
Abstract
The goal of the Statistical Assessment of the Modeling of Proteins and Ligands (SAMPL) challenge is to improve the accuracy of current computational models to estimate free energy of binding, deprotonation, distribution and other associated physical properties that are useful for the design of new pharmaceutical products. New experimental datasets of physicochemical properties provide opportunities for prospective evaluation of computational prediction methods. Here, aqueous pKa and a range of bi-phasic logD values for a variety of pharmaceutical compounds were determined through a streamlined automated process to be utilized in the SAMPL8 physical property challenge. The goal of this paper is to provide an in-depth review of the experimental methods utilized to create a comprehensive data set for the blind prediction challenge. The significance of this work involves the use of high throughput experimentation equipment and instrumentation to produce acid dissociation constants for twenty-three drug molecules, as well as distribution coefficients for eleven of those molecules.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Drug discovery and development processes are under increased pressure to deliver medicines and vaccines to patients faster than ever. The demand to have robust and efficient clinical chemistry, manufacturing, and control (CMC) strategies is the main driving factor in the implementation of new approaches which allow for faster experimentation without sacrificing the quality of the results. Inspired by biological screening, chemical development of new active pharmaceutical ingredients (APIs) has been leveraging parallel experimentation over the past few decades to disrupt the approach that scientists adopt to investigate the chemical and formulation space. Design of Experiments and advanced statistical tools are essential to design and evaluate results in an efficient manner [1]. In fact, the results of these studies generate comprehensive datasets across multiple continuous variables and factors which can be fed to modeling algorithms [2,3,4,5]. This provides further insight and knowledge on the effect of multiple variables on the target process and can help identify critical operating parameters. Combining High Throughput Experimentation (HTE) with computational modeling may prove to be an effective tool for visualizing and reporting results with a fully traceable and consistent methodology [6,7,8]. As these benefits are realized, the reach of HTE has extended into other areas of research such as chemical synthesis optimization [4, 9] and conducting drug solubility assessments in various media [8, 10,11,12,13,14].
The determination of API partitioning in aqueous and organic media is one of the key steps in developing new synthetic routes and to determine the bioavailability of the drug substance upon administration to the patient [15]. When investigating new chemical processes, the partitioning of impurities and active ingredients is sometimes the costliest unit operation in chemical development [16]. For this reason, the determination and modeling of this parameter during the early phase of drug development can accelerate and simplify the control strategy for process quality and robustness.
Within this context, Statistical Assessment of the Modeling of Proteins and Ligands (SAMPL) is a series of blind challenges that bring together scientists on a global scale to improve the capability of current computational methods in drug discovery. This collaborative approach aims to better facilitate development of the next-generation computational models that can be used as predictive tools in drug discovery. Various iterations of SAMPL over the last decade have focused on evaluating how well physical and empirical modeling methodologies can predict several physicochemical properties of drugs that can be used to aid in drug discovery, such as hydration free energies, acid dissociation, and partition and distribution coefficients [17,18,19,20,21,22,23,24,25,26,27]. The aim of the SAMPL8 challenge is to assess quantitative accuracies of current methods and isolate deficiencies with the advantage of access to a larger database of pharmaceutical compounds provided by GlaxoSmithKline, which created a comprehensive data set to be used for evaluating new prediction methods. In this study, research was focused on creating a standard data set of solubility-based pKa and pH-dependent distribution coefficients for various immiscible solvent combinations by exploiting laboratory automation and HTE.
Distribution coefficients are values which describe the behavior of solutes in two immiscible liquids, and account for the total concentration of ionized and unionized drug in both the aqueous and organic phases [28]. The distribution coefficient is often used to understand whether a drug is more hydrophilic (drawn to aqueous systems) or hydrophobic (drawn to organic or lipophilic systems). This in turn, helps predict the movement of the drug through the lipid bi-layer for absorption into the bloodstream. The distribution coefficient (logD) is defined as the ratio of the sum of concentrations of both charged and neutral species in the organic and aqueous phases. This differs from the partition coefficient (logP) since the latter only accounts for the ratio of neutral species in organic and aqueous phases. The differences can be seen below in Eqs. (1) and (2), describing the two quantities.
Equation 1. Partition Coefficient
Equation 2. Distribution Coefficient
Prior to beginning the process of estimating distribution coefficients, the pKa of each compound was first determined using an optimized HTE workflow. pKa is the acid dissociation constant which is used to estimate the pH at which a compound will be optimally dissolved [29]. The pKa of a compound affects the fraction of molecules being ionized, which in turn affects the solubility of the compound in aqueous media since ionized molecules are more soluble in aqueous media than neutral molecules. Using the Henderson-Hasselbalch equation, a relationship between the solubility and the pKa can be established:
where S0 is the solubility of the neutral compound. Using the above equations, the macroscopic pKa can be derived for any compound as a function of the solubility. It also demonstrates that solubility is highly dependent on the pH of the solvent. The pKa can hence be used to determine the pH of aqueous phase during the computation of distribution coefficients, since it ensures solubility of the compound in aqueous phase.
Materials and methods
Compound nomenclature
F simplicity, each compound that is referred to in this manuscript is identified by the following nomenclature: “SAMPL8-X”. SAMPL refers to the entirety of the Statistical Assessment of the Modeling of Proteins and Ligands challenges. The number “8” denotes that this is the eighth SAMPL challenge iteration. A number follows, in place of the “X”, to identify the unique compound, or drug molecule. As will be described below, there are a total of twenty-three compounds analyzed in this investigation. They are listed in no particular order, and are numbered sequentially from 1 through 23, as “SAMPL8-1” through “SAMPL8-23”.
