
Abstract
The directed synthesis of asymmetrically substituted porphyrins—tetraphenylporphyrine derivatives containing amino acid residues as a functional group, which can be used as “anchor” groups, for incorporation into the structure of a protein molecule, was carried out. The obtained compounds were characterized by a number of spectral methods confirming their structure and purity. Their base and acid ionization constants in the AN–HClO4 and DMSO–potassium cryptate (KOH[222])—systems were measured by spectrophotometric titration. It was found that the asymmetric substitution architecture contributed to the stabilization of the protonated forms of porphyrins. This allowed, for the first time, to extract and spectrally characterize the mono- and doubly-protonated forms (H3P+ and H4P2+) of each ligands in the AN–HClO4 system. The analysis of the spectral changes of the monoamino derivative led to identify three stages of the protonation, which first involves the nitrogen atom of the periphery substituent (pKb = 13.26) and then the central nitrogen atoms of the macrocycle (pKb1 = 11.50; pKb2 = 9.65; pKb1,2 = 21.15). The relative stability of the first intermediate is assumed to be caused by charge delocalization. The electron-donor nature of the solvent in the DMSO–KOH [222] system led to the leveling of the ionization constants for the first and second stages, which allowed us to determine only the total values for the porphyrins studied. The successive stages of acid–base interactions were analyzed in the article.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Porphyrins and metalloporphyrins are widespread in nature and are of great biological value. Studies of the unique properties of porphyrins can become the basis not only for solving the fundamental problems (the identification of the energy conversion mechanisms in photosynthesis, obtaining synthetic oxygen carriers, etc.), but also for handling a lot of issues of practical importance, such as creating new generation drugs, semi-conductor, sensor and catalytically active materials. The diverse useful properties of porphyrins and their analogues are the result of their structural features [1,2,3,4,5,6].
Since the reaction centre of the porphyrin molecule contains pyrrolenine (–N=) and pyrrole (–NH–) nitrogen atoms, porphyrins can be considered typical amphoteric compounds possessing both basic (N-base) and very weak acidic (NC-acids) properties. Porphyrins, evidently, can both bond one or two protons through the pyrrolenine nitrogen atoms forming a mono- (H3P+) or a di-cation (H4P2+) and donate one or two pyrrole protons forming a mono- (HP−) and a di-anion (P2−), respectively. The equilibrium studies of porphyrins (H2P) and their structural analogues in solutions of poly-centered acids and bases play a special role in the development of the modern acid–base interaction theory [7,8,9]. The introduction of substituents of different nature makes it possible to vary the physicochemical properties of the porphyrin class of compounds within a wide range of values [10,11,12]. Finding solutions to the problems of fundamental studies and practical application of this class of compounds directly depends on the optimization of the porphyrin synthesis methods and possibility to modify the macrocycle periphery chemically [13, 14]. The most available and well-studied synthetic porphyrin—5,10,15,20-tetraphenylporphyrin (H2TPP) can be a suitable object for further modification of its periphery. One of the most relevant methods of H2TPP molecule functionalization is bonding of one or several “anchor” groups responsible for the high value of the constant of specific bonding, a property that allows the porphyrin molecule to penetrate cell membranes and is quite useful for biochemistry and medicine [15]. However, structural modification of a molecule is always accompanied by electronic effects of substitution causing redistribution of the electron density between the macrocycle and the newly bound molecular fragments, which certainly affects the chemical and photochemical properties of the obtained compounds. Besides, the asymmetric character of the substitution produces a specific effect [16] on all the physicochemical properties of the compounds.
This work presents the results of the synthesis and study of the spectral and acid–base properties of asymmetrically substituted derivatives of 5,10,15,20-tetraphenylporphyrin—H2P1, H2P2, H2P3 (Scheme 1)—prepared by bonding substituents in the para-position of one of the phenyl fragments.

