurgy 187, 81–100 (2019)
Kesler, S.E., Gruber, P.W., Medina, P.A., Keoleian, G.A., Everson, M.P., Wallington, T.J.: Global lithium resources: relative importance of pegmatite, brine and other deposits. Ore Geol. Rev. 48, 55–69 (2012)
Kaunda, R.B.: Potential environmental impacts of lithium mining. J. Energy Nat. 38(3), 237–244 (2020)
Reck, B.K., Graedel, T.E.: Challenges in metal recycling. Science 337(6095), 690–695 (2012)
Martin, G., Rentsch, L., Höck, M., Bertau, M.: Lithium market research—global supply, future demand and price development. Energy Storage Mater. 6, 171–179 (2017)
Swain, B.: Separation and purification of lithium by solvent extraction and supported liquid membrane, analysis of their mechanism: a review. J. Chem. Technol. Biotechnol. 91(10), 2549–2562 (2016)
Pedersen, C.J.: Cyclic polyethers and their complexes with metal salts. J. Am. Chem. Soc. 89(10), 2495–2496 (1967)
Frensdorff, H.K.: Stability constants of cyclic polyether complexes with univalent cations. J. Am. Chem. Soc. 93(3), 600–606 (1971)
Izatt, R.M., Pawlak, K., Bradshaw, J.S., Bruening, R.L.: Thermodynamic and kinetic data for macrocycle interactions with cations and anions. Chem. Rev. 91(8), 1721–2085 (1991)
Glue, K. (ed.): Macrocyclic chemistry—current trends and future perspectives. Springer, Netherlands (2005)
Bianchi, A., Bowman-James, K., García-España, E. (eds.): Supramolecular chemistry of anions. Wiley-VCH, New York (1997)
Katsuta, S., Imoto, T., Kudo, Y., Takeda, Y.: Selective extraction of lithium with a macrocyclic trinuclear complex of (1,3,5-trimethylbenzene)ruthenium(II) bridged by 2,3-dioxopyridine. Anal. Sci. 24(10), 1215–1217 (2008)
Katsuta, S., Nomura, H., Egashira, T., Kanaya, N., Kudo, Y.: Highly lithium-ion selective ionophores: macrocyclic trinuclear complexes of methoxy-substituted arene ruthenium bridged by 2,3-pyridinediolate. New J. Chem. 37(11), 3634–3639 (2013)
Gutsche, C.D., Muthukrishnan, R.: Calixarenes. 1. Analysis of the product mixtures produced by the base-catalyzed condensation of formaldehyde with para-substituted phenols. J. Org. Chem. 43(25), 4905–4906 (1978)
Gutsche, C.D. (ed.): Calixarenes revisited. The Royal Society of Chemistry, Cambridge (1998)
Asfari, Z., Böhmer, V., Harrowfield, J., Vicens, J. (eds.): Calixarenes 2001. Kluwer Academic Publishers, Netherlands (2001)
Lumetta, G.J., Rogers, R.D., Gopalan, A.S.: Calixarenes for separations. American Chemical Society, Washington, DC (2000)
Ohto, K.: Review of the extraction of metal cations with calixarene derivatives. Solvent Extr. Res., Dev. Jpn. 17, 1–18 (2010)
Ohto, K.: Review on adsorbents incorporating calixarene derivatives for metals recovery and hazardous ions removal—concept of adsorbent design and classification of adsorbents. J. Incl. Phenom. Macrocycl. Chem. 101(3), 175–194 (2021)
Ohto, K.: Chap. 3 Molecular design and metal extraction behavior of calixarene compounds as host extractants. Ion Exch. Solv. Extr. 21, 81–127 (2013)
Ohto, K., Furugou, H., Morisada, S., Kawakita, H., Isono, K., Inoue, K.: Stepwise recovery of molybdenum, tungsten, and vanadium by stripping using an amino-type “Trident” molecule. Solvent Extr. Ion Exch. 39, 512–532 (2021)
Ohto, K.: Research on various structural effects of calix[4]arene derivatives on extractive separation behavior of metal ions. J. Ion Exch. 30(2), 17–28 (2019)
Sadamatsu, H., Morisada, S., Kawakita, H., Ohto, K.: Allosteric coextraction of sodium and metal ions with calix[4]arene derivatives 3. Effect of propyl groups on size-discrimination for the second coextracted ion. Solvent Extr. Ion Exch. 33(3), 264–277 (2015)
Sadamatsu, H., Hanada, T., Morisada, S., Kawakita, H., Ohto, K.: Comprehensive comparison of alkali metal extraction with a series of calix[4]arene derivatives with propyl and/or acetic acid groups. J. Incl. Phenom. Macrocycl. Chem. 84, 87–97 (2016)
Ohto, K., Yano, M., Inoue, K., Yamamoto, T., Goto, M., Nakashio, F., Shinkai, S., Nagasaki, T.: Solvent extraction of trivalent rare earth metal ions with carboxylate derivatives of calixarenes. Anal. Sci. 11(6), 893–902 (1995)
Gutsche, C.D., Levine, J.A.: Calixarenes. 6. Synthesis of a functionalizable calix[4]arene in a conformationally rigid cone conformation. J. Am. Chem. Soc. 104(9), 2652–2653 (1982)
Ohto, K., Fuchiwaki, N., Morisada, S., Kawakita, H., Weigand, J.J., Marco, W.: Comparative extraction of aluminum group metals using acetic acid derivatives with three different-sized frameworks for coordination. Separations 8(11), 211 (2021)
Ohto, K.: unpublished data, in CDCl3 at 20 °C (1995)
Acknowledgements
This work was financially supported in part by the Sasakawa Scientific Research Grant No. 25-301 from “The Japan Science Society” and by JSPS KAKENHI Grant Number (Grant-in-Aid for “Scientific Research (C) 25410192) from JSPS”. The NMR measurements were conducted at Analytical Research Center for Experimental Sciences, Saga University supported by “Project for Promoting Public Utilization of Advanced Research Infrastructure”. We would like to thank Editage (www.editage.com) for English language editing.
Funding
This study was funded by Japan Society for the Promotion of Science, KAKENHI (C) 25410192, Sasakawa Scientific Research Grant, 25-301.
Author information
Authors and Affiliations
Contributions
KO planned this research work, elucidated the extraction mechanism, and wrote the paper. HS prepared most of the reagents, measured spectra data, and carried out extraction. TH prepared one of the reagents, measured spectra data, and carried out extraction. SM discussed with spectra data of the reagents. HK supported for the extraction reaction and for drawing the theoretical lines to estimate the extraction constants and separation factors.
Corresponding author
Ethics declarations
Conflict of interest
There are no conflict to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ohto, K., Sadamatsu, H., Hanada, T. et al. Li-selective calix[4]arene with trialkyl-monoacetic acid groups: effect of three alkyl branches and t-octyl groups at p-position on selectivity for Li extraction. J Incl Phenom Macrocycl Chem 104, 185–197 (2024). https://doi.org/10.1007/s10847-024-01233-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10847-024-01233-5