Compound selection
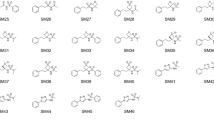
To assemble the set of compounds for this study, drug molecules registered by GlaxoSmithKline were identified as those associated with a compound collection enhancement project code (i.e., purchasable compounds) but not with an active program code. An additional requirement was that a minimum of 100 mg of solid was available in the compound stores. From this set of ~ 77,000 compounds, 88 were selected which contained two widely separated polar groups (separated by greater than three bonds), scaffolds often found in screening hits, and/or the presence of sulfonamide or sulfone (due to a lack of public ΔGtransfer data for such compounds [30]). Three of the selected compounds were matched molecular sets (SAMPL8-7, 8–9, and 8–17), with the intention of determining if there is a measurable role that small changes on a given scaffold would have on the experimental data. The entire list of compounds selected had a molecular weight ranging from 165 to 403 Dalton (Table 4) and zero to six rotatable bonds. Of these 88 compounds, some failed with visually observable degradation, while many others, which did progress to HTE testing, failed to exhibit a measurable pKa. Further to that, additional molecules were not progressed because they would not dissolve in any of the solvents selected for this study. The final list of 23 compounds is shown below (Fig. 1).

Molecules Used in the SAMP8 pKa Challenge
Buffer systems
For the pH-solubility studies, Britton-Robinson buffers were used (Ricca Chemical Company, Arlington, TX, USA). The buffers, listed below in Table 1, have an ionic strength of 0.1 M.
Solvent combination selection
By collecting logD measurements of small molecules in bi-phasic systems with a variety of organic solvents, the aim was to develop an opportunity to evaluate the performance of computational techniques for modeling solvation effects in different solvent environments. We reasoned that a common solute set measured in different solvent pairs will be helpful to understand which solvent systems can be modeled accurately by current computational methods, and which solvents may need more thorough selection to improve the experimental design approach.
Octanol–water is the most common bi-phasic system for logD measurements, and it has been used as a lipophilicity metric that predicts membrane partitioning of pharmaceutical compounds [31]. The octanol phase is known to be challenging for physical modeling techniques due to its conformational flexibility and tendency to form a heterogenous solvent phase with hydrophobic pockets (composed of lipophilic octyl tails) and hydrophilic pockets (composed of polar head groups and water molecules) [32, 33]. In the past, simpler solvents with more restricted conformational ensembles such as cyclohexane were preferred as a modeling test system with intermediate complexity to see the underlying capability of computational techniques when conformational sampling problem of the solvent is largely mitigated. The SAMPL5 cyclohexane-water logD prediction challenge, and the SAMPL6 octanol–water logP prediction challenge for physical modeling techniques, resulted in very different prediction accuracies. One examined that the SAMPL6 octanol–water logP predictions were more accurate in general [27, 34]. However, due to differences in predicted values (logP versus pH-dependent logD that depends on pKa predictions) and the number and identity of compounds in datasets, it was not possible to investigate where these performance differences stem from. This motivated the desire to collect a logD dataset of a common set of solutes with a variety of organic solvents-water pairs which will enable investigation of how well models can capture solvation in different organic solvents and how the chemical properties of organic solvents can impact the accuracy of logD predictions.
In the partitioning studies, seven organic solvents were selected. These solvents are immiscible with water to ensure that bi-phasic partitioning conditions could be met: octanol (OCTL), cyclohexane (CYHL), ethyl acetate (ETAC), heptane (HP), methyl ethyl ketone (MEK), tert butyl methyl ether (TBME) and dimethylformamide (DMF). Comparison of cyclohexane-water vs. heptane-water logD can show the effect of conformational flexibility [28]. We can learn about how modeling accuracy is affected by homogeneous and heterogeneous organic solvent phase by comparing the prediction performance for cyclohexane –water and heptane-water logD values to octanol–water logD values. Comparative evaluation of ethyl acetate, MEK, and TBME-water logD predictions can lead to conclusions about how models handle solvents with different polarity and hydrogen bond acceptor groups.
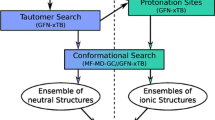
The goal was to employ an automated approach to measure the pKa as well as the distribution coefficient as visualized by the flowchart in Fig. 2. Customized experimentation was avoided in favor of developing standardized workflows due to the large number of compounds and solvent combinations that were selected for testing. As will be described in the section below, there were a substantial amount of experimental data generated in support of this investigation. For context, this publication provides details on the methods and analysis of more than 250 data points for the pH-solubility (pKa) portion, and slightly less than 1,000 data points for the logD portion.

Overview of the experimental steps involved in the computation of the distribution coefficient
Analytical method development
Solubility data used to obtain the acid dissociation and distribution coefficient data for the compounds was acquired using High Performance Liquid Chromatography (HPLC) analytical instrument. Hence the first phase involved development of analytical methods for HPLC. An Agilent 1290 HPLC instrument (Agilent Technologies, Santa Clara, CA, USA) was used to quantify the amount of solute present in different solutions. This was used in the measurement of the analyte in the different phases for distribution coefficient computation and for calculating the experimental pKa as well. A Waters X-Select Charge Surface Hybrid (CSH) C18, 2.1 mm × 30 mm, 5 μm column was used in the HPLC in a gradient elution mode. Standard solutions were prepared for each compound using a backing solvent consisting of 62.5% acetonitrile, 25% tetrahydrofuran, and 12.5% HPLC-grade water v/v to a target of 1 mg/mL concentration. Serial dilution was performed for the following calibration standards with the goal of having a total of five standards per curve at 1.0 mg/mL, 0.5 mg/mL or 0.3 mg/mL, 0.1 mg/mL, 0.01 mg/mL, and 0.001 mg/mL.
Chromatographic data is analyzed via Agilent ChemStation software with the offline data analysis version. The Unchained Labs CM3 platform Library Studio software communicates directly to the Agilent HPLC and prepares the chromatography plate sequence based on the library design. The HPLC sequence is initiated by an instruction from Unchained Labs Automation Studio software. Details of the chromatography data from the entire sequence, such as the retention time, peak height, integrated peak area, and the corresponding drug concentration are stored in ChemStation software where it can be further curated by the analyst.
pKa determination
The pKa was calculated by measuring the concentration of the compounds in Britton-Robinson buffers of various pH [2,3,4,5,6,7,8,9,10,11,12]. 1 mg of drug substance was added to 500 µL of buffer, with the overall workflow shown in Fig. 3. The experiments were primarily carried out in a high throughput manner on the Unchained Labs Freeslate CM3 robotic platform (Unchained Labs, Pleasanton, CA, USA) in high throughput microtiter plates (MTP).