Molecular structure of porphyrin objects
Materials and methods
Synthesis of 5-(4′-nitrophenyl)-10,15,20-triphenylporphyrin (H2P4)
Sodium nitrite (13.8 mg, 0.16 mmol) and H2TPP (123 mg, 0.16 mmol) was stirred in 5 ml trifluoroacetic acid. After 5 min of stirring, the reaction mixture was quenched with 20 ml ice water and neutralized with aqueous ammonia. The precipitate was filtered and dried at 90 °C. Column chromatography on silica gel with a dichloromethane-hexane (7:3) mixture showed that there were three zones: the first corresponded to H2TPP, the second one—to the target 5-(4′-nitrophenyl)-10,15,20-triphenylporphyrin (4), and the third—no separable mixture dinitrophenylporphyrins. Yield: 73 mg (63%). 1H NMR (CDCl3) δ, ppm.: 8.88 (d, 3 J = 4.25 Hz, 2H, β-H), 8.85 (s, 4H, β-H), 8.72 (d, 3 J = 4.25 Hz, 2H, β-H) 8.62 (d, 3 J = 8.55 Hz, 2H, 3′,5′-PhNO2), 8.38 (d, 3 J = 8.55 Hz, 3H, 2′,6′-PhNO2), 8.20 (d, 3 J = 6.7 Hz, 6H, 2′,6′-Ph) 7.83–7.70 (m, 9H, 3′,4′,5′-Ph), − 2.81 (s, 2H, NH). UV/Vis (CHCl3) λmax, nm (lgε): 517 (4.24), 554, (3.97), 592 (3.79), 650 (3.72).
Synthesis of 5-(4′-aminophenyl)-10,15,20-triphenylporphyrin (H2P1)
SnCl2 × 2H2O (350 mg, 1.5 mmol) and 5-(4′-nitrophenyl)-10,15,20-triphenylporphin (H2P4) (345 mg, 0.5 mmol) was stirred in 15 ml of 35% HCl. The reaction mixture was heated at 65 °C for 1 h. Then 25 g of ice was added and the reaction mixture was neutralized with aqueous ammonia. The precipitate was filtered, dried at 90 °C and purified by column chromatography on silica gel. H2P1 was obtained as the second fraction eluted by dichloromethane. Yield: 224 mg (71%). 1H NMR (CDCl3) δ ppm: 8.93 (d, 3 J = 4.25 Hz, 2H, β-H); 8.82 (s, 6H, β-H); 8.20 (d, 3 J = 6.7 Hz, 6H, 2′,6′-Ph); 7.98 (d, 3 J = 7.95 Hz, 2H, 2′,6′-PhNH2); 7.67–7.82 (m, 9H, 3′,4′,5′-Ph); 7.05 (d, 3 J = 7.95 Hz, 2H, 3′,5′-PhNH2); 4.01 (s, 2H, –NH2); − 2.75 (brs, 2H, –NH). UV/Vis (CHCl3) λmax, nm (log ε): 420 (5.43); 516 (4.27); 554 (4.12); 648 (3.98).
Synthesis of 5-(4′-N-tertbutyloxycarbonylglicinamidophenyl)-10,15,20-triphenylporphyrin (H2P3)
A mixture of 5-(4′-aminophenyl)-10,15,20-triphenylporphin (H2P1) (250 mg, 0.4 mmol), di-tert-butyldicarbonate (154 mg, 0.9 mmol), 1-(3′-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (173 mg, 0.85 mmol) and triethylamine (0.1 ml, 0.44 mmol) was stirred in 80 ml dichloromethane at 0 °C for 1 h. The reaction mixture was heated to room temperature for 3 h. The solvent was evaporated and the residue was purified by flash chromatography using dichloromethane as the eluent. Yield: 310 mg (75%). UV/Vis (CHCl3) λmax, nm (log ε): 420 (5.05), 516 (3.85), 551 (3.69), 591 (3.59), 647 (3.59); 1H NMR (CDCl3) δ ppm: 8.89–8.82 (m, 8H, β-H); 8.25–8.15 (m, 6H, 2′,6′-Ph, 2H, 2′,6′-PhGly); 7.93 (d, 3 J = 8.25 Hz, 2H, 3′,5′-PhGly); 7.80–7.71 (m, 9H, 3′,4′,5′-Ph); 5.58 (brs, 1H, –NHCO); 4.11, 3.95 (d, 2H, –CH2–); − 2.78 (s, 2H, –NH). MS (MALDI TOF) m/z: Calcd. 786.33 for C51H42N6O3. Found experimentally: 787.22 [M + H]+. (The 1H NMR spectra of the compounds are presented on Fig. ESM_1; MALDI TOF spectra are presented on Fig. ESM_2 of the Supplementary material).
Synthesis of 5-(4′-glycinacylamidophenyl)-10,15,20-triphenylporphyrin (H2P2)
5-(4′-N-tertbutoxycarbonylglycinamidophenyl)-10,15,20-triphenylporphin (H2P3) was dissolved in a mixture of 2 mL dichloromethane and 2 mL trifluoroacetic acid. The mixture was kept at room temperature for 4 h. The reaction mixture was poured onto ground ice and neutralized with an aqueous ammonia solution. The target product was extracted by 3 × 25 mL of dichloromethane. The organic extract was dried over Na2SO4, the solvent was removed by vacuum distillation. The product was purified by column chromatography on silica gel (with dichloromethane as the eluent). Yield: 91%. UV/Vis (CHCl3) λmax, nm (log ε): 419 (5.16), 514 (4.07), 551 (4.03), 591 (3.94), 647 (3.95); 1H NMR (CDCl3) δ ppm: 8.94–8.83 (m, 2H, β-H); 8.86–8.79 (m, 6H, β-H); 8.61–8.44 (m, 2H, 2′,6′-PhGly); 8.21 (d, 6H, 3 J = 8.00 Hz, 2′,6′-Ph); 7.99 (d, 3 J = 8.00 Hz, 2H, 3′,5′-PhGly); 7.81–7.70 (m, 9H, 3′,4′,5′-Ph); 5.57 (brs, 1H, –NHCO); 4.02 (s, 2H, –CH2–); − 2.78 (s, 2H, –NH). MS (MALDI TOF) m/z: Calcd. 686.28 for C46H34N6O. Found experimentally: 686.23 [M + H]+. (The 1H NMR spectra of the compounds are presented on Fig. ESM_3; MALDI TOF spectra are presented on Fig. ESM_4. of the Supplementary material).
The solvents (concentrated perchloric acid, and acetonitrile, DMSO, 4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo[8.8.8]hexacosane (cryptand [222])) and KOH were purchased from Sigma-Aldrich Co. and used without further purification (Scheme 1).
General experimental methods and instrumentation
The NMR spectra were obtained on a Bruker Avance 500 MHz spectrometer. Tetramethylsilane (TMS) was used as an internal standard. The mass spectra were recorded using an HP5989A apparatus (CI and EI, 70 eV ionisation energy) with an Apollo 300 data system or a Thermo Finnigan LCQ Advantage apparatus. A Shimadzu UV-1800 spectrophotometer was used to obtain the ground state absorption spectra, carry out the spectrophotometric titration experiments, and make the kinetic measurements. The methods of the titration procedure and protocols of experimental data analysis were described in detail in our previous papers [7, 8, 11]
Results and discussion
Free base porphyrins synthesis
The synthesis of substituted porphyrins consists of several stages. Aminophenylporphyrins is almost impossible to obtain directly through aminobenzaldehyde condensation with pyrrole because the initial aminobenzaldehydes are very unstable. That is why asymmetric aminophenylporphyrins (H2P1) are normally obtained by reduction of the corresponding nitrophenylporphyrins (Scheme 2), which, in turn, are prepared with quite high yields through benzaldehyde and nitrobenzaldehyde condensation with pyrrole by the method similar to the one described in [17] or, in case of the p-isomer, by H2TPP nitration [18].

Synthesis of 5-(4′-N-tertbutyloxycarbonylglycinamidophenyl)-10,15,20-triphenylporphin (H2P3). Reagents and conditions: (i) NaNO2, TFA, rt, 5 min; (ii) SnCl2, HCl, 65 °C, 1 h; (iii) di-tertbutyl-dicarbonate, 1-(3′-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride, triethylamine, DCM, 0 °C 1 h, rt 3 h
Tin bichloride in hydrochloric acid or in polar solvents usually plays a role of the reducing agent in this reaction [19, 20]. The rather mild reduction conditions of mono-nitrophenylporphyrins are required. That is why in this case, the reduction is carried out in methanol at a temperature of 65 °C, according to [16,17,18,19,20,21], with complete reduction of the initial nitrophenylporphyrins and formation aminophenylporphyrins. Aminophenylporphyrins can be easily acylated by acid chlorides of acids [16] with the formation of acylamino derivatives. So, we obtained Boc-protected acylaminoporphyrin (H2P3) using this procedure.
The removal of the H2P3 porphyrin Boc-protection was carried out in a mixture of dichloromethane and trifluoroacetic acid (Scheme 3). The process was controlled by thin-layer chromatography. Since there were significant differences in the mobility of the initial H2P3 porphyrin and the H2P2 product, we were able not only to clearly identify the hydrolysis completion point, but also to successfully separate the obtained reaction mixture from the trace amounts of the initial H2P3 porphyrin and products with a nonporphyrin structure during the chromatographic purification.

Removal of the protective group in the H2P3 porphyrin
Formation of mono- and doubly-protonated porphyrins species
The concept of acid–base properties of tetrapyrrole macrocycles includes the formation of anionic and cationic forms of an acid–base nature, proceeding with a charge change due to proton exchange in alkaline or acidic media.
The dissociation processes of the cationic forms of the synthesized H2P1, H2P2, and H2P3 porphyrins in an acidic medium were studied in an acetonitrile (AN)—perchloric acid system (0.01 mol/L solution in acetonitrile) at 298 K. In these conditions, HClO4 with a high value of the dissociation constant in acetonitrile [22] is in a completely ionized form, and the protonation is carried out through a solvated proton. The equilibria in the solution (without taking into account the solvent) are described by Eqs. 1 and 2:
Here H2P, H3P+, and H4P2+ are the free base, mono- and di-protonated forms of the H2P1, H2P2, and H2P3 porphyrins, respectively.
The total value of the base ionization constant for the studied compounds in the AN–HClO4 system at 298 K was calculated by Eq. (3):
Here Kb is Kb1 or Kb2—protonation constants of the first and second stages, Ind is the [H2P]/[H3P+] or [H3P+]/[H4P2+] indicator ratio, pH is the analytical value of the solution acidity created by the titrant. The values were determined based on the earlier obtained data of the spectropotentiometric study of the pH-function of a glassy electrode [23]. These data were used to calculate the protonation constants. The calculation error did not exceed 3–5%.
The UV/Vis spectral changes during the titration process and titration curves of the porphyrins under study are shown in Figs. 1–8, Table 1.