Flow chart of the different steps involved in calculation of experimental pKas of compounds
The 96-well MTP plates contain 1 mL vials according to the layout in Fig. 4. A target of 1 mg of drug substance was weighed into 1 mL 96-well plate vials using a Mettler-Toledo Quantos QX96 automated powder dispensing platform (Mettler-Toledo GmbH, Greifensee, Switzerland). One Teflon-coated flea stir-bar was added to each vial to facilitate mixing of the constituents. 500 µL of buffer media was then added to each individual vial using a Rainin multichannel pipette, ensuring that the drug substance was in a saturated state before continuation. The vials were capped, and the entire 96-well plate was placed on an Unchained Labs Freeslate CM3 platform and stirred for 24 h at 500 RPM with temperature controlled at 22 °C. At the conclusion of stirring, the magnetic stir bars were removed from the vials and the supernatant from the samples were filtered using the Hamilton Microlab NIMBUS liquid handler (The Hamilton Company, Boston, MA, USA) through a Millipore 0.45-micron hydrophilic filter plate by way of plate centrifugation. A Thermo Lynx 4000 plate centrifuge was set for 5 min at 3500 RPM under controlled temperature conditions of 22 °C. The filtrate was diluted from the source plate using a Hamilton NIMBUS Microlab pipetting robot to prepare dilutions at 10X and 100X with (50:50 v/v) acetonitrile/water mixture as a diluent. An appropriate dilution factor was applied to ensure that the final concentration of the sample was within the linear range of the calibration curves collected, between 0.001 mg/mL to 1 mg/mL. At this point, the samples were ready for chromatography analysis on an Agilent 1290 HPLC. At the conclusion of each HPLC run, the plates were returned to the Freeslate CM3 to measure the final pH of the solutions for confirmation.

Typical MTP plate design for automated pH-Solubility experiments. Different colors along the columns represent the pH2-12 Britton-Robinson buffers. Six different compounds were added to the vials, one per row
pH-solubility was plotted using Synergy Software's Kaleidagraph data analysis application (Synergy Software, Reading PA) based on the chromatography results, and a curve was fitted for each compound, with a respective R2, using the Henderson-Hasselbalch solubility equations [35, 36]. The approach used for modeling the pKa was originally established by Jagannadham and Sanjeev, and later confirmed by Bahr et al., whereby the final pH of the solution is plotted against the log of the concentration, and the resulting plot is modeled against the solubility equations provided in Table 2 [13, 37]. Each curve was unique to the nature of the compound, for example there were unique equations for acids, bases and ampholytes as well as for mono- and di-protic acids and bases. The model curve for the pKa determination, chosen by the software, was based on the compound’s acidic or basic functional groups. The curve fitting equations used to solve the optimization and determine the pKa for each drug molecule are shown in Table 2.
The five equations listed in Table 2 are the Kaleidagraph software iterations of the Henderson-Hasselbalch solubility equation. The constants correspond to different ionization states of the compound (pKa values), while the pS0 term represents the solubility of neutral species.
Distribution coefficient protocol
Once the experimental pKa was determined for each molecule, automated experiments to measure the distribution coefficient could then be progressed as demonstrated in the flowchart illustrated in Fig. 5.

Flow chart of the different steps involved in determination of experimental distribution coefficients of compounds
The diagram shown in Fig. 6 illustrates the experimental design on the HTE platform. Each organic solution is placed on the Freeslate CM3 deck in 20 mL vials on an 8-well plate, along with dispensing heads for each of the drug substances (labeled “API”). Since a greater volume of aqueous buffer is used across all samples in this design, a larger 125 mL glass container holds the aqueous medium.

Depiction of a typical automated distribution coefficient design. Refer to the list of abbreviations for the names of the solvents used in the experiment. The 8-well plates contain 20 mL vials while the 2-well plate on the bottom left contains 125 mL vials. The solvents and the compounds are added to 24-well plates on the top containing 8 mL vials. Extracted samples are transferred into a 96-well plate containing 1 mL vials
As previously mentioned, seven different solvent combinations were selected. The Mettler Toledo Quantos was used to dispense powder into 8 mL vials which were assembled onto a 24-well plate. After each compound was dispensed into the vials, the organic and the aqueous phases were added respectively.
3 mL of each solvent phase was added to the vials containing compounds. The samples were vortexed for 30 min and then allowed to settle for 60 min. Once the solutions reached equilibrium, they were checked for any particulate in both the top and bottom phases to ensure that the drug was in solution. If particulate was still observed in the phases, the vials would be vortexed for an additional 30 min, then allowed to reach equilibrium. 500 µL of solution was drawn from the upper and lower phases of the 8 mL vials (Fig. 7) by way of a 22 gauge syringe needle attached to the Unchained Labs CM3 platform. This narrow-gauge needle is beneficial to reducing potential error due to the low surface area of the exposed needle, the positive air gap inside the needle capillary, and the presence of a septum on the vial cap which wipes away any errant solvent when the needle is withdrawn from the vial. The aliquots of solution were then individually transferred into separate 1 mL vials on the 96-well plate for HPLC analysis using the Agilent 1290 auto-sampler. The sampling height was optimized so that the needle withdraws the liquid at the mid-height position for each of the phases to avoid cross contamination due to liquid eddies that may potentially form due to the sampling needle creating turbulence at the solution interface. This ensured consistent and reproducible sampling conditions for every sample analyzed.