Changes in the electronic absorption spectra (A) and spectrophotometric titration curve (λ 415 nm) (B) for H2P1 in the AN–HClO4 system (Cporph = 1.21·10–5 mol/L; CHClO4 = 0 ÷ 1.51·10–4 mol/L), 298 K

Changes in the electronic absorption spectra in the H2P1–AN–HClO4 system (A), dependence of the indicator ratio on the perchloric acid concentration logarithm (B) (CHClO4 is from 5.01·10–7 to 3.16·10–6 mol/L)

Changes in the electronic absorption spectra in the H2P1–AN–HClO4 system (A), dependence of the indicator ratio on the perchloric acid concentration logarithm (B) (CHClO4 is from 2.51·10–6 to 7.94·10–6 mol/L)

Changes in the electronic absorption spectra in the H2P1–AN–HClO4 system (A), dependence of the indicator ratio on the perchloric acid concentration logarithm (B) (CHClO4 is 7.94·10–6 ÷ 1.25·10–5 mol/L). A detailed analysis of the changes in the EAS of the H2P2 and H2P3 compounds has shown that they form only mono- and di-deprotonated forms

Changes in the electronic absorption spectra (A) and curve of spectrophotometric titration (λ 415 nm) (B) for H2P2 in the AN–HClO4 system, (Cporph is 6.05·10–6 mol/L; CHClO4 is 0 ÷ 1.70·10–5 mol/L), T = 298 K. One proton is involved in each stage of the reaction. As in case of the H2P1 porphyrin, the difference between the constants of basic dissociation of the protonated H2P2 forms is within one order of magnitude

Changes in the electronic absorption spectra in the H2P2-10,15,20-triphenylporphin–AN–HClO4 system (A), dependence of the indicator ratio on the perchloric acid concentration logarithm (B) (CHClO4 is 3.47·10–6 ÷ 1.70·10–5 mol/L)

Changes in the electronic absorption spectra (A) and curve of spectrophotometric titration (λ 414 nm) (B) H2P3 in the AN–HClO4 system (Cporph is 1.31·10–5 mol/L; CHClO4 is 0 ÷ 1.26·10–2 mol/L), T = 298 К

Changes in the electronic absorption spectra in the H2P3–AN–HClO4 system (A), dependence of the indicator ratio on the perchloric acid concentration logarithm (B) (CHClO4 is 1.17·10–5 ÷ 2.57·10–4 mol/L)
As expected, the protonation of two imine nitrogen atoms of the porphyrin macrocycle leads to significant changes of its optical spectra, namely a bathochromic shift of the Soret band, a decrease in the number of bands to two for the dication in the visible part of the spectrum, and an increase in the intensity of band I.
Spectrophotometric studies of asymmetrically-substituted H2TPP derivatives in the AN–HClO4 system have shown that at a higher concentration of perchloric acid, the electronic absorption spectra reflected the appearance of several families of spectral curves, each with its own set of isosbestic points, which indicated that the protonation process consisted of multiple stages. The constants of base ionization and spectral characteristics of molecular and ionized forms of the H2P1, H2P2, and H2P3 porphyrins at T = 298К are given in the Table 1.
5-(4′-aminophenyl)-10,15,20-triphenylporphine H2P1 was used as the reference compound to evaluate the effect of asymmetric substitution on the base properties of the porphyrin macrocycle because there are data on its basicity in a variety of solvents obtained by different methods in the literature [24, 25]. However, in the binary AH—HClO4 solvent, we observed interesting features when studying the basic properties of H2P1 (Fig. 1).
We were able to separate the processes of formation of the H2P1 ionized forms.
The electronic absorption spectra and titration curves were gradually considered depending on the formation of isosbestic point families. We identified the families of the spectral curves and evaluated the clarity of the isosbestic points in order to determine the constants of monoprotonated forms.
An analysis of the UV/Vis spectral changes (presence of several families of isosbestic points) and the titration curve (with three regions in it) indicates that the substituent nitrogen atoms first involves in the protonation (pKb = 13.26) (Fig. 2), and then the central atoms of the macrocycle (pKb1 = 11.50; pKb2 = 9.65; pKb1,2 = 21.15) (see the Table 1).
The spectral changes occurring during the acid–base interaction at the macrocycle periphery can be represented by Eq. (4):
This hypothesis was also confirmed by the changes in the band intensity of the electronic absorption spectra, whereas the spectrum type itself did not change, which is, evidently, associated with the preservation of the compound initial symmetry. The linear logarithmic dependence of the indicator ratio on CHClO4 with the curve slope remains equal to 1, which indicates the macromolecule interaction with one proton (Fig. 2, Fig. ESM_5 of the Supplementary material).
To exclude acid–base interactions involving protons of the reaction center, the palladium complex of 5-(4′-aminophenyl)-10,15,20-triphenylporphyrin was titrated in the same concentration intervals. At the same time, similar spectral changes were observed. The observed changes clearly indicate acid–base interactions at the periphery of the macrocycle (Fig. ESM_6 of the Supplementary material).
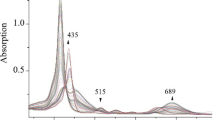
At even higher concentrations of perchloric acid, the “depth of protonation” is likely to increase, which leads to the appearance of a new isosbestic points family (at λ = 435, 501, 605 nm), corresponding to a series of spectral curves (Fig. 3). At the same time, the intensity of the Soret band at λ 415 nm becomes lower, a new peak appears at λ 453 nm, and in the region of Q bands—a high-intensity band appears at λ 689 nm and the intensity of the bands at 500–600 nm decreases.
The absorption band I is especially sensitive to structural changes. It is known that in a number of porphyrins, the shift of band I in the UV/Vis spectrum in the protonation can be used to characterize the extension of its chromophore system [26]. For example, it can be applied to evaluate the degree of involvement of the π-electrons of the meso-substituents in the general conjugation circuit if such conjugation is at all possible. In the case of 5-(4′-aminophenyl)-10,15,20-triphenylporphine H2P1, the bathochromic shift of band I may indicate the presence of the mesomeric effect between the phenyl fragment containing the protonated amino-group and the porphyrin macrocycle and, consequently, the extension of the π-electronic system of the H2P1 compound.
The process shown in Fig. 3, evidently, corresponds to the formation of a monoprotonated form of H3P1+ (Scheme 4):

Molecular structure of the porphyrin macrocycle and protonated forms corresponding to it. The bold line shows the main isoelectronic system of conjugation the dashed line denotes the intramolecular hydrogen bonds, the compound symmetry type is given in brackets [1]
The main isoelectronic systems of conjugation of the neutral (H2P) and protonated (H3P+ and H4P2+) forms of the porphyrin macrocycle include 18 π-electrons but, as Scheme 3 shows, differ from each other in their size, charge, and symmetry [23, 27]. Therefore, all the acid–base forms of the porphyrins are intensely coloured and have typical differences in the UV/Vis spectrum [1, 26]. For example, when the molecule becomes more symmetric, the spectrum shows two long-wave bands in the Q-range instead of four.
Further acid titration of H2P1 leads to the formation of a diprotonated form at a higher titrant concentration, which is confirmed by the appearance of a third series of spectral curves with its own family of isosbestic points (Scheme 4, Fig. 4).
From the shape of the H2P2 and H2P3 titration curve, one can distinguish two stages during spectrophotometric titration (Figs. 5, 6, 7, and 8, the Table 1, Fig. ESM_7, ESM_8 of the Supplementary material) characterizing the formation of mono- and di-cationic forms (processes (1) and (2)).
It is known that the introduction of electron-acceptor groups to the molecule periphery (directly or through the phenyl rings) affects the electron density of the transannular nitrogen atoms. Thus, protonated amino-groups reduce the electron density on the pyrrolenine nitrogen atoms due to the negative induction effect, which weakens the nitrogen–hydrogen interaction and acid properties of the protonated forms of porphyrin molecules [27]. The asymmetric character of substitution facilitates the spectral separation of charged forms. That is why it is quite natural that the base properties of H2P1 are weaker than those of H2P2 by about 3.5 orders of magnitude. The lower values of the protonation constants of H2P3 are evidently associated with the possibility to form intramolecular hydrogen bonds inside the substituent fragment, which reduces the electron-donor effect of the substituent and electron density in the molecule reaction centre. Similar porphyrin structures are described in detail in work [28].
Formation of mono- and doubly-deprotonated porphyrins species
The choice of a medium is decisive of the anionic interactions study. The regions containing molecules in anionic forms can differ significantly from each other, depending on the solvent nature, and therefore the acidity scale.
An analysis of the literature and our own data related to acidic properties of macrocycles [27,28,29] shows that dimethyl sulfoxide (a dipolar aprotic solvent) is the most promising solvent for studying deprotonation processes. In this work, the processes of acid dissociation of the synthesized H2P1, H2P2, and H2P3 porphyrins in an acidic medium were studied in a potassium cryptate (KOH[222])–DMSO system (a 0.01 mol/L solution) at 298 K. The KOH[222] solution preparation procedure was similar to the one described in [29]: KOH granules were dissolved in DMSO in the presence of 4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo[8.8.8]hexacosane (cryptand [222]).
The equilibria in the solution (without taking into account the solvent) are described by Eqs. 5 and 6:
Here H2P, HP− and P2− are the free base, mono- and di-protonated forms of the H2P1, H2P2, and H2P3 porphyrins, respectively.
The total value of the acid dissociation constant of the studied compounds in the KOH[222])–DMSO system at 298 K was calculated by Eq. (7):
Here Ka, Ka1 and Ka2 are, respectively, the total deprotonation constant of the first and second stages and those of the first and second stages separately; Ind is the [HP−]/[H2P] or [P2−]/[H2P2] indicator ratio, lgCKOH[222] is the analytical basicity value of the KOH[222] titrant solution in DMSO. These data were used to calculate the deprotonation constants. The calculation error did not exceed 3–5%.
The changes in the electronic absorption spectra of the studied porphyrins during the titration, values of the acid dissociation constants, titration curves, and dependences of the indicator ratio on the КOH[222] concentration logarithm are given in Figs. 9, 10, and 11A, B and in the Table 1.