Image of an 8 mL vial showing the organic (top) and aqueous (bottom) phases
The aqueous phase used in the above experiments involved the use of Britton-Robinson buffers. The pH of the buffers used in the solution mixture was selected based on the pKa of the compound. This approach was necessary due to the low aqueous solubility of the drug substances in their neutral state, hence it was necessary to determine logD at a pH that the molecules were significantly ionized in the aqueous state to ensure total dissolution of the drug within the bi-phasic solvents. The samples were analyzed using the HPLC analytical instrument, which included a needle-wash for the HPLC injection needle to eliminate cross-contamination, and the distribution coefficient was computed using the equation below (Eq. 3):
Equation 3. Distribution Coefficient (logD)
In the case of cyclohexane and dimethylformamide, the cyclohexane was taken as the top phase and the dimethylformamide was taken as the bottom phase.
Results
As described in the Methods section, final pH was measured for each aqueous sample and was plotted against the logC (concentration). This data was fitted to the Henderson-Hasselbalch equation to determine the pKa. An example is shown in Fig. 8. The experimental pKa for the example of SAMPL8-7 was calculated to be 6.63 (R2 = 0.997).

Example table listing the data points obtained experimentally using pH-solubility experiments for a single compound (SAMPL8-7) and a plot of the data fitted with a monoprotic base version of the Henderson-Hasselbalch Equation to obtain the pKa of the compound
Settimo et al. reported that pKa predictors may provide a degree of inaccuracy due to a significant molecular weight difference between the generic organic molecules used in modeling software and the larger and more complex molecules typically found in drug research [38]. So for the investigation presented here, the authors believe that while predicted values are excellent starting points, they are not based on experimentally measured values, and therefore it is important to confirm the acid dissociation experimentally.
For each molecule studied in this investigation, ChemAxon JChem software with the pKa Plugin (ChemAxon, Budapest, Hungary) was used to predict pKa values based on the structure and functional groups of each molecule. The pH range of viability for modeling the pKa, based on the Henderson-Hasselbalch equation, is pH 5–9 [36]. Despite this, much of our experimental data provided sufficient information to fit calculated pKa values with a high R2 that were outside of that pH range.
In some cases of the molecules investigated, the experimentally determined pKa did not match the predicted values, and this was likely due to pKa values that were at the extreme ends of the upper or lower pH scale and therefore could not be experimentally measured. An example of this is SAMPL8-13, which had a predicted pKa of 13.86, and could not be confirmed experimentally.
Reijenga et al. investigated several methods historically used for pKa determination, and concluded that HPLC analysis is a strongly favorable approach with good precision, but is limited at the far ends of the lower and upper pH scale [39]. Their research also asserted that the use of HPLC instrumentation is a time-consuming experimental approach that is only effective if the analyte has a chromophore. For the work presented here, it was confirmed that all of the drug molecules did present a chromophore and therefore could be assayed using HPLC.
In pre-candidate selection experimental methodology at GSK, it is acceptable to perform high throughput studies without replicates when access to drug substance is limited. This experimental approach may typically sacrifice statistical significance in favor of generating volumes of data over a larger span of experimental design. For our research, the collaborators agreed to focus on as many bi-phasic solvent combinations as possible. The novelty of this work is in the use of automation to streamline the data collection process. So this same approach was used for all twenty-three compounds, with the results provided in Table 4.
We acknowledge that a lack of replicates could present a risk for uncertainty in the reported pKa values. Given the lack of replicates, it is difficult to estimate any uncertainty associated with the pKa values obtained. So a robustness study was performed with the distribution coefficient samples as mentioned above in the methods. This accounted for the error and variability associated with any measurement performed by the HPLC instrument and hence can be associated with both the pKa measurements as well as the distribution coefficient measurements. Table 3 below lists the mean absolute deviation (MAD) (Fig. 4) and the standard mean error (Eq. 5) for the three compounds that were subjected to replicate HPLC injections to ensure robustness in the chromatography instrumentation. The authors are confident that every opportunity was taken to ensure accurate sample preparation and data collection.
Equation 4. Mean Absolute Deviation.
Equation 5. Standard Mean Error.
where \(\sigma\) is the standard deviation, n is the total number of samples, X is an individual sample and \(\mu\) is the mean.
As described in the Compound Selection section in Materials in Methods, twenty-three compounds were ultimately investigated in the work. Calculated acid dissociation results are provided in Table 4 along with a standardized compound identifier (beginning with “SAMPL8-X”), the molecular scaffolding structure, molecular weight, the pH range tested for the drug substance, and the R2 (calculated values produced by Kaleidagraph).
From the twenty-three molecules that were tested for pH-solubility to determine pKa, automated logD experiments were successfully conducted for eleven molecules. The eleven that were successful are presented in Table 5 and are composed of four weakly acidic compounds (SAMPL8-1, 3, 5, 6) and seven weakly basic compounds (SAMPL8-7, 9, 10, 12, 14, 16, 17). For the weak acids, Britton-Robinson pH 8 buffer (identified in the table as BR-8) was selected for the aqueous phase to ensure that the drug was in a protonated state, whereby the drug would be in a concentration below saturation for proper partitioning. Similarly, for the weak bases, Britton-Robinson pH 3 buffer (identified as BR-3) was selected for the aqueous phase to ensure the drug was de-protonated for partitioning with the organic phase. In each experiment, the final pH of the aqueous phase was measured, and is included in Table 5 where appropriate. In many cases, either the drug did not adequately dissolve in the organic phase, or the measured concentration was below the limit of quantification. In those instances, logD cannot be calculated from an indeterminate fraction, and is therefore represented by a “- “.
Discussion
Several drug substances initially selected for this study failed to progress through the screening process. This was mainly due to a few factors; the first being that there were multiple compounds for which a calibration curve for the analytical HPLC method could not be established. This was caused by a lack of solubility of these compounds in the solvent used for preparation of standards which meant that a standard curve for measurement of solubility of these compounds could not be applied. An often standardized solvent for dissolving poorly-soluble molecules is dimethyl sulfoxide (DMSO); considered a universal solvent because it can dissolve both polar and non-polar molecules [40]. However, since the melting point of pure DMSO is 19 °C, it can pose a risk when running high throughput experiments near room temperature, as was the case for the work presented here. As a result, the HTE lab at GSK standardizes on a common “backing solvent” consisting of 62.5% acetonitrile, 25% tetrahydrofuran, and 12.5% HPLC-grade water v/v for all high-throughput experiments on the Unchained Labs CM3 platforms. This backing solvent serves multiple purposes in the HTE lab, and is the primary diluent of choice. The use of this backing solvent has proven beneficial in nearly all applications in GSK’s HTE lab, with few exceptions. Using a DMSO-based solution in place of backing solvent would not likely have improved the outcome, since the few compounds that were excluded due to low solubility were not soluble at the lower concentrations of 0.001 mg/mL. One of the goals of this research was to develop a standardized automated approach to measuring the ionization constant and distribution coefficients of a large number of molecules. The utility of the backing solvent selected for this work extends beyond the experiments described here. This solvent mixture is employed in a variety of applications throughout the lab, and is used as the primary diluent for the majority of our experiments, by default. For this reason, we elected not to complicate any aspects of the experimental design by using customized solutions for each individual molecule.
The second reason that some of the molecules from the initial group were rejected was due to an inability to estimate the pKa of the compound due to a lack of trends shown in their respective pH-solubility curves. In other words, across the pH 2–12 range, an ionization state was not observed, indicating that the actual pKa was either outside of the test limits, or that the molecule was indeed a non-ionizable species. The third possible reason for rejecting a drug substance was due to the inability of the compound to dissolve completely in either phase of a bi-phasic mixture. Hence all three of the above-mentioned factors are related to poor solubility of certain compounds under specific conditions, and cause for removal from the study.
High throughput pH-solubility assessment
Accurate measurement of aqueous solubility across a range of pH provides an ideal starting point for ultimately determining the distribution coefficient of a drug substance. Without first knowing the pH-solubility profile, the appropriate pH of the aqueous phase for the aqueous/organic bi-phasic mixture would be in question. The research presented here initially focuses on the development of an automated approach to determine pH-solubility profiles for a variety of drug substances with a wide range of physicochemical properties such as molecular weight, scaffolding, and tendencies for protonation/deprotonation. The experimental designs leveraged several HTE robotic platforms to enable the development of aqueous solubility profiles. Because of the efficiency of these automated platforms, the pH-solubility studies were conducted with minimal demand on resources for the investigators, so it was determined early in the project to include these studies as part of the experimental approach. At the onset of this portion of work, the investigators assumed that specific pH buffers would be required for each drug molecule, with the goal of being at least 3 pH away from the measured pKa in order to ensure that the molecule was fully dissolved in the aqueous phase. However, after the data was collected and analyzed, it was recognized that the distribution coefficient experiments could standardize on either pH 3 or pH 8 buffers as the aqueous phase, depending on the ionic state of each molecule.
Ultimately, twenty-three compounds were successfully measured for pH-solubility using an HTE approach. These included weak acids, weak bases, amphoteric, and (apparently) non-ionizable molecules. The primary goal was to efficiently conduct the experiments with a simplified and standardized design, while also ensuring accurate data capture for the range of Britton-Robinson buffers selected. A primary limitation to consider was a lack of abundant drug substance availability, so it was determined that experiments which utilized a 96-well plate were ideal for this first portion of the study. The limitation of available drug substance also prevented any possibility of running these experiments with replicate samples. Following sample preparation, the vials were mixed for 24 h at room temperature to ensure that full drug saturation was achieved. A standardized analytical HPLC method was developed with the intent of using the same primary method for all drug substances investigated in this study, with the exception of establishing the appropriate wavelength and retention time for each drug substance. The final pH of each sample was collected from the multi-tip pH probe configuration on the Unchained Labs CM3 platform. This automated pH measurement process includes a water bath followed by blow-drying each pH probe in between measurements. It is possible that some error is introduced into the final pH reading, if there remains a small droplet of water on the pH probe when it is being inserted into the 500 µL volume sample. This is likely not to be a considerable introduction of possible error, but it needs to be included as a possible source if one exists.
This experimental approach seemed ideally suited for pKa determination. With solubility data that was collected, ionization constants were computed using Kaleidagraph software [37]. Once established, the ionization constants were then used to confirm at which pH the appropriate aqueous buffer would be selected for the subsequent distribution coefficient experiments. Predicted pKa values are provided by ChemAxon/JChem, and are not based on experimental data, but rather from models that calculate all possible ionization constants based on the molecular structure. Three of the molecules from this set of 23 were selected because of their commonality as matched sets (SAMPL8-7, 8–9, and 8–17). These three weak bases are all benzimidazole scaffolds with molecular weights between 324.2 and 340.2 dalton. One reason for including these three was to ascertain how closely their JChem predicted pKa values align with the experimentally determined pKa values. The JChem predicted pKa values for these three molecules were close together, and averaged 7.56. The experimentally determined pKa’s for these three molecules, as reported in Table 4, average 6.43. The experimentally determined pKa’s were 85% less than the predicted values, and provide support to the decision for measuring the ionization constants rather than relying exclusively on the JChem predicted values. The original intent was to select individual pH buffers as the aqueous media depending on the experimentally determined pKa. However, after evaluation of the complete data set, it was concluded that the distribution coefficient experiments could be conducted with standardized pH buffers in groupings. This resulted in running entire sets of distribution coefficient experiments with either pH 3 or pH 8 buffers. This significantly simplified the experimental process, and conveniently eliminated any additional complexity in the automated design.