Changes in the electronic absorption spectra (A, C) and curve of spectrophotometric titration (λ 420 nm) (B) for H2P1 in the DMSO–КOH[222] system (Cporph is 1.34·10–5 mol/L; CKOH[222] is 0 ÷ 3.16·10−3 mol/L), 298 К

Changes in the electronic absorption spectra (A, C), curve of spectrophotometric titration (λ 420 nm) (B) for H2P2 in the DMSO–КOH[222] system (Cporph is 0.95·10–5 mol/L; CKOH[222] is 0 ÷ 6.03·10−3 mol/L), 298 К

Changes in the electronic absorption spectra (A, C), curve of spectrophotometric titration (λ 420 nm) (B), and dependence of the indicator ratio on the KOH[222] concentration logarithm (D) for H2P1-H2P3 in the DMSO–КOH[222] system (Cporph is 1.31·10–5 mol/L; CKOH[222] is 0 ÷ 2.19·10−2 mol/L), 298 K
Spectrophotometric studies of asymmetrically substituted tetraphenylporphyrin derivatives in the KOH[222]–DMSO system have shown that at higher KOH[222] concentrations, the electronic absorption spectra have several families of spectral curves, each with its own set of isosbestic points (for the H2P1: 436, 471, 659 nm and 439, 473, 662 nm; for H2P2: 379, 429, 557 nm and 382, 431, 560 nm; for H2P3: 345, 486, 598, 733 nm and 373, 430, 628, 669 nm), which also confirms that the acid dissociation of the protonated forms represents a two-stage process.
However, the spectrophotometric titration curves based on the experimental data cannot be clearly distinguished into stages, which do not mean that the ionization consisted of one stage but suggests that the values of the protonation constants of the equilibria were close to each other. The presence of isosbestic points and the character of changes in the absorption spectra indicate that as the concentrations of the two centers of the porphyrin molecule absorption changed, the ratio between the ionized forms during the porphyrin deprotonation remained the same. The linear dependence of the indicator ratio on the lgCKOH[222] with the slope tg ~ 2 indicates the separation of two protons from one ligand macromolecule (Fig. 11B). The calculated acidity constants for H2P1, H2P2, and H2P3 in the KOH[222]–DMSO system had similar values of the total pKa1,2 deprotonation constant (see the Table 1).
The difference between the acid dissociation constants is within one order of magnitude (as shown above, the maximum difference between the basicity constants in the studied porphyrins is 3.5 orders of magnitude) because acid–base interactions with the functional groups at the macrocycle periphery are impossible. In this case, evidently, the effect of DMSO (DNDMSO = 29.8, compared with DNAN = 14) [30]) on the deprotonation equilibria is rather controversial because DMSO does not only act as the medium, but also competes with the titrant anion forming stable associates with the protons of the neutral ligand reaction centre [31]. It should be said that the solvent has practically no additional stabilizing effect on HP− in the DMSO–KOH[222] system due to the electron-donor nature of the solvent (similar results were obtained by the authors of [27] when studying tetraphenylporphyrin). Thus, the pKa1 and pKa2 values are similar in the studied H2P1, H2P2, H2P3 ligands, which has allowed us to determine only the total acidity constant value for the two ionization stages.
Conclusion
We have carried out targeted synthesis of asymmetrically tetraphenylporphyrin derivatives substituted with amino-acid residues acting as the functional group. Such residues can be used as “anchor” groups for building into the structure of the protein molecule. Both well-known literature methods and our own original techniques were employed in the multi-stage synthesis, which allowed us to achieve high yields of the target products. The obtained compounds were characterized by a number of spectral methods confirming their structure and purity. We have measured the base and acid ionization constants of 5-(4′-aminophenyl)-10,15,20-triphenylporphyrin (H2P1), 5-(4′-glycinacylamidophenyl)-10,15,20-triphenyl-porphyrin (H2P2) and 5-(4′-N-tertbutoxycarbonylglycinamidophenyl)-10,15,20-triphenylporphyrin (H2P3) by the spectrophotometric titration method. It has been established that protonation of 5-(4′-aminophenyl)-10,15,20-triphenylporphyrine in the AN–HClO4 system first