Determination of logD Values
The acid dissociation and distribution coefficient measurements prepared for this study were entirely solubility-based. Solubility workflows are easily adaptable to the current automated platforms available for sample preparation and high throughput chromatography for determining drug concentrations in a variety of solutions. Utilizing an HTE approach ensured that a multitude of drug substances and solvent systems could be analyzed in a rapid manner, with limited availability of raw materials. The conventional shake-flask method continues to remain as the gold standard for traditional distribution coefficient measurements, despite the high drug substance demand for experiments involving large volumes of solvents [41, 42]. The automated method presented here for determining logD, has similarities to the shake-flask method yet was performed at a significantly lower volume. However, instead of manually shaking the flask, the sample vials were vortexed for no less than 30 min, and then allowed to reach equilibrium. The traditional shake-flask approach for partition coefficient studies that use octanol and water may sometimes involve pre-saturation of the biphasic systems for 72 h, primarily because water is 20% soluble in octanol [43]. However, due to the high-throughput nature of the experiments presented here and the number of solvent mixtures investigated, our experiments did not pre-saturate all of the solvent combinations. It is possible that the lack of pre-saturated solvents may introduce error in the solubility readings, which should be considered when performing final data analysis. Since traditional experiments typically focus on octanol/water, the benefits of the approach presented in this manuscript include the ability to perform experiments at a smaller scale using glass vials to explore a multitude of solvent combinations, and to allow the SAMPL participants an opportunity to determine if the range of solvent combinations are beneficial to data science and modeling.
The automated approach used here overcomes certain limitations by reducing the time required to prepare and execute the experiment while also providing for an opportunity to create the volume of samples per compound that were desired for this iteration of the SAMPL challenge. This approach extended into the chromatography analysis by way of an autosampler and a high-throughput sequence on the HPLC instrumentation for data collection. Because the experimental design was automated, the goal was to prepare samples that ensured the solute would go completely into solution, thereby avoiding the need for determining mass balance.
The pH of the aqueous phase of bi-phasic mixture was selected according to the pKa of the compound being used. This was done to ensure that the entirety of solid drug substance would go into solution for the analysis of the bi-phasic mixture, to provide for the computation of the distribution coefficient. Typically, logD measurement experiments are performed at a specific pH of interest, such as physiological pH. In this study which aims to create a benchmark dataset for evaluating computational predictions, there wasn’t a need to focus on a particular pH. We had the freedom to select any pH that would make the logD measurements easier and more accurate by ensuring adequate aqueous solubility. If any solid particles were to be found in either phase, it could hinder the chromatography analysis which may contribute to a significant error when computing logD.
Limits of detection
As can be observed in Table 5, there are several instances of data for logD that could not be computed since the logarithm of zero is undefined. This result is determined by the lowest concentration that could be detected on the HPLC instrument. Chromatography instruments are very precise and can calculate an analyte to a high degree of accuracy. However, there are limits of detection (LoD) based on the inherent molar absorptivity of the compounds and the dynamic range of the photo-diode array detector used to analyze the compounds. Additionally, the precision of the balances and pipettes that are used for sample preparation have a role in assessing the LoD. In the experiments presented here, it was determined that any chromatography data that presented an area below 5 milli-absorbance units (mAU), which equates to concentrations at or below 5 µg/mL, could not justifiably be provided.
Experimental design considerations
Given the large number of compounds and experiments that were investigated, standardization of the experimental design was very beneficial wherever possible, given that one of the goals was to utilize high-throughput instrumentation. While this approach enabled experiments to be conducted with a high degree of efficiency, and produced accurate data for analysis, it was noted during the investigation that improvements could be made for future work. The inclusion of replicates for future experiments would be beneficial, since this could establish standard errors associated with either human error or with sample preparation and would allow for statistical data analysis. Further improvements to the experimental design could be accommodated using larger vials to possibly improve dissolution of the samples and utilizing light scattering as a means of monitoring the presence of undissolved particles. To ensure full dissolution of the drug particles in the bi-phasic sample vials, the vials could possibly be mixed for a longer time period, and analyzed at various timepoints, to ensure equilibrium is reached.
Uncertainty analysis
From the robustness study that was performed, it is evident that Mean Absolute Deviation and Standard Mean Error values for the three samples (SAMPL8-16, SAMPL8-17 and SAMPL8-14) that contained replicate measurements were very similar and demonstrated that the measured drug concentrations of those three samples were repeatable. However, it should be noted that these replicates were sampled from the same sample vial, which may imply that the MAD and SME are measures related to the sampling capabilities of the robotic platform rather than the actual samples themselves. To improve upon the uncertainty analysis, replicate sample vials should be prepared, and replicate measures should be drawn from each individual vial.
Conclusions
The investigations described here provide a collection of data intended for use in the SAMPL8 Physical Properties Challenge (https://doi.org/10.5281/zenodo.4245127) [44]. The zenodo link provides a presentation from the SAMPL satellite conference at the 2020 German Conference on Cheminformatics. The presentation describes the automated approaches taken to determine the distribution coefficients and pKa for the set of GSK compounds used in this investigation. This challenge is composed of two distinct components: the pKa challenge and the logD challenge. The data was generated predominantly using high-throughput experimentation platforms and instrumentation. pKa values were determined for 23 compounds, and logD values were determined for 11 compounds in a variety of bi-phasic systems with an Unchained Labs Freeslate CM3 robotic platform and an Agilent 1290 HPLC with auto-sampler. The logD for these compounds was determined using the following bi-phasic mixtures: aqueous-octanol, aqueous-cyclohexane, aqueous-ethyl acetate, aqueous-heptane, aqueous-MEK, aqueous-TBME, and cyclohexane-DMF. Not all combinations of distribution coefficient are available because we experienced compound solubility issues below the limit of detection in several of the different phases which resulted in incalculable distributions due to an undefined logarithm. At the onset of the experimental design, we did not anticipate that some of the solvent combinations would eventually result in incalculable distributions, but the investigators favored the inclusion of any data that could be provided rather than eliminating any solvent combination data series (such as CYHL/BR8) despite the presence of only a single data point being available. There were several integratable peaks in some of the data; however, the limit of detection restricts the authors from publishing those values.
During this work, we determined that several areas for improvement could be implemented to enhance the volume of data collected. Of those, we recognize that two calibration curves – one for the organic, one for the aqueous – would greatly improve the logD calculations by producing appropriate quantification limits on the chromatography instrumentation. Streamlining this process could be realized by employing emerging technologies such as an online mini-LC to reduce sampling time, resulting in the ability to screen more compounds [45]. This process can be further enhanced and automated with the deployment of imaging tools and imaging analysis software packages. In addition, further insight may be gained from future studies if the analytical approach included the use of LC–MS/MS [46]. LC–MS/MS is highly sensitive and selective and can provide insight into the ionization state which may not be possible with the chromatography approach presented here [47].
The experimental data collected could potentially be used in future SAMPL blind prediction challenges as the data sets continue to grow and provide more information that is useful in building accurate and comprehensive drug substance prediction models.
Data availability
The datasets generated during and/or analyzed during the current study are available in the GitHub repository, https://github.com/samplchallenges/SAMPL8. As of the time of this writing, only input data will be available, but at the close of the SAMPL8 challenge, measured values will also be released.
Abbreviations
- OCTL:
-
Octanol
- CYHL:
-
Cyclohexane
- ETAC:
-
Ethyl acetate
- HP:
-
Heptane
- MEK:
-
Methyl ethyl ketone
- TBME:
-
Tert butyl methyl ether
- DMF:
-
Dimethylformamide
- BR:
-
Britton Robinson
- API:
-
Active pharmaceutical ingredient
References
Asli N, Sergio C, Taosheng C (2013) Data analysis approaches in high throughput screening. Drug Discov. https://doi.org/10.5772/52508
Coley CW, Eyke NS, Jensen KF (2020) Autonomous discovery in the chemical sciences Part I: progress. Angew Chem Int Ed Engl 59(51):22858–22893
Rosso V, Albrecht J, Roberts F, Janey JM (2019) Uniting laboratory automation, DoE data, and modeling techniques to accelerate chemical process development. Reac Chem Eng 4(9):1646–1657
Nunn C, DiPietro A, Hodnett N, Sun P, Wells KM (2017) High-throughput automated design of experiment (DoE) and kinetic modeling to aid in process development of an API. Org Process Res Dev 22(1):54–61
Coley CW, Eyke NS, Jensen KF (2020) Autonomous discovery in the chemical sciences Part II: outlook. Angew Chem Int Ed Engl 59(52):23414–23436
Selekman JA, Qiu J, Tran K, Stevens J, Rosso V, Simmons E et al (2017) High-throughput automation in chemical process development. Annu Rev Chem Biomol Eng 8(1):525–547
Fridgeirsdottir GA, Harris R, Fischer PM, Roberts CJ (2016) Support tools in formulation development for poorly soluble drugs. J Pharm Sci 105(8):2260–2269
Bahr MN, Modi D, Patel S, Campbell G, Stockdale G (2019) Understanding the role of sodium lauryl sulfate on the biorelevant solubility of a combination of poorly water-soluble drugs using high throughput experimentation and mechanistic absorption modeling. J Pharm Pharm Sci 22(1):221–246
Rubin AE, Tummala S, Both DA, Wang C, Delaney EJ (2006) Emerging technologies supporting chemical process R&D and their increasing impact on productivity in the pharmaceutical industry. Chem Rev 106(7):2794–2810
Thygs FB, Merz J, Schembecker G (2016) Automation of solubility measurements on a robotic platform. Chem Eng Technol 39(6):1049–1057
Alsenz J, Kansy M (2007) High throughput solubility measurement in drug discovery and development. Adv Drug Deliv Rev 59(7):546–567
Bahr MN, Damon DB, Yates SD, Chin AS, Christopher JD, Cromer S et al (2018) Collaborative evaluation of commercially available automated powder dispensing platforms for high-throughput experimentation in pharmaceutical applications. Org Process Res Dev 22(11):1500–1508
Bahr MN, Angamuthu M, Leonhardt S, Campbell G, Neau SH (2021) Rapid screening approaches for solubility enhancement, precipitation inhibition and dissociation of a cocrystal drug substance using high throughput experimentation. J Drug Deliv Sci Technol 61:102196
Bahr MN, Morris MA, Tu NP, Nandkeolyar A (2020) Recent advances in high-throughput automated powder dispensing platforms for pharmaceutical applications. Org Process Res Dev 24(11):2752–2761
Utsey K, Gastonguay MS, Russell S, Freling R, Riggs MM, Elmokadem A (2020) Quantification of the impact of partition coefficient prediction methods on physiologically based pharmacokinetic model output using a standardized tissue composition. Drug Metab Dispos 48(10):903–916
Selekman JA, Tran K, Xu Z, Dummeldinger M, Kiau S, Nolfo J et al (2016) High-throughput extractions: a new paradigm for workup optimization in pharmaceutical process development. Org Process Res Dev 20(10):1728–1737
Mobley DL, Chodera JD, Isaacs L, Gibb BC (2016) Advancing predictive modeling through focused development of model systems to drive new modeling innovations. In: UC Irvine: Department of Pharmaceutical Sciences U, editor
Isik M, Levorse D, Rustenburg AS, Ndukwe IE, Wang H, Wang X et al (2018) pKa measurements for the SAMPL6 prediction challenge for a set of kinase inhibitor-like fragments. J Comput Aided Mol Des 32(10):1117–1138
Isik M, Levorse D, Mobley DL, Rhodes T, Chodera JD (2020) Octanol-water partition coefficient measurements for the SAMPL6 blind prediction challenge. J Comput Aided Mol Des 34(4):405–420
Nicholls A, Mobley DL, Guthrie JP, Chodera JD, Bayly CI, Cooper MD et al (2008) Predicting small-molecule solvation free energies: an informal blind test for computational chemistry. J Med Chem 51(4):769–779