proceed at the molecule periphery and then the central nitrogen atoms of macrocycle. The asymmetric character of substitution contributed to the stabilization of the protonated forms, which allowed us to identify and spectrally characterize H3P+ and H4P2+ for each of the porphyrins. The acid ionization constants were determined in the DMSO–KOH[222] system. The electron-donor nature of the solvent of this liquid-phase system levelled off the ionization constants of the first and second stages, which allowed us to determine only their total values for the H2P1, H2P2, and H2P3 porphyrins. The obtained results are useful for a better understanding of the proton transfer processes involving porphyrins in natural systems. They broaden the fundamental concepts of bioporphyrins in vivo functioning and enable the simulation of biochemical processes with porphyrins in a complex multi-component donor–acceptor environment based on them.
References
Berezin, B.D.: Coordination Compounds of Porphyrins and Phthalocyanines. Wiley, New York (1981)
Buchler, J.W.: Synthesis and properties of metalloporphyrins. In: Dolphin, D. (ed.) Porphyrins. Academic Press, New York (1978)
Röder, B., Büchner, M., Rückmann, I., Senge, M.O.: Correlation of photophysical parameters with macrocycle distortion in porphyrins with graded degree of saddle distortion. Photochem. Photobiol. Sci. (2010). https://doi.org/10.1039/C0PP00107D
Hambright, P.: Chemistry of water soluble porphyrins. In: Porphyrin Handbook. Acad. Press, New York (2000)
Rocheva, T.K., Shevchenko, O.G., Mazaletskaya, L.I., Sheludchenko, N.I., Belykh, D.V.: The antioxidant properties of asymmetrically substituted tetra(meso-aryl)porphyrins with one phenolic substituent: the contribution of phenol and porphyrin fragments to antioxidant activity. Macroheterocycle. (2018). https://doi.org/10.6060/mhc170302b
Figueira, F., Pereira, P.M.R., Silva, S., Cavaleiro, J.A.S., Tome, J.P.C.: Porphyrins and phthalocyanines decorated with dendrimers: synthesis and biomedical applications. Curr. Org. Synth. (2014). https://doi.org/10.2174/15701794113106660089
Dao, T.N., Ivanova, Yu.B., Puhovskaya, S.G., Kruk, M.M., Syrbu, S.A.: Acid-base equilibria and coordination chemistry of the 5,10,15,20-tetraalkyl-porphyrins: implications for metalloporphyrin synthesis and sensor design. J. RSC Adv. (2015). https://doi.org/10.1039/c5ra01323b
Pukhovskaya, S., Ivanova, Yu., Dao, T.N., Vashurin, A., Golubchikov, O.: Coordination and acid-base properties of meso-nitro derivatives of β-octaethylporphyrin. J. Porphyrins Phthalocyanines. (2015). https://doi.org/10.1142/S1088424615500649
Kruk, M.M., Pukhovskaya, S.G., Ivanova, Yu.B., Koifman, O.I.: Enthalpy-entropy compensation upon metal ion coordination with porphyrins: generalization for the free bases and doubly deprotonated macrocycles. Russ Chem Bull (2020). https://doi.org/10.1007/s11172-020-2868-6
Senge, M.O.: Exercises in molecular gymnastics bending, stretching and twisting porphyrins. Chem. Commun. (2006). https://doi.org/10.1039/b511389j
Kielmann, M., Senge, M.O.: Molecular engineering of free base porphyrins as ligands—the N–H… X binding motif in tetrapyrroles. Angew. Chem. Int. l Edition. (2019). https://doi.org/10.1002/anie.201806281
Dao, T.N., Pukhovskaya, S.G., Ivanova, Y.B., Liulkovich, L.S., Semeikin, A.S., Syrbu, S.A., Kruk, M.M.: Porphyrin acidity and metal ion coordination revisited: electronic substitution effects. J. Inclusion Phenom. Macrocycl. Chem. (2017). https://doi.org/10.1007/s10847-017-0758-9
Vicente, M.G.H., Smith, K.M.: Syntheses and functionalizations of porphyrin macrocycles. Curr. Org. Synth. (2014). https://doi.org/10.2174/15701794113106660083
Syrbu, S.A., Ageeva, T.A., Kolodina, E.A., Semeykin, A.S., Koifman, O.I.: Strategies for the synthesis of porphyrin monomers. J. Porphyrins Phthalocyanines (2006). https://doi.org/10.1142/S1088424606000235
Ogashi, N., Nomura, A., Kodera, M., Hitomi, Y.: Structurally simple cell-permeable porphy rins: efficient cellular uptake and photo-toxicity of porphyrins with four peripheral primary-amine-terminated oligo(ethylene oxide) chain. Chem. Lett. (2017). https://doi.org/10.1246/cl.170821