Skillman AG, Geballe MT, Nicholls A (2010) SAMPL2 challenge: prediction of solvation energies and tautomer ratios. J Comput Aided Mol Des 24(4):257–258
Geballe MT, Guthrie JP (2012) The SAMPL3 blind prediction challenge: transfer energy overview. J Comput Aided Mol Des 26(5):489–496
Geballe MT, Skillman AG, Nicholls A, Guthrie JP, Taylor PJ (2010) The SAMPL2 blind prediction challenge: introduction and overview. J Comput Aided Mol Des 24(4):259–279
Guthrie JP (2014) SAMPL4, a blind challenge for computational solvation free energies: the compounds considered. J Comput Aided Mol Des 28(3):151–168
Mobley DL, Wymer KL, Lim NM, Guthrie JP (2014) Blind prediction of solvation free energies from the SAMPL4 challenge. J Comput Aided Mol Des 28(3):135–150
Muddana HS, Fenley AT, Mobley DL, Gilson MK (2014) The SAMPL4 host-guest blind prediction challenge: an overview. J Comput Aided Mol Des 28(4):305–317
Bannan CC, Burley KH, Chiu M, Shirts MR, Gilson MK, Mobley DL (2016) Blind prediction of cyclohexane-water distribution coefficients from the SAMPL5 challenge. J Comput Aided Mol Des 30(11):927–944
Bannan CC, Calabro G, Kyu DY, Mobley DL (2016) Calculating partition coefficients of small molecules in octanol/water and cyclohexane/water. J Chem Theory Comput 12(8):4015–4024
Di L, Kerns EH (2016) pKa. In: Di L, Kerns EH (eds) Drug-like properties. Academic Press, Boston, pp 51–59
Mobley DL, Guthrie JP (2014) FreeSolv: a database of experimental and calculated hydration free energies, with input files. J Comput Aided Mol Des 28(7):711–720
Arnott JA, Planey SL (2012) The influence of lipophilicity in drug discovery and design. Expert Opin Drug Discov 7(10):863–875
Linkov I, Ames MR, Crouch EA, Satterstrom FK (2005) Uncertainty in octanol-water partition coefficient: implications for risk assessment and remedial costs. Environ Sci Technol 39(18):6917–6922
Schönsee CD, Bucheli TD (2020) Experimental determination of octanol-water partition coefficients of selected natural toxins. J Chem Eng Data 65(4):1946–1953
Isik M, Bergazin TD, Fox T, Rizzi A, Chodera JD, Mobley DL (2020) Assessing the accuracy of octanol-water partition coefficient predictions in the SAMPL6 Part II log P Challenge. J Comput Aided Mol Des 34(4):335–370
Avdeef A (2012) Solubility. In: Avdeef A (ed) Absorption and drug development. Wiley, New York, pp 251–318
Po HN, Senozan NM (2001) The Henderson-Hasselbalch equation: its history and limitations. J Chem Educ 78(11):1499
Jagannadham V, Sanjeev R (2012) Playing around with “Kaleidagraph” program for determination of pKa values of mono, di and tri basic acids in a physical-organic chemistry laboratory. Creat Educ 3(3):380–382
Settimo L, Bellman K, Knegtel RMA (2014) Comparison of the accuracy of experimental and predicted pKa values of basic and acidic compounds. Pharm Res 31(4):1082–1095
Reijenga J, van Hoof A, van Loon A, Teunissen B (2013) Development of methods for the determination of pKa values. Anal Chem Insights 8:53–71
Bavishi DD, Borkhataria CH (2016) Spring and parachute: HOW cocrystals enhance solubility. Prog Cryst Growth Charact Mater 62(3):1–8
Poulsen CE, Wootton RC, Wolff A, deMello AJ, Elvira KS (2015) A microfluidic platform for the rapid determination of distribution coefficients by gravity-assisted droplet-based liquid-liquid extraction. Anal Chem 87(12):6265–6270
Matter H. Drug Design Strategies: Quantitative Approaches. Edited by David J. Livingstone and Andrew M. Davis. ChemMedChem. 2012;7(7):1295–6.
Montalbán MG, Collado-González MM, Trigo R, DíazBaños FG, Víllora G (2015) Experimental measurements of octanol-water partition coefficients of ionic liquids. J Adv Chem Eng 5:1000133
Nandkeolyar A, Bahr M (2020) Automated high throughput pKa and distribution coefficient measurements of pharmaceutical compounds for SAMPL8 Blind Prediction Challenge: Zenodo. https://doi.org/10.5281/zenodo.4245127
Pham M, Foster SW, Kurre S, Hunter RA, Grinias JP (2021) Use of portable capillary liquid chromatography for common educational demonstrations involving separations. J Chem Educ 98(7):2444–2448
Ediage EN, Aerts T, Lubin A, Cuyckens F, Dillen L, Verhaeghe T (2019) Strategies and analytical workflows to extend the dynamic range in quantitative LC–MS/MS analysis. Bioanalysis 11(12):1187–1204
Page JS, Kelly RT, Tang K, Smith RD (2007) Ionization and transmission efficiency in an electrospray ionization—mass spectrometry interface. J Am Soc Mass Spectrom 18(9):1582–1590
Acknowledgements
We appreciate the National Institutes of Health for its support of the SAMPL project via R01GM124270 to David L. Mobley (UC Irvine). The authors would also like to acknowledge Lisa McQueen and Alan Graves (formerly of GSK) for their contributions during the early stages of establishing the partnership, collecting the materials necessary for the study, and providing chemometrics insight. Lastly, we acknowledge the guidance and support from Kenneth Wells of GSK.
Author information
Authors and Affiliations
Contributions
This manuscript was written through the contributions from each of the authors MNB, AN. Each author has given approval to the final version of the manuscript.
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bahr, M.N., Nandkeolyar, A., Kenna, J.K. et al. Automated high throughput pKa and distribution coefficient measurements of pharmaceutical compounds for the SAMPL8 blind prediction challenge. J Comput Aided Mol Des 35, 1141–1155 (2021). https://doi.org/10.1007/s10822-021-00427-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10822-021-00427-0