Lyubimtsev, A., Semeikin, A., Zheglova, N., Sheinin, V., Kulikova, O., Syrbu, S.: Synthesis and photophysicalproperties of low symmetrical porphyrin-amino acid conjugates and their Zn complexes. Macroheterocycles (2018). https://doi.org/10.6060/mhc171151l
Gerasimova, O.A., Milaeva, E.R., Shpakovsky, D.B., Semeykin, A.S., Syrbu, S.A.: Synthesis of 5-(4-hydroxyphenyl)-10,15,20-tris[3,5-di(tert-butyl)-4- hydroxyphenyl ]porphine and 5-(4-palmitoyloxyphenyl)-10,15,20-tris[3,5-di(tert-butyl)-4- hydroxyphenyl]porphine and generation of phenoxyl radicals from them. Russ. Chem. Bull. Int. Ed. (2007). https://doi.org/10.1007/s11172-007-0124-y
Luguya, R., Jaquinod, L., Fronczek, F.R., Vicente, M.G.H., Smith, K.M.: Synthesis and reactions of meso-(p-nitrophenyl)porphyrins. Tetrahedron (2004). https://doi.org/10.1016/j.tet.2004.01.080
Collman, J.P., Gagne, R.R., Halbert, T.R., Marchon, J.C., Reed, A.C.: Reversible oxygen adduct formation in ferrous complexes derived from a picket fence porphyrin. Model for oxymyoglobin. J. Am. Chem. Soc. (1973). https://doi.org/10.1021/ja00804a054
Meng, S., Xu, Z., Hong, G., Zhao, L., Zhao, Z., Guo, J., Ji, H., Liu, T.: Synthesis, characterization and in vitro photodynamic antimicrobial activity of basic amino acid–porphyrin conjugates. Eur. J. Med. Chem. (2015). https://doi.org/10.1016/j.ejmech.2014.12.029
Palka, A., Czuchajowski, L.: Porphyrins containing aziridinyl-p-benzoquinone substituents. Chem. Lett. (1994). https://doi.org/10.1246/cl.1994.547
Kolthoff, I.M., Chantooni, M.K., Sadhana, I.R.: Titration of bases in acetonitrile. Anal. Chem. (1967). https://doi.org/10.1021/ac50156a039
Pukhovskaya, S.G., Ivanova, Yu.B., Nam, D.T., Vashurin, A.S.: Dependence of the basic properties of meso-nitro-substituted derivatives of β-octaethylporphyrin on the nature of substituents. Russ. J. Phys. Chem. (2014). https://doi.org/10.7868/S004445371410032X
Gündüz, N., Gündüz, T., Hayvali, M.: Titrations in non-aqueous media: potentiometric investigation of symmetrical and unsymmetrical tetra-aryl porphyrins with 4-nitrophenyl and 4-aminophenyl substituents in nitrobenzene solvent. Talanta (1999). https://doi.org/10.1016/s0039-9140(98)00222-7
Ermolina, E.G., Kuznetsova, P.T., Semenishin, N.N.: Acid-base properties and phototransformations of complexone(ate)-substituted tetraphenylporphyrin. Russ. J. Phys. Chem. A. (2012). https://doi.org/10.1134/S0036024412090026
Andrianov, V.G., Malkova, O.V.: Acid-base properties of porphyrins in non-aqueous solution. Macroheterocycles. (2009). https://doi.org/10.6060/mhc2009.2.130
Vitasovic, M., Gouterman, M., Linschitz, H.: Calculations on the origin of hyperporphyrin spectra in sequentially protonated meso-(dimethylaminophenyl) porphyrins. J. Porphyrins Phthalocyanines (2001). https://doi.org/10.1002/jpp.309
Sweed, A.M.K., Senge, M.O., Atta, S.M.S., Farrag, S.D., Abdel-Rahman, H., Shaker, Y.M.: Synthesis of amphiphilic meso-tetrasubstituted porphyrin-L-amino acid and -heterocyclic conjugates based on m-THPP. J. Porphyrins Phthalocyanines (2018). https://doi.org/10.1142/S1088424618500979
Sheinin, V.B., Ivanova, Y.B.: The acid properties of benzodiamyloxyl and thiadiazole porphyrazine derivatives in the H2L-(K[2.2.2])OH-DMSO system. Russ. J. Phys. Chem. A. (2007). https://doi.org/10.1134/S0036024407080134
Burger, K.: Solvation, ionic reactions and complexation in non-aqueous media. Moscow (1984)
Stuzhin, P.A., Malyasova, A.S., Kokareva, E., Tarakanov, P.A., Koifman, O.I., Sheinin, V.B.: Acid-base properties of tetrapyrazinoporphyrazines. 1. Deprotonation of octaethyltetrapyrazinoporphyrazine in CH2CL2, THF, DMSO and pyridine the crucial role of water. Dyes Pigm. (2017). https://doi.org/10.1016/j.dyepig.2016.12.047
Acknowledgements
The work was financially supported by the Russian Foundation for Basic Research, grant No.19-03-00214 A, and was carried out on the equipment of the Upper Volga Region Center of Physico-Chemical Research.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ivanova, Y.B., Pukhovskaya, S.G., Lyubimtsev, A.V. et al. Spectral studies of protonated and anionic forms of porphyrins with an asymmetric substitution system. J Incl Phenom Macrocycl Chem 102, 493–505 (2022). https://doi.org/10.1007/s10847-022-01131-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10847-022-01